
Edited by Zachary Fisk, University of California, Irvine, CA, and approved January 7, 2021 (received for review December 9, 2020)
Author contributions: L.V.P. and S.K. designed research, performed research, analyzed data, and wrote the paper.
2L.V.P. and S.K. contributed equally to this work.
Second-order phase transitions in solids occur due to spontaneous symmetry breaking with an order parameter continuously emerging from the disordered high-temperature phase. In some materials, the phase transitions are clearly detected in thermodynamic functions (e.g., specific heat), but the microscopic order parameters remain “hidden” from researchers, in some cases for decades. Here, we show how such hidden-order parameters can be unambiguously identified and the corresponding ordered phase fully described using a first-principles many-body linear response theory. Considering the canonical “hidden-order” system neptunium dioxide, we also identify an unconventional mechanism of spontaneous multipolar exchange striction that induces an anomalous volume contraction of the hidden-order phase in NpO2.
The nature of order in low-temperature phases of some materials is not directly seen by experiment. Such “hidden orders” (HOs) may inspire decades of research to identify the mechanism underlying those exotic states of matter. In insulators, HO phases originate in degenerate many-electron states on localized f
Spontaneous symmetry breaking, or a phase transition, in extended systems is associated with emergence of a macroscopic order parameter, which is the statistical average over some physical observable. In some systems, the onset of order is clearly observed in the behavior of thermodynamic functions; however, the order parameter is not detectable by standard probes like neutron scattering or magnetic susceptibility measurements. In metals, such phenomena are typically associated with strongly correlated heavy-fermion behavior, as in the case of enigmatic URu2Si2 (12–3). In correlated insulators, hidden order (HO) phases typically originate in high-rank multipolar degrees of freedom on localized -and -electron shells. It was realized a long time ago (4) that in correlated magnetic insulators with strong spin-orbit coupling, apart from the ordinary Heisenberg interaction between localized spins, there may also exist intersite interactions coupling higher-order spin operators (magnetic multipoles). If those interactions are essentially large, this might lead to a new state of matter—a multipolar order without any associated magnetic order (56–7). Such purely multipolar orders have been observed in various -electron (2, 8910–11) and transition metal-correlated systems (1213–14). Owing to numerous competing order parameters and small relevant energy scales, quantitative description of the HO represents a formidable challenge for theory. First-principles approaches to HO are typically restricted to simulations of a few likely phases inferred experimentally and do not attempt to explore the full space of possible multipolar orders (1, 15).
Difficulties of identifying the physical order parameter in a vast phase space of possible HOs are exemplified by the case of cubic NpO2 (8). A sharp second-order phase transition at 26 K was detected in NpO2 more than a half-century ago (16), with no evidence for underlying magnetic order and structural transformations (1718–19), apart from a small anomalous contraction of the cubic unit cell volume observed (20) below . At the same time, NMR measurements (21) detect two inequivalent oxygen sites in the unit cell below , due to lowering of the cubic symmetry from high-temperature to by a longitudinal order of Np quadrupoles. However, a primary quadrupolar order parameter initially suggested (22) is excluded since muon spin-rotation measurements detect a nonzero magnetic density (23). Moreover, the crystal-field (CF) ground-state (GS) quadruplet is split in the HO phase into two singlets and a doublet, suggesting a time-odd primary order parameter (19, 24, 25). There is a multitude of possible high-rank odd order parameters realizable within the = 9/2 ground-state multiplet of Np4+ ( = 3, 5, 7; i.e., octupolar, triakontadipolar, etc.). A lot of effort has been directed over the last two decades (21, 23, 24, 262728–29) to identify a possible primary order able to reconcile various experimental facts. In particular, assumption of a triakontadipolar antiferromagnetic (AF) 3 order of the symmetry was shown (24, 25, 30) to lead to the best agreement with X-ray scattering and inelastic neutron scattering (INS) spectra. The same hypothesis is also strongly supported by estimates of the relative strength of odd multipole moments on the Np shell in the presence of CF splitting (24, 31).
Although there is substantial experimental evidence to support the 3 rank 5 order in NpO2, the mechanism of its formation is still not well understood. The simplest possible form of the superexchange Hamiltonian, consisting of diagonal nearest-neighbor (NN) interactions between three triakontadipoles and between three dipole moments, has been employed in analysis of low-temperature susceptibility and INS data (24, 25). The full structure of this Hamiltonian cannot be extracted from experiment due to a large number of possible superexchange interactions (SEIs). The measured CF splitting of 55 meV between the ground state and first exited CF level (8, 9, 32) is much larger than , suggesting SEI between states on neighboring Np ions as the origin of its exotic ordered phase. Such low-energy superexchange Hamiltonian has not been so far derived theoretically. Previous theoretical density functional theory (DFT) + U studies have confirmed the stability of a triakontadipolar order in NpO2 (15, 33); however, they imposed an initial symmetry breaking consistent with the 3 rank 5 order inferred experimentally.
In this work, we apply a first-principles many-body framework to the problem of “hidden” multipolar orders in correlated insulators as exemplified by NpO2. It consists of evaluating the full low-energy superexchange Hamiltonian from an ab initio description of the symmetric paramagnetic phase. We start with charge self-consistent DFT + dynamical mean-field theory (DMFT) (3435–36) calculations for paramagnetic NpO2 treating Np 5 within the quasiatomic Hubbard-I (HI) approximation (37) (this method is abbreviated below as DFT + HI) to obtain its electronic structure and the composition of a CF-split Np 5 shell. A force theorem within HI (FT-HI) approach (38) is subsequently employed to derive SEIs between the calculated CF ground-state quadruplets. In summary, we perform an ab initio electronic structure calculation of a complete superexchange Hamiltonian for high-rank magnetic multipoles in an -electron crystalline material. By solving this Hamiltonian, we find a 3 rank 5 primary magnetic multipolar order (MMO) accompanied by a secondary longitudinal quadrupole order. The calculated time-odd splitting of and magnetic excitation spectra are in a good agreement with experiment. The lattice contraction effect (20) is shown to be not related to quadrupole ordering as suggested before (39) but rather, to stem from the volume dependence of leading time-odd SEIs. This multipolar “exchange spring” effect is quite unique and has not been, to our awareness, discussed previously in the literature. Overall, we show that within our first-principles scheme, which treats all competing order parameters on the equal footing, high-rank multipolar orders in correlated insulators can be captured both qualitatively and quantitatively.
We start with evaluating the Np CF splitting in paramagnetic NpO2; as discussed above, this splitting determines the space of low-energy states forming the MMO. The CF splitting of the Np ground-state multiplet = 9/2 calculated by DFT + HI is shown in Fig. 1A. The ground-state quartet is separated from another excited quartet by 68 meV, in agreement with the experimental range for this splitting, 30 to 80 meV, inferred from INS measurements (19, 32, 40). The broad experimental range for the excited energy is due to the presence of a dispersive phonon branch in the same range [this overlap has been a major source of difficulties for the phenomenological analysis of NpO2 in the framework of effective CF model model (8)]. The third exited level, doublet, is located at much higher energy above 300 meV (see Fig. 1A).

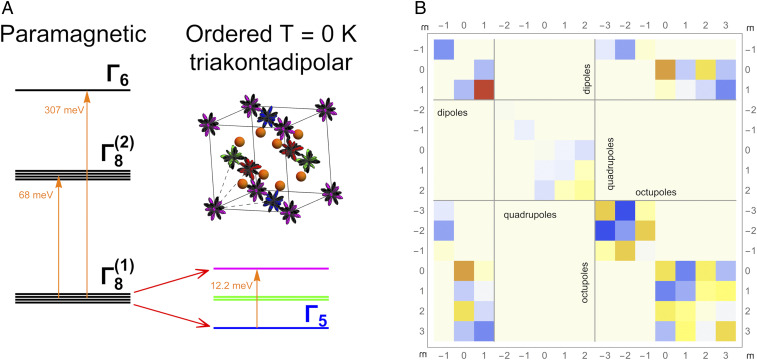
Np 5 on-site splitting and Np–Np intersite interactions in NpO2. (A) Calculated CF spiting of Np J = 9/2 multiplet in paramagnetic state (Left) and exchange splitting of the ground-state quartet in the predicted ordered phase at zero temperature (Right). Note the energy rescaling for the level on the CF splitting plot. (Inset) The crystal structure of the NpO2. Positions of the Np atoms are surrounded by the polar plot of the calculated primary order parameter (triakontadipoles) in the ground state. Different colors indicate four nonequivalent Np positions in the ordered state. The orange spheres are oxygen atoms. (B) SEI matrix between NN Np in NpO2. These SEIs couple multipolar operators in the = 3/2 space of the CF ground state. Their values in the local coordinate system (with z axis directed along the given bond direction and y axis along the edge of the face-centered cubic lattice) are presented as a temperature map, with the warm and cool colors representing antiferromagnetic and ferromagnetic coupling of the corresponding multipoles, respectively.
Our calculated CF corresponds to = −0.54 parameterizing the relative magnitude of the four- and six-order contributions to the cubic CF (41). Our values are in good agreement with = −0.48 inferred from INS measurements (19) and analysis of CF excitation energies along the actinide dioxide series (31). The CF-level energies calculated within DFT + HI are also in good agreement with previous DFT + DMFT calculations by Kolorenč et al. (42) employing an exact diagonalization approach to Np 5. The calculated wave functions (WFs) of the CF ground-state quartet (SI Appendix, Table S1) feature a small admixture of the excited = 11/2 and 13/2 multiplets.
The space of CF GS quadruplet is conveniently represented by the effective angular momentum quantum number = 3/2, with the WFs labeled by the corresponding projection and the phases of WFs chosen to satisfy the time-reversal symmetry (SI Appendix, Table S1). The on-site degrees of freedom within the GS quadruplet are (pseudo-)dipole, quadrupole, and octupole moments defined for = 3/2 in the standard way (8). Hence, the most general form for a superexchange coupling between quadruplets on two Np sites reads
We employed the FT-HI approach (38) to evaluate all interactions from the converged DFT + HI NpO2 electronic structure for the several first Np coordination shells. Only NN SEIs are significant, with longer-distance ones being more than an order of magnitude smaller. All SEIs for a given bond form a matrix, designated below as the SEI matrix, with the rows and columns labeled by the moments and on the sites and , respectively. The NN SEI matrix is graphically represented in Fig. 1B (SI Appendix, Table S2) using a local coordinate system with the quantization axis . This matrix is of a block-diagonal form since the interactions between time-even and time-odd moments are zero by symmetry. It can thus be separated into the dipole–dipole (DD), quadrupole–quadrupole (QQ), octupole–octupole (OO), and dipole–octupole (DO) blocks. Despite this simplification, the SEI matrix can in principle contain 70 distinct elements. The number of distinct nonzero matrix elements in , while reduced by the cubic symmetry to 38, remains rather large.
Our calculations predict the largest values for the diagonal DD (AF, 1.6 meV) SEI. However, the off-diagonal OO to (ferro, −1.5 meV) and DO to (AF, 0.95 meV) couplings are of about the same magnitude as the DD one. Overall, the calculated matrix shown in Fig. 1B features many nonnegligible DD, OO, and DO interactions of a comparable magnitude. The QQ interactions are weaker, reflecting the secondary nature of the quadrupole order. Our calculations thus predict a complex and frustrated superexchange in NpO2, which may give rise to multiple competing time-odd orders. Therefore, as has been previously noted (8, 25), extracting a full superexchange Hamiltonian of NpO2 from experimental (e.g., INS) data is virtually impossible due to a large number of parameters entering into the fit of an excitation spectra. The same difficulty is encountered by ab initio approaches based on total energy calculations for symmetry broken phases (15, 33, 43), which require a large number of very precises calculations to extract multiple nonnegligible matrix elements of , with the magnitude of 0.5 meV and above. Within the present framework, all interactions are extracted from a single ab initio calculation for paramagnetic NpO2.
The calculated superexchange Hamiltonian for NpO2 reads
The resulting GS order of NpO2 in the space consists of a primary (pseudo-)DO order combined with secondary (pseudo-)quadrupole one (SI Appendix, Table S3 lists the values of all order parameters; see also SI Appendix, Fig. S1). The pseudodipole order is a complex 3 planar AF structure, with four inequivalent simple-cubic sublattices forming two pairs with different moment magnitude; the moments of those two pairs are aligned along the and directions in the fcc lattice, respectively. The origin of this inclined AF structure of pseudodipoles is in the DO SEI, similar to physical AF magnetic orders found in some materials with large spin-orbit coupling that are likely induced by higher-order magnetic multipolar interactions. The pseudoctupoles order is also oriented in nonsymmetrical directions. The secondary pseudoquadrupole order is of a 3 type, which we analyze in detail below.
We subsequently mapped the moments calculated in the space into the observable multipolar moments that are defined in the physical space of Np 5 GS multiplet (Methods). The calculated physical multipole order of NpO2 is displayed in Fig. 2. Notice that observable moments up to = 7 can exist on an -electron shell (8); we show the largest primary (odd) and secondary (even) order parameters as well as the physically important quadrupole order. All nonzero multipole moments are listed in SI Appendix, Table S4. The physical dipole magnetic moments are found to completely vanish since their contribution into the and pseudodipole moments is exactly canceled by that due to the and pseudoctupoles. The primary order parameter is of rank 5 (triakontadipole); the physical octupole moment is an order of magnitude smaller, and the magnitude of rank 7 moments is about 1/3 of that for triakontadipoles, in agreement with previous estimates for the relative contribution of those multipoles into the MMO order in NpO2 (24). The magnetic triakontadipoles on different sublattices are oriented in four different directions (forming mutual angles corresponding to the angles between the cube’s main diagonals), thus structured similarly to the 3-AFM dipole order in UO2 (Fig. 2A). The secondary order is dominated by hexadecapole (rank 4) (Fig. 2B). The ordered quadrupole moments (Fig. 2C) are roughly twice smaller. The quadrupole order is directly related to the pseudoquadrupole one since the (or ) pseudoquadrupoles directly map into the physical ones, apart from swapping and . The resulting physical quadrupole order can be represented, in the space of quadrupoles [,,] by four directions [11], [11], [11], and [111] for four inequivalent Np sublattices [0,0,0], [1/2,1/2,0], [1/2,0,1/2], and [0,1/2,1/2], respectively. These quadrupoles can be depicted as moments with the principal axes along the corresponding direction at each given site (Fig. 2C). One sees that the ordered quadrupoles on the four Np sites forming the tetrahedron around each oxygen can either have their principal axes directed along the Np–O bonds toward the central O or form the same angle of with respect to those bonds. In both cases, the tetrahedron symmetry is preserved. The first case is realized for two O along one of the principal cubic diagonals, while the second one is found for six others, resulting in the lowering of NpO2 symmetry to from without any distortion of the cubic structure. This longitudinal 3 quadrupole order, previously proposed on the basis of resonant X-ray scattering (30) and subsequently confirmed by the splitting of 17O-NMR spectra in ordered NpO2 (21), is thus predicted by our ab initio superexchange Hamiltonian (2).
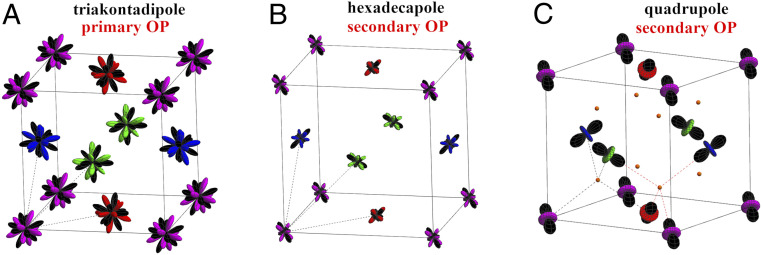
Calculated multipolar order in NpO2. This order of the physical multipoles in NpO2 is derived by mapping of the = 3/2 space to the full = 9/2 space. The physical magnetic dipoles (magnetic moments) are exactly canceled on all Np sites, resulting in a purely multipolar order. (A) The primary physical order parameter (OP), = 5 triakontadipole. (B) The largest secondary “slave” OP, = 4 hexadecapole. The displayed isovalue for the primary and secondary OPs is normalized with respect to the maximum possible value of a given moment in the CF ground-state quartet. Hence, the relative size of plotted moments indicates their relative magnitude. Black dashed lines are the primitive lattice translations of the original Np cubic face-centered sublattice, which, in the ordered phase, connects inequivalent Np sublattices. (C) Longitudinal 3 order of slave quadrupole moments, which are scaled on the plot by a factor of four. Np–O bonds for two inequivalent O sites (small orange balls) in ordered NpO2 are indicated by dashed lines. One sees that in the Np tetrahedron around each O, all ordered Np quadrupoles either are directed along the Np–O bonds or form the same angle with those bonds. This longitudinal 3 quadrupolar order preserves the cubic positions of oxygen sites in the ordered phase of NpO2, in contrast to the transverse 3 quadrupole order in UO2.
Having obtained the MMO of NpO2, we subsequently calculated its excitation spectra, which have been previously measured, in particular, by INS (19, 25).
The MMO lifts the degeneracy of CF GS quartet; the resulting exchange splitting calculated from the ab initio superexchange Hamiltonian (2) is depicted in Fig. 1 A, Right. The ground state is a singlet with the first excited doublet found at 6.1 meV above the GS singlet and the second excited level, singlet, located at 12.2 meV. The calculated position of first excited doublet is in excellent agreement with the location of a prominent peak in INS spectra at about 6.4 meV (19) in the ordered phase; another broad excitation was observed in the range of 11 to 18 meV (25) (see below).
Previously, an exchange splitting of the was obtained in ref. 24 assuming a diagonal uniform SEI between triakontadipoles. The value of this SEI was tuned to reproduce the experimental position of the excited doublet; the energy for the excited singlet calculated in ref. 24 is in agreement with our ab initio result.
We also calculated the theoretical INS cross-section, , where () is the energy (momentum) transfer; ; and is the dynamical magnetic susceptibility. The latter was evaluated within the random-phase approximation (RPA) from the full calculated ab initio superexchange Hamiltonian (2) (Methods). The resulting INS spectrum for along high-symmetry directions of the fcc lattice is displayed in Fig. 3A. Along the path, it is similar to that previously calculated from the simplified empirical superexchange Hamiltonian of ref. 24. The structure is richer along other directions showing multiple branches in the range from 4 to 8 meV. The experimental INS spectrum has not been measured so far due to lack of large single-crystal samples (8); hence, our result represents a prediction for future experiments. The calculated spherically averaged INS cross-section is compared in Fig. 3B with that measured (25) on powder samples at the same = 2.5 Å−1. The theoretical INS spectrum exhibits prominent peaks at around 6 meV and at about 12.5 meV, corresponding to the transition from the GS to the excited doublet and singlet, respectively.
![INS spectra in ordered NpO2. (A) INS cross-section S(q,E) with the momentum transfer q along a high-symmetry path in the fcc BZ. The special points are Γ = [0,0,0], X = [1,0,0], W = [1,12,0], and L = [12,12,12], in units of 2π/a. (B) Powder (spherically averaged) INS cross-section vs. energy transfer E for |q| = 2.5 Å−1. The theoretical spectra were broadened with the Gaussian resolution function of 1.5 meV. The experimental points are from Magnani et al. (25). (C) Ratio of the spectral weights of the low-energy (around 6-meV) and high-energy (10- to 20-meV) features vs. momentum transfer in the powder INS spectra (B). The experimental points are from Magnani et al. (25). Theoretical points from the same work are calculated with a semiempirical superexchange Hamiltonian (in the text).](/dataresources/secured/content-1766030616610-52eff468-71f5-41c6-962e-e53dc682a286/assets/pnas.2025317118fig03.jpg)
INS spectra in ordered NpO2. (A) INS cross-section with the momentum transfer along a high-symmetry path in the fcc BZ. The special points are = [0,0,0], X = [1,0,0], W = , and L = , in units of /a. (B) Powder (spherically averaged) INS cross-section vs. energy transfer for = 2.5 Å−1. The theoretical spectra were broadened with the Gaussian resolution function of 1.5 meV. The experimental points are from Magnani et al. (25). (C) Ratio of the spectral weights of the low-energy (around 6-meV) and high-energy (10- to 20-meV) features vs. momentum transfer in the powder INS spectra (B). The experimental points are from Magnani et al. (25). Theoretical points from the same work are calculated with a semiempirical superexchange Hamiltonian (in the text).
The theoretical low-energy feature agrees very well with the measured INS spectra, after experimental broadening is taken into account (Fig. 3B). The high-energy feature, however, is clearly split in experiment into two broad peaks centered at about 12 and 16 meV. In order to understand whether the relative weights of the low- and high-energy features are captured in the theory, we employed the same analysis as Magnani et al. (25). Namely, we evaluated, as a function of the momentum transfer, the ratio of high-energy feature spectral weight to that of the low-energy one. The calculated ratio is in excellent agreement with experiment up to = 2.5 Å−1. As noticed in ref. 25, a phonon contribution to INS appears below 18 meV for large , thus rendering the separation of magnetic and phonon scattering less reliable for 2 Å−1. The splitting of high-energy peak was clearly observed at all measured ; it was not reproduced by the simplified semiempirical SEI employed by Magnani et al. (25). They speculated that this splitting might stem from complex realistic Np–Np SEIs, which they could not determine from experiment. In the present work, we determined the full superexchange Hamiltonian for the GS CF quadruplet. Hence, the fact that the splitting of INS high-energy feature is still not reproduced points out to its origin likely being a superexchange coupling between the ground-state and first excited quadruplets (a significant contribution of the very high-energy CF doublet is unlikely). This interquadruplet coupling can be in principle derived using the present framework; we have not attempted to do this in the present work.
Alternatively, lattice-mediated interactions might be also considered as the origin of the high-peak splitting. Those interactions couple time-even moments (i.e., the quadrupoles within the space). However, the QQ coupling is rather expected to impact the shape of the low-energy peak in Fig. 3B because the corresponding lowest on-site excitation in the ordered phase (Fig. 1 A, Inset) is due to reverting of the on-site quadrupole moment (24). Indeed, we recalculated the theoretical INS spectrum (Fig. 3B) with the magnitude of QQ block in the SEI (Fig. 1B) scaled by a factor from zero to five. These variations of the QQ coupling strength do modify the shape of the low-energy peak but have no impact on the high-energy one. Since the lattice-mediated coupling is expected to modify exclusively the QQ block, it is thus quite unlikely to be the origin of the splitting.
The onset of the HO phase NpO2 is marked by an anomalous volume contraction vs. decreasing temperature (anti-Invar anomaly). The estimated total volume contraction in the ordered state as compared with the paramagnetic phase is 0.018% at zero temperature (20). This effect cannot be attributed to the conventional volume magnetostriction since ordered magnetic moments are absent in NpO2. Hence, this anomaly was speculated (39) to be induced by a (secondary) quadrupole order coupling to the lattice. As we show below, this is not the case, and the volume contraction in NpO2 rather stems from the volume-dependent SEI coupling high-rank magnetic multipoles.
In order to make a quantitative estimation for this anomaly, we evaluated the volume dependence of NpO2 ordering and elastic energies. To that end, we adopted the elastic constants calculated for NpO2 in the framework of the DFT + U approach (45): = 404 GPa, = 143 GPa. The corresponding parabolic volume dependence of elastic energy , where is the volume contraction and is the NpO2 equilibrium volume (45), is depicted in Fig. 4. The dependence of MMO energy vs. volume was obtained by calculating the SEIs at a few different volumes and then evaluating the MF order and ordering energy vs. volume expansion or contraction. The superexchange ordering energy remains linear vs. in a rather large range (1%); its dependence upon for the relevant range of small is thus easily obtained.
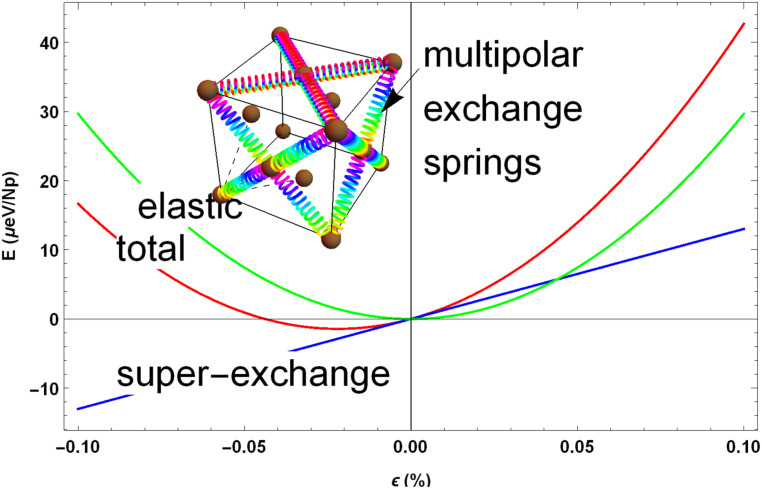
Multipolar exchange striction in NpO2. The curves represent the DFT + U elastic energy (green) and the ordering energy of the multipolar ground state vs. volume (blue); the latter is calculated from the volume-dependent ab initio SEIs. The multipolar order is seen to induce the contraction of the equilibrium volume (total energy; red curve) due to the two-site multipolar striction effect, analogously to the two-site volume magnetostriction in conventional magnetically ordered materials. The “springs” in Inset schematically illustrate the action of the intersite superexchange energy upon the onset of NpO2 multipolar order.
As is seen in Fig. 4, the superexchange contribution shifts the equilibrium volume in the ordered state toward smaller volumes. The negative slope for the ordering energy vs. volume is expected as the SEIs become larger with decreasing Np–Np distance. Thus, the multipolar SEIs act as springs (scheme in Fig. 4, Inset) pulling Np atoms closer as the order parameters increase below . Our approach is thus able to qualitatively capture this very small in magnitude subtle effect: The calculated spontaneous multipolar exchange striction is 0.023% (Fig. 4) at zero as compared with the experimental estimate of 0.018% (20, 39).
We also performed the same calculations suppressing the QQ superexchange, obtaining only a very minor change, by about 0.5%, in the slope of MMO energy vs. volume. Hence, the secondary quadrupole order plays virtually no role in the anomalous volume contraction. The physical origin of this effect is the volume dependence of leading, time-odd SEIs.
To analyze the temperature dependence of the anomalous contraction, one may recast the linear in volume MMO energy (Fig. 4) into a general form of , where and are primary and secondary order parameters, respectively, and and are the slopes of volume dependence for the corresponding contributions to MMO energy. The temperature dependence of the anomalous volume contraction is thus given by that of squares of the order parameters, and . As shown in SI Appendix, Fig. S2, secondary quadrupole exhibits a smooth evolution across and rather slowly increases vs. decreasing , while primary time-odd features a discontinuity in the slope at , as expected, with a rapid growth for , reaching about 60% of total magnitude at . This behavior of is in a perfect agreement with the shape of temperature dependence of the volume anomaly (20), thus confirming that it is induced directly by the time-odd primary order.
In conclusion, we have applied an advanced ab initio framework to the HO phase of neptunium dioxide NpO2. Our framework is based on the density functional + DMFT (DFT + DMFT) in conjunction with a quasiatomic approximation to local correlations on Np 5. Its crucial part is a force theorem method (38) that we employ to calculate SEIs between all multipole moments of the Np lowest CF manifold. From the resulting superexchange Hamiltonian, we derive all order parameters of the HO phase, its ordering temperature, magnetic excitations, and volume effect. In fact, numerous properties of the NpO2 HO phenomenon that have been painstakingly determined in experiments over several decades—absence of conventional magnetic order, the CF-level scheme, the primary triandicapole order and secondary longitudinal 3 quadrupole order, the singlet–doublet–singlet exchange splitting of the CF ground state, the two-peak structure of the magnetic excitation spectra—are reproduced by our calculations that contain essentially no adjustable parameters. Therefore, the present scheme is shown to provide a full, parameter-free, and quantitatively correct description of superexchange in complex realistic correlated insulators. On the basis of our theory, we may also identify the features that are not stemming from the intersite coupling between the ground-state CF levels. For example, the splitting of the high-energy peak in NpO2 INS spectra likely stems from an SEI with excited CF levels. We also uncover an unconventional mechanism for the anomalous volume contraction observed in ordered NpO2, which is induced not by the secondary quadrupole order coupling to the lattice as previously assumed but rather, by the volume dependence of leading time-odd SEIs.
This first-principles methodology—a dynamical MF treatment of symmetric paramagnetic phase combined with the force theorem extraction of the full complex intersite exchange responsible for the spontaneous symmetry breaking—can be applied to a wide range of rare earth, actinide, and heavy-transition metals correlated systems (2, 8, 9, 46), in which the interplay of a large spin-orbit coupling with crystalline environment gives rise to a large degeneracy of the CF ground state and high-rank multipole moments. HOs, stemming from coupling between those moments, can be predicted and their interplay with various parameters—external or chemical pressure, applied field, lattice distortions—identified, thus opening up an avenue for theoretical search of new exotic phases of matter.
Our charge self-consistent DFT + DMFT calculations using the HI approximation for Np 5, abbreviated as DFT + HI, were carried out for the CaF2-type cubic structure of NpO2 with the experimental lattice parameter Å. We employed the Wien-2k full-potential code (47) in conjunction with “TRIQS” library implementations for the DMFT cycle (36, 48) and HI. The spin-orbit coupling was included in Wien2k within the standard second-variation treatment. The Brillouin zone (BZ) integration was carried out using 1,000 k points in the full BZ, and the local density approximation (LDA) was employed as DFT exchange correlation potential.
The Wannier orbitals representing Np 5 states were constructed by the projective technique of refs. 49 and 50 using the Kohn–Sham bands enclosed by the energy window eV around the Kohn–Sham Fermi energy; this window thus encloses all Np 5-like bands. The use of a narrow window enclosing only the target (5-like) band for the construction of local orbitals results in a so-called “extended” Wannier basis. By employing such a basis within DFT + HI, one may effectively include the contribution of hybridization to the CF splitting on localized shells, as discussed in ref. 51. The same choice for the local 5 basis was employed in our previous DFT + HI study (52) of UO2, resulting in a good quantitative agreement of the calculated CF splitting with experiment.
The on-site Coulomb interaction between Np 5 was specified by the Slater parameter = 4.5 eV and the Hund’s rule coupling = 0.6 eV; the same values were previously employed for UO2 (52). The double-counting correction was computed using the fully localized limit (53) with the atomic occupancy of Np shell (54). The DFT + DMFT charge self-consistency was implemented as described in ref. 55. In our self-consistent DFT + HI calculations, we employed the spherical averaging of the Np 5 charge density, following the approach of Delange et al. (56), in order to suppress the contribution of LDA self-interaction error to the CF. The DFT + HI calculations were converged to 5 Ry in the total energy.
The self-consistent DFT + HI calculations predict a CF split 4I9/2 atomic multiplet to have the lowest energy, in agreement with Hund’s rules for an shell; the calculated spin-orbit coupling = 0.27 eV. The CF splitting of the 4I9/2 multiplet predicted by these calculations is shown in Fig. 1A, and the corresponding CF WFs are listed in SI Appendix, Table S1. The cubic CF parameters— = −152 meV, , = 32.6 meV, and —were extracted by fitting the converged DFT + HI one-electron 5-level positions (56).
The states of the CF ground-state quadruplet were labeled by projection of the pseudoangular quantum number = 3/2 as specified in SI Appendix, Table S1. We subsequently employed the FT-HI method (38) to evaluate all SEIs between the quadruplet for several first Np–Np coordination shells. Previously, the FT-HI method was applied to systems with conventional magnetic primary order (52, 57). Within this method, matrix elements of intersite coupling for the Np–Np bond read
We solved the obtained superexchange Hamiltonian (2) using the numerical MF package MCPHASE (44) including all 1 structures up to 4 4 4 unit cells. We have also verified the numerical solution by an analytical approach. Namely, the MF equations read
This order in the space was subsequently mapped into the physical space. The density matrix on the inequivalent site reads , where are the corresponding (pseudo-)moments. This density matrix is upfolded into the one in the space, neglecting small contributions of the excited and multiplets and renormalizing the CF states accordingly, as , where is the CF GS basis, with WFs written as columns in the order of . The physical moments are then calculated in the standard way, .
In order to calculate the dynamical magnetic susceptibility , we implemented a general RPA approach (59). Namely, the general susceptibility matrix in the space reads
The local susceptibility for the inequivalent site is calculated from the MF eigenvalues and eigenstates :
After the susceptibility matrix is calculated, it is “upfolded” to the physical space similarly to the density matrix as described above. The magnetic susceptibility is given by the DD blocks of the upfolded summed over the sublattice indices.
L.V.P. acknowledges support from European Research Council Grant ERC-319286-“QMAC” and the computer team at CPHT. S.K. thanks Ecole Polytechnique for financial support and Centre de Physique Théorique (CPHT) for its hospitality.
All relevant data are available within the manuscript and SI Appendix. Raw computational data have been also deposited in the publicly accessible Materials Cloud Archive (10.24435/materialscloud:12-7q) (60)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
 Hidden order and multipolar exchange striction in a correlated f-electron system
Hidden order and multipolar exchange striction in a correlated f-electron system
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

