 Turn-on mode diarylethenes for bioconjugation and fluorescence microscopy of cellular structures
Turn-on mode diarylethenes for bioconjugation and fluorescence microscopy of cellular structures
Proceedings of the National Academy of Sciences of the United States of America


Contributed by Stefan W. Hell, February 22, 2021 (sent for review January 5, 2021; reviewed by Jean-Marie P. Lehn and Atsushi Miyawaki)
Author contributions: M.L.B., M.I., V.N.B., and S.W.H. designed research; K.U., A.A., and M.L.B. performed research; K.U., A.A., M.L.B., M.I., and V.N.B. analyzed data; and K.U., A.A., M.L.B., M.I., and V.N.B. wrote the paper.
Reviewers: J.-M.P.L., University of Strasbourg; and A.M., RIKEN Center for Brain Science.
1K.U. and A.A. contributed equally to this work.

- Altmetric
In superresolution fluorescence microscopy, employing synthetic dyes that can be reversibly photoswitched between a nonfluorescent (“dark”) and a fluorescent (“bright”) state has been an attractive alternative to using photoswitchable fluorescent proteins. However, employing such synthetic dyes has been elusive because they have defied reliable attachment to proteins and required UV light for photoswitching. Here we prepared “turn-on mode” fluorescent diarylethenes (fDAEs) that are switchable with visible rather than UV light and blink between a bright fluorescent and a dark state in aqueous buffers. Moreover, our thienyl-substituted fDAEs effectively labeled two thiol groups on nanobodies bearing a single maleimide tag. With these small-sized probes, we acquired superresolution images of vimentin filaments in cells by applying just yellow (561 nm) light.
The use of photoswitchable fluorescent diarylethenes (fDAEs) as protein labels in fluorescence microscopy and nanoscopy has been limited by labeling inhomogeneity and the need for ultraviolet light for fluorescence activation (on-switching). To overcome these drawbacks, we prepared “turn-on mode” fDAEs featuring thienyl substituents, multiple polar residues, and a reactive maleimide group in the core structure. Conjugates with antibodies and nanobodies displayed complete on-switching and excitation with violet (405 nm) and yellow-green (<565 nm) light, respectively. Besides, they afforded high signal-to-noise ratios and low unspecific labeling in fluorescence imaging. Irradiation with visible light at 532 nm or 561 nm led to transient on-off switching (“blinking”) of the fDAEs of double-labeled nanobodies so that nanoscale superresolution images were readily attained through switching and localization of individual fluorophores.
Diarylethenes (DAEs) are photochromic molecules that reversibly switch between two stable states upon irradiation with light. Their fatigue-resistant and efficient photoswitching reactions involve the so-called open-form (OF) and closed-form (CF) isomers (SI Appendix, Scheme S1). These isomers are thermally stable, and their absorption bands are well separated, so that the intermittent irradiation with two distinct wavelengths results in a full transition to each state (1234–5).
The combination of photoswitching and fluorescence is an attractive yet uncommon feature in synthetic organic molecules. Therefore, merging photochromic and fluorescent properties in a single entity attracted a lot of attention over past decades (2, 678–9). Photoresponsive fluorescent DAEs (fDAEs) are particularly advantageous in life (101112131415–16) and material science (171819202122–23) applications. For example, fDAEs are very promising on−off switching fluorophores for superresolution fluorescence microscopy or nanoscopy, as these methods rely on transitions between dark (off) and fluorescent (on) states for distinguishing neighboring fluorescent molecules at subdiffraction length scales (<200 nm) (242526–27). A crucial factor is the way the optical transitions between the on and off states are induced, controlled, and detected. Therefore, it is not surprising that the advent of fluorescence nanoscopy bolstered the interest in novel switchable fluorophores. Fortunately, synthetic organic chemistry allows creating “tailor-made” compounds with novel features that set the pace in this rapidly growing field.
As most photochromic compounds (PCs) have nonfluorescent isomeric states, early approaches toward fluorescence switching were based on binding a PC, in a dyad style, with a fluorescent molecule. If the photochromic and fluorescent parts are connected via linkers, the emission is modulated via fluorescence resonance energy transfer (17, 28, 29) or photoinduced electron transfer (30) in parallel with the progress of the photochromic reaction. This molecular design is rather complex and leads to turn-off fluorescence switches; note that the initial state (OF) is fluorescent. More recently, the photochromic and fluorescent parts were combined in one core, and these new fDAEs boosted the development of synthetic probes for nanoscopy. They have much simpler structures based on “oxidized” benzothiophene units and were called “turn-on mode” fDAEs, because their initial state (OF) is nonfluorescent (31323334–35). Besides, they possess two outstanding properties: an emissive CF with high brightness (product of the fluorescence quantum yield and absorption coefficient at the excitation wavelength) and a large Stokes shift, which are rarely found in combination. Recently, we reported turn-on mode water-soluble fDAEs decorated with numerous carboxylic acid residues and demonstrated their applications in fluorescence nanoscopy, particularly in the methods called reversible saturable optical linear fluorescent transitions (36, 37) and STORM (stochastic optical reconstruction microscopy) (38).
The drawbacks limiting the performance of current turn-on mode fDAEs in bioimaging and nanoscopy are 1) the need for phototoxic and background-generating ultraviolet (UV) light for the on-switching (365 nm), as well as blue light for excitation and off-switching (488 nm) and 2) unspecific bioconjugation in which only in situ and unselectively generated N-hydroxysuccinimidyl (NHS) esters are applied (3637–38). The latter drawback is particularly challenging, because it requires redesign of the structure and synthesis.
Here we introduce thienyl-substituted, red-emitting fDAEs bearing a single maleimide tag at the central reactive carbon atom and six carboxylic acid residues attached to primary amides (Scheme 1 and SI Appendix, Scheme S1). Only the ring closure reaction is induced with 405-nm light. The closed-ring isomers are highly fluorescent under irradiation with blue to green light and afford large Stokes shifts. The presence of thienyl groups dramatically enhances the Urbach tail effect (39), which allows single molecule on−off “blinking” with only one irradiation source (<565 nm). The single reactive group (maleimide) simplifies bioconjugation and allows control of the degree of labeling (DOL). We decorated nanobodies with two dye residues, applied bioconjugates as photoswitchable markers in confocal and STORM superresolution microscopy (26, 38), and traced the effect of their size on the imaging of the sample at the nanoscale.

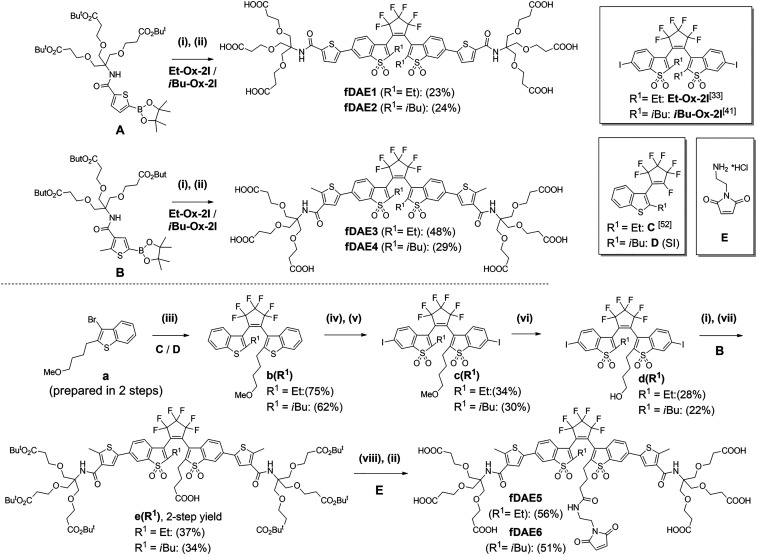
Synthesis of symmetric (fDAE1-4) and asymmetric hexa-carboxylic acids (fDAE5-6) with a maleimide group. Reagents and conditions: (i) Pd(dba)2, Sphos, K2CO3, aqueous (aq.) THF, reflux, 30 min; (ii) CF3CO2H/CH2Cl2 (1/1, vol/vol), room temperature (r.t.), 1 h; (iii) nBuLi, −90 °C to r.t., 12 h; (iv) 30% aq. H2O2, CH3COOH, reflux, 1 h; (v) I2/H5IO6, concentrated H2SO4, 0 °C, 30 min; (vi) BBr3, DCM, −10 °C to r.t. 24 h; (vii) TEMPO/BAIB, ACN/H2O (1/1, vol/vol), r.t., 30 min; and (viii) TSTU/DIPEA, r.t., 10 min, then E/DIPEA, r.t., 30 min.
Results and Discussion
Dye Design and Synthesis of Thienyl-Substituted fDAEs.
We reported 1,2-bis(2-ethyl-1-benzothiophen-3-yl)perfluorocyclopentenes with oxidized and nonoxidized benzothiophene cores, as well as additional thiophene rings (one or two) attached to C-6 and/or C-6′ in a symmetric or asymmetric fashion (33, 40). The oxidized compounds with thienyl substituents underwent ring closure reactions to produce fluorescent CFs upon irradiation with UV (365 nm, 405 nm) as well as visible (470 nm) light. Remarkably, the on−off switching with visible light—alternating blue (470 nm) and green (530 nm) irradiation—demonstrated the Urbach tail effect (40). To impart water solubility and improve the photofatigue resistance, we incorporated the branched linkers with terminal carboxylic acids into the structures of fDAEs (3637–38, 41). The photostability in aqueous solutions was observed for primary amides with N-tert-alkyl-substitutes capped with polar groups and attached to the α- or β-positions of the thienyl groups (41). Methyl (Me), ethyl (Et), and isobutyl (iBu) groups were introduced to the central carbon atoms (C-2 and C-2′), and the influence of these groups on the emission efficiency and the cyclization/cycloreversion reactions was studied (35, 41). First, we prepared four symmetric fDAE1-4 (Scheme 1). Thienyl boronic esters A and B having N-(tert-alkyl)carboxamide moieties at α- and β-positions were synthesized (see SI Appendix, Scheme S2) and coupled by a Suzuki−Miyaura reaction with aromatic diiodides Et-Ox-2I (33) or iBu-Ox-2I (41). Deprotection of the intermediate tert-butyl esters with trifluoroacetic acid (TFA) gave symmetric hexa-carboxylated fDAE1-4. Based on the photophysical properties (see the next section), we used fDAE3 and fDAE4 as prototypes for the design of asymmetric fDAE5 and fDAE6 decorated with the maleimide residue which reacts with thiol groups (e.g., cysteine moieties in proteins not forming disulfide bonds) (42). Asymmetric precursors b(R1) (R1= Et or iBu) with methoxyalkyl linkers were prepared from a and compounds C and D. The nonfluorescent DAEs b(Et) and b(iBu) were converted to fluorescent compounds c(Et) and c(iBu) by oxidation to bis(benzothiophene S,S-dioxides) with aqueous hydrogen peroxide in refluxing acetic acid followed by diiodination at positions 6/6′ with I2/H5IO6 in sulfuric acid. Then the methoxyalkyl group in compounds c(Et) and c(iBu) was cleaved by treating with excess of BBr3. Next, the obtained hydroxyalkyl compounds d(Et) and d(iBu) (with aromatic iodide sites) were coupled with thienyl boronic ester (B) by a Suzuki−Miyaura reaction. After that, the hydroxyalkyl groups were oxidized to carboxylic acids by using the 2,2,6,6-Tetramethylpiperidyl-1-oxyl/(Diacetoxyiodo)benzene (TEMPO/DAIB) reagent (43), and intermediates e(Et) and e(iBu) were isolated in moderate yields. The maleimide-containing residue was introduced via the coupling of the in situ generated NHS ester N,N,N',N'-tetramethyl-O-succinimidyluronium tetrafluoroborate/diisopropylethylamine (TSTU/DIPEA) with amine E. Finally, the cleavage of tert-butyl esters with TFA gave the target compounds (fDAE5 and fDAE6; see Scheme 1 and SI Appendix, Scheme S3) in good yields (see full procedures and NMR spectra in SI Appendix, Figs. S1–S22).
Photophysical Characterization.
The photophysical properties of fDAEs (Table 1) were studied in acetonitrile and buffered aqueous solutions (phosphate buffer, 100 mM, pH = 7), under irradiation with violet (405 nm) and green (505 nm or 530 nm) light. The absorption spectra of OF and CF and normalized fluorescence spectra of CF are shown in Fig. 1A and SI Appendix, Fig. S28. In fDAE1-2, the electron-accepting amido groups are in the α-positions of the thiophene rings. Compounds fDAE3-6 with these groups as β-substituents and the donor methyl groups as α-substituents have the red-shifted absorption and emission bands. The changes are more pronounced in the CFs. The CFs of the α- and β-substituted (in respect of the amido groups) thieno-fDAEs may be readily excited with 532- and 561-nm lasers, respectively. Importantly, the cycloreversion quantum yields of the β-substituted dyes with α-methyl groups were particularly low (10−4 to 10−6), which is advantageous if the single-molecule detection is required (44).

| λmaxabs [nm]/ε × 10−3 [M-1cm−1]* | λmaxem λfl,max[nm] | Φfl† CF | Φisom/MeCN | Φisom/Aq. buffer | Φfl/ΦCF→OF | |||||
| OF* | CF | CFΦOF→CFc | MeCN | Aq. buffer | OF→CFΦOF→CF‡ | CF→OFΦCF→OF§ | OF→CFΦOF→CF‡ | CF→OFΦCF→OF§ | Aq. buffer | |
| fDAE1 | 360/27.9 | 487/50.0 | 578 | 0.63 | 0.36 | 1.0 × 10−2 | 1.0 × 10−5 | 2.7 × 10−3 | 1.0 × 10−5 | 34,500 |
| fDAE2 | 360/30.6 | 491/54.3 | 575 | 0.61 | 0.37 | 5.3 × 10−3 | 2.5 × 10−4 | 2.7 × 10−3 | 2.9 × 10−4 | 1,270 |
| fDAE3 | 369/19.3 | 504/43.7 | 608 | 0.65 | 0.34 | 9.2 × 10−4 | 3.3 × 10−6 | 2.9 × 10−4 | 3.8 × 10−6 | 88,400 |
| fDAE4 | 368/23.6 | 506/52.8 | 605 | 0.59 | 0.33 | 1.1 × 10−3 | 5.2 × 10−5 | 3.0 × 10−4 | 5.3 × 10−5 | 6,180 |
| fDAE5 | 370/19.1 | 503/41.9 | 606 | 0.48 | 0.29 | 4.6 × 10−4 | 1.1 × 10−5 | 4.0 × 10−4 | 4.3 × 10−6 | 67,700 |
| fDAE6 | 372/25.1 | 505/49.6 | 606 | 0.51 | 0.27 | 1.2 × 10−3 | 1.9 × 10−5 | 3.2 × 10−4 | 1.6 × 10−5 | 17,400 |
* Lowest energy absorption peak.
† Fluorescence quantum yield.
‡ Measured at 405 nm or 365 nm.
§ Measured at 505 nm or 530 nm (see SI Appendix for details).
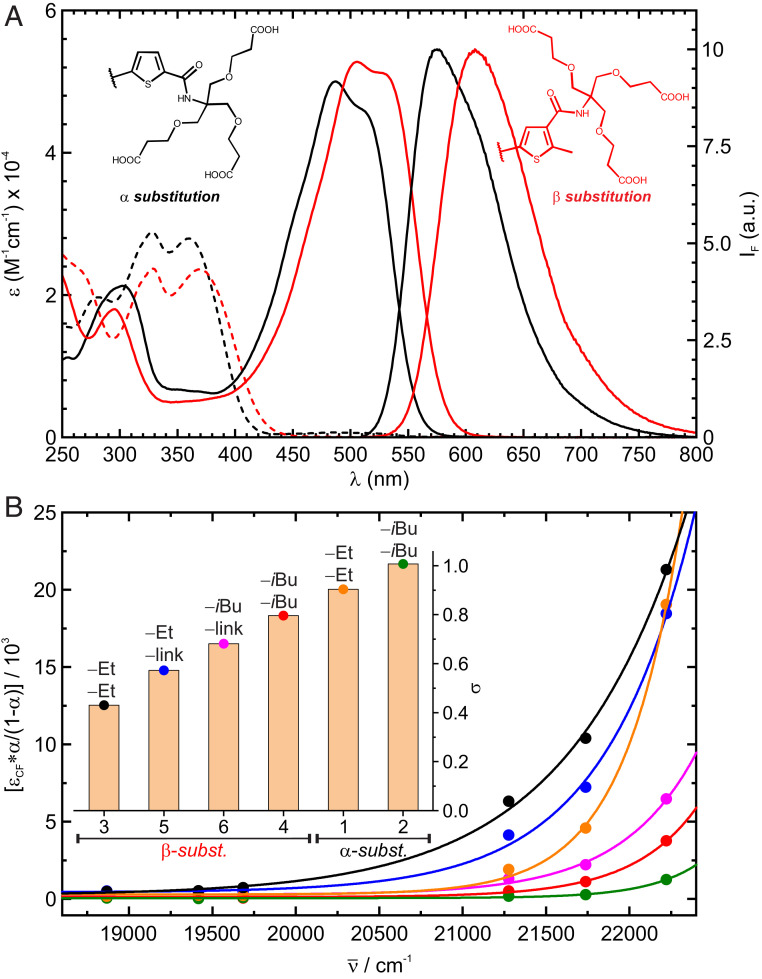
(A) Absorption spectra of fDAE1 (black) and fDAE4 (red) in OF (dashed lines) and CF (solid lines), as well as substitution pattern of compounds and emission of the CF in acetonitrile. (B) Conversion (α/1-α) through Urbach tail excitation of the OFs with the corresponding fit according to Eq. 4. (Inset) Calculated steepness parameters (σ); the substituents at the central positions (as in Scheme 1 and Fig. 1) are indicated.
When solutions of the compounds containing only the OF were exposed to blue to green light (i.e., 450 nm to 530 nm), a photoequilibrium containing a relatively large fraction of the CF was reached, although only the CF absorbs in this region. This indicates the Urbach tail effect: a cyclization reaction induced by absorption of light by weak “hot” bands of the OF (39). To study this effect, a solution of each compound was irradiated with light of different energies, until the corresponding photostationary state (PSS) was reached. Total conversion, proven by high performance liquid chromatography (HPLC) measurements, was observed under irradiation with 405 nm, or 365 nm for compounds fDAE1 and fDAE2. Then the conversion to the CF at each PSS () was calculated as the ratio of the absorbances at the maximum of the CF observed by irradiation at and 405 nm or 365 nm,
To avoid the multiplication of errors in the calculation of isomerization quantum yields (, ), and absorption coefficients (, ), Eq. 2a was rearranged into Eq. 2b and combined with Eq. 3 to obtain Eq. 4, where all constants are combined in the amplitude parameter A′ under the assumption that and do not depend on the irradiation wavelength. Thus, we can expect an exponential dependency between the left term in Eq. 4 and the energy of the irradiation light,
Bioconjugation and Fluorescence Microscopy.
The conjugation of water-soluble polycarboxylated fDAEs with proteins was previously reported (3637–38). Using fDAE1-4 in an excess (ca. 5 eq to 10 eq of dye per protein) afforded antibodies with DOL (dye-to-protein ratios) broadly distributed in the range one to four (Fig. 2A and SI Appendix, Fig. S23 and Table S1). Thiol-reactive maleimides fDAE5 and fDAE6 can react with proteins incorporating a certain number of cysteine residues, which enables site-specific conjugation and defined degrees of labeling. To test the utility of this approach, we selected nanobodies containing two cysteine residues. Nanobodies are particularly challenging as recognition units, due to the small size with respect to the fluorescent dyes, while the cysteine−maleimide strategy has been reported to result in better-preserved epitope recognition (46). A lower excess of fDAEs (ca. 1.5 eq to 2 eq per cysteine residue) afforded efficient conjugation and easier separation of unreacted dyes from the conjugates. All nanobodies labeled with compounds fDAE5 and fDAE6 retained high specificity against the corresponding targets. The method resulted in bioconjugates with a defined DOL value of two (Fig. 2B and SI Appendix, Fig. S24).
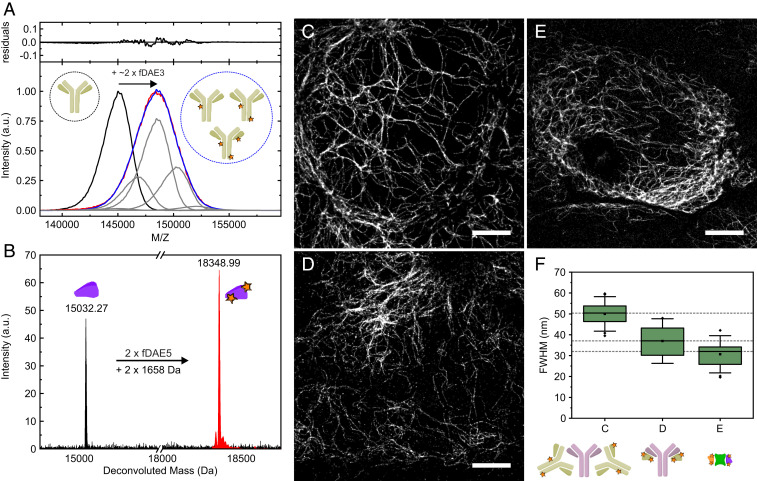
Mass analysis and imaging applications of antibodies and nanobodies labeled with fDAEs. (A) Matrix-assisted laser desorption ionization mass spectrometry analysis of a polyclonal secondary antibody labeled with fDAE3. (Lower) The spectra of the unlabeled antibody (black) and the labeled one (red) are shown along with a fit (blue) to a distribution of the shifted curves (representing the addition of dye molecules) with various amplitudes (gray). (Upper) The residuals of the fit. (B) Electrospray ionization mass spectrometry of an anti-GFP nanobody (NBg2a) containing two cysteine residues. The spectra of the unlabeled nanobody (black) and the labeled one (red) are presented, and the molecular mass of the dye (fDAE5) is indicated. (C–E) Superresolution images of U2OS cells expressing rsEGFP2 in vimentin and labeled with a primary and a secondary antibody (C), a primary antibody and a secondary (NBr) nanobody (D), and two anti-GFP nanobodies (NBg2a and NBg2b, 1:1 mixture, in E). The primary antibody used in C and D is the same. (F) (Upper) Average widths of single filaments; each boxplot was calculated from 20 independent measurements. Boxes show the first and third quartiles of data values; the line and the square show the median and the mean values, respectively. Error bars cover the mean value plus and minus 2 times the SD. (Scale bars, 4 μm.) In panels A, B, and F, the schematic representations of the antibodies, nanobodies, GFP, and the fluorescent labels are shown.
The staining and imaging suitability of the prepared bioconjugates was first assessed by confocal microscopy (SI Appendix, Fig. S25). Images were acquired upon activation at 405 nm and excitation with a 561-nm laser. The activation/excitation sequence was applied pixel by pixel. In a wide-field microscope, irradiation with a green laser (532 nm or 561 nm) led to sparse activation of single molecules via Urbach tail absorption (Movie S1). The activation sufficed to acquire thousands of frames, after which the 405-nm activation light was switched on to speed up acquisition (SI Appendix, Fig. S26). Fluorescence nanoscopy images of tubulin filaments stained with all prepared bioconjugates are presented in SI Appendix, Fig. S27. Remarkably, the samples were mounted in phosphate-buffered saline, without any additives. Compounds fDAE3 and fDAE5 were imaged with a 561-nm laser, but compounds fDAE1-2, fDAE4, and fDAE6 required illumination with a 532-nm laser to achieve a reasonable activation rate and thus an acceptable imaging time. These results are in accordance with the tendencies observed for the steepness parameters (Fig. 1B) measured in bulk experiments.
To assess the performance of fDAE in bioconjugates, we measured the width of single vimentin filaments. Compounds fDAE3 and fDAE5 with stronger Urbach tail effect (Fig. 1B) and higher expected number of photons (Table 1) were chosen to provide a quantitative comparison of three different staining strategies on U2OS cells expressing rsEGFP2 in vimentin: primary/secondary antibodies, primary antibody/secondary nanobodies, and GFP/anti-GFP nanobodies (Fig. 2). To this end, we evaluated and compared the apparent optical resolution observed in each strategy by comparing the widths of vimentin filaments (Fig. 2 C–F), calculated from Gaussian fits (average of 20 single filaments). We achieved a clear reduction in the apparent size of single filaments from 51 nm with antibody complexes to 37 nm with antibody/nanobody complexes, and further to 31 nm with fluorescent protein/nanobody complexes. This improvement did not correlate with any noticeable changes in image quality and is not due to different localization accuracy, as the three images in Fig. 2 have approximately the same mean number of photons per burst (∼450). The width of vimentin filaments devoid of decorations was measured by electron microscopy to be 10 nm (47).
Conclusions
We designed and synthesized photoswitchable fDAEs exhibiting bright orange to red emission and applied their bioconjugates in fluorescence microscopy. The synthesis is based on asymmetric functionalization and introduction of a single reactive maleimide tag to the central fDAE core which is independent from the surrounding polar groups. We succeeded to control bioconjugation and prepared fDAE−nanobody assemblies with a defined DOL, specifically of two. Importantly, the conjugates retained the fluorescence and photoswitching properties of the fDAEs, as well as the affinity and specificity of nanobodies. With these conjugates, we successfully targeted primary antibodies (against tubulin and vimentin) and the fluorescent protein rsEGFP2 (expressed on vimentin). The fDAEs underwent photoswitching in aqueous solutions, under irradiation with violet light (405 nm) and visible light (>450 nm), without addition of thiols and blinking buffers, which are essential for the popular cyanine dyes routinely used for superresolution STORM. By contrast, our fDAEs do not require strict chemical control of the imaging conditions (48, 49). The Urbach tail absorption of the OFs allowed us to apply just a 532- or 561-nm laser in order to induce single-molecule photoswitching (blinking) and acquire superresolution images. The apparent width of tubulin and vimentin filaments correlated with the size of the fluorescent assembly. Our results also highlight the advantages of small-sized bioconjugates prepared from functional fDAE under strict control of the DOL. Last but not least, our findings have also implications for synthesizing novel photoswitchable fluorophores for emerging MINFLUX (MINimal photon FLUX) nanoscopy (5051–52).
Materials and Methods
Abbreviations, full descriptions of the synthesis, compounds’ spectra, bioconjugation procedures, fluorescence microscopy, and image analysis are given in SI Appendix.
Acknowledgements
This work was supported by Bundesministerium für Bildung und Forschung (Germany), Grant FKZ 13N14122. We thank J. Bienert (MPI-BPC), Dr. H. Frauendorf, Dr. M. John, and coworkers (Institut für Organische und Biomolekulare Chemie, Georg-August-Universität, Göttingen, Germany) for recording mass and NMR spectra. We thank the Mass Spectrometry Core Facility in MPI for Medical Research (Heidelberg) for recording mass spectra of proteins. We thank Prof. S. Jakobs (MPI-BPC) for kindly providing the genetically modified (rs-EGFP2) cell line.
Data Availability
All study data are included in the article and/or supporting information.
References
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51