 Gene duplication at the Fascicled ear1 locus controls the fate of inflorescence meristem cells in maize
Gene duplication at the Fascicled ear1 locus controls the fate of inflorescence meristem cells in maize
Proceedings of the National Academy of Sciences of the United States of America


Edited by Jane A. Langdale, University of Oxford, Oxford, United Kingdom, and approved January 13, 2021 (received for review September 11, 2020)
Author contributions: S.H. and Z.Z. designed research; Y.D., C.L., Y.L., and D.J. performed research; Y.D., C.L., Y.L., and Z.Z. analyzed data; and Y.D., C.L., and S.H. wrote the paper.
1Y.D., C.L., and Y.L. contributed equally to this work.

- Altmetric
The maize ear is unbranched and terminates in a single point. The ear and tassel inflorescences of Fascicled ear mutants fail to grow as a single point and instead are branched. This phenotype results from the misexpression of duplicated transcription factors, ZMM8 and DRL2. We hypothesize that these gene rearrangements create regulatory sequences that cause misexpression in early inflorescence meristems, thus activating a laminar program, ablating the meristem, and producing branches. This work demonstrates that zmm8 and drl2 must be restricted from the inflorescence meristem to maintain its terminal point, and conversely, a mechanism by which branching may be imposed. Manipulation of these genes can be used to alter plant architecture, potentially to improve agronomic traits.
Plant meristems are self-renewing groups of pluripotent stem cells that produce lateral organs in a stereotypical pattern. Of interest is how the radially symmetrical meristem produces laminar lateral organs. Both the male and female inflorescence meristems of the dominant Fascicled ear (Fas1) mutant fail to grow as a single point and instead show deep branching. Positional cloning of two independent Fas1 alleles identified an ∼160 kb region containing two floral genes, the MADS-box gene, zmm8, and the YABBY gene, drooping leaf2 (drl2). Both genes are duplicated within the Fas1 locus and spatiotemporally misexpressed in the mutant inflorescence meristems. Increased zmm8 expression alone does not affect inflorescence development; however, combined misexpression of zmm8, drl2, and their syntenic paralogs zmm14 and drl1, perturbs meristem organization. We hypothesize that misexpression of the floral genes in the inflorescence and their potential interaction cause ectopic activation of a laminar program, thereby disrupting signaling necessary for maintenance of radially symmetrical inflorescence meristems. Consistent with this hypothesis, RNA sequencing and in situ analysis reveal altered expression patterns of genes that define distinct zones of the meristem and developing leaf. Our findings highlight the importance of strict spatiotemporal patterns of expression for both zmm8 and drl2 and provide an example of phenotypes arising from tandem gene duplications.
Plant architecture results from the activities of meristems, groups of totipotent cells that maintain a central population of indeterminate cells while initiating organs peripherally. The types of organs formed by the meristem depend on whether the plant is in its vegetative or reproductive phase. Leaves form during the vegetative phase, subtending suppressed axillary meristems. However, in the reproductive phase, leaves are reduced, and axillary meristems predominate, producing branches or flowers, depending on the species (1).
Meristems are radially symmetrical with a central/peripheral axis. During initiation of determinate lateral organs of the shoot, including leaves and floral organs, radial symmetry is broken with the onset of asymmetric patterning (2). The localized planar growth of lateral organs is derived from their positional relationship to the meristem (34–5) in which the adaxial surface faces the meristem while the abaxial surface faces away from the meristem (2). Thus, the central/peripheral axis continues into the adaxial/abaxial axis of shoot lateral organs (2).
Laminar outgrowth of lateral organs results from boundaries created by the juxtaposition of cells expressing antagonistic abaxial and adaxial fate regulatory factors (6, 7). This same juxtaposition exists within the radial shoot axis, with adaxial genes expressed in the center and abaxial genes expressed in the periphery (8). Abaxial genes are excluded from the center of the meristem (910–11). The genetic basis of polarity establishment in Arabidopsis involves several distinct transcription factor families and small regulatory RNAs (12). The CLASS III Homeodomain-Leucine Zipper (HD-ZIPIII) family is responsible for adaxial cell fate; KANADI and YABBY gene families and Auxin Response Factors (ARF3 and ARF4), together with the microRNA miR166, all contribute to abaxial cell fate identity in Arabidopsis (13, 14).
Multiple levels of regulation exist to maintain the meristem so that it is not consumed in organogenesis nor increased in size beyond what is needed for growth of the plant. The well-studied pathway for meristem maintenance is the CLAVATA3 (CLV3)–WUSCHEL (WUS) negative feedback loop, through which the balance of stem cell fate occurs as WUS activates CLV3 expression, and the CLV3 signal is transmitted to the central domain to restrict the expression of WUS, through leucine rich repeat receptors CLV1 and CLV2 (1516–17). The CLV-WUS pathway is well conserved (16), and mutant phenotypes have been described for maize orthologs of CLV1, CLV2, and CLV3 (171819–20). Maize contains two distinct inflorescence meristems (IMs): the apical male tassel and the lateral female ear, which forms in the axil of a leaf (21). The loss-of-function CLV phenotypes include enlarged IMs with both fasciated tassels and ears because of interference with regulation of IM development (171819–20).
Fascicled ear1 (Fas1) is a dominant maize mutant, displaying reiterated bifurcation of both the tassel and ear (22). Fascicled, which means “bundled” or “bunched” in Latin, distinguishes Fas1 from the large class of fasciated ear mutants that are not branched. Here, we describe positional cloning and analysis of the Fas1 locus. A tandem duplication of the Zea mays MADS8/drooping leaf2 (zmm8/drl2) gene pair causes the dominant Fas1 phenotype through spatiotemporal misregulation of both genes. Genes that regulate central-/peripheral-zone cell fates are differentially expressed. We propose a model in which early misexpression of zmm8/drl2 in the IMs affects the regulation of central/peripheral zone identity, leading to punctate domains of meristem termination, followed by ectopic proliferation of lateral domain cells, resulting in repeated bifurcation of male and female IMs. These data indicate that maintenance of central/peripheral identity is required within cells of the IM to sustain its single, terminal central axis.
Results
Fas1 Is a Dominant Mutation with Repeated Bifurcation in the Tassel and Ear.
Fascicled ear1 (Fas1) mutants were discovered as naturally occurring mutations in two separate breeding lines: Fas1-R (23) and Fas1-2. The Fas1 mutation affects both male and female inflorescences and has no effect on vegetative plant architecture. Mutants fail to maintain a single, terminal IM and instead display deep branching caused by continuous bifurcations along the axis of the rachis (Fig. 1 A–D). In Fas1-2 in the B73 inbred background, the tassel lacks a central rachis, resulting in many long branches (Fig. 1 A and E and SI Appendix, Table S1). Fas1-R tassels in either the A188 or B73 inbred backgrounds display a central rachis that splits several times leading to multiple terminal points instead of one (ranging from two to six) (Fig. 1 B and F and SI Appendix, Table S1). Whereas normal ears are unbranched, Fas1-2 ears undergo two or three bifurcations but maintain normal kernel development (Fig. 1 C and G and SI Appendix, Table S2). Fas1-R ears show variable bifurcation numbers leading to many terminal points (Fig. 1 D and H and SI Appendix, Table S2). Tassel length and tassel branch number show no dosage effects, but kernel row number and length–width ratio of the ear are intermediate in heterozygotes (SI Appendix, Tables S1 and S2).

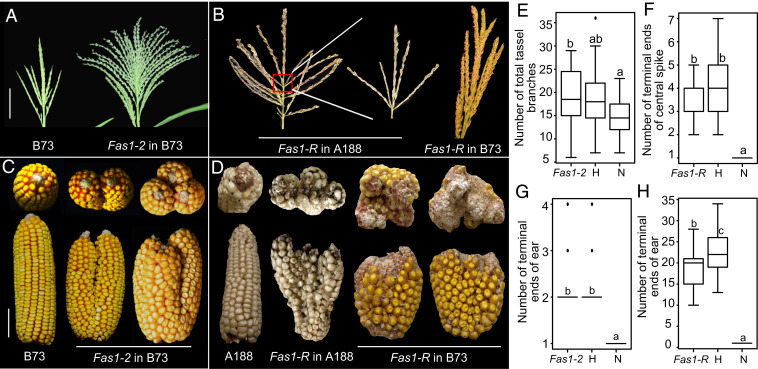
Inflorescence architecture of Fas1 mutants. (A, B) Tassels of Fas1-2 and Fas1-R in B73 or A188. (Scale bar, 5 cm.) (C, D) Ear bifurcations of Fas1-2 and Fas1-R in B73 or A188. (Scale bar, 5 cm.) (E, F) A statistical analysis of the number of tassel branches or terminal ends of the central rachis of Fas1-2 in B73. Fas1-2 and Fas1-R refer to Fas1-2/Fas1-2 and Fas1-R/Fas1-R homozygotes, respectively; H refers to Fas1-2/+ or Fas1-R/+ heterozygotes; and N refers to the nonmutant sibling. (G, H) Box and whisker plots of ear bifurcations for Fas1-2 in B73, differences in letter designations denote differences in means as measured by Student’s t test, P < 0.05. Genotype designations as in E and F.
Scanning Electron Microscopy Reveals Initial Events that Lead to Bifurcation.
To better understand early inflorescence organogenesis in Fas1-2 and Fas1-R mutants, young (1- to 2-mm) ears and tassels were visualized using scanning electron microscopy (SEM). In wild type of all genetic backgrounds, after the transition from vegetative meristem to the IM, the tassel and ear IMs initiate spikelet pair meristems (SPMs), which each initiate two spikelet meristems (SMs), that in turn initiate two floral meristems (FMs) (Fig. 2 A–F). Throughout development, the IM maintains a single organizing center, terminating in a single tassel rachis or ear tip.
Inflorescence developments in Fas1 mutants. (A–F) Ear and tassel development in wild type (B73). (G–J) Ear development in Fas1-2; the double arrow segment in (G) marks increased IM diameter, and the arrows and arcs in H and J mark central/peripheral axis, respectively. (K–O) Ear development in Fas1-R; the double arrow segment in (K) marks increased IM diameter, and the arrows in (L–N) mark central axis, 1° and 2° in O refers to primary and secondary bifurcations. (P–S) Tassel development in Fas1-2; the arrows in P refer to branch meristem primordia. (T–W) Tassel development in Fas1-R; the double arrow segment in T marks increased IM diameter, the arrows in (U) and (V) mark central axis, and the arcs in (V) mark peripheral axis. IM, inflorescence meristem; BM, branch meristem; SPM, spikelet pair meristem; SM, spikelet meristem; FM, floral meristem. (Scale bar, 200 μm.)
In the early stage of Fas1-2 ear (B73) development, the IM becomes wider, and the peripheral region grows while the central region ceases. Two or three independent, ear-like structures arise. Each newly formed IM initiates a normal progression of SPMs, SMs, and FMs (Fig. 2 G–J); in A188, the Fas1-R ear IM displays line fasciation and undergoes repeated rounds of bifurcations leading to multiple branch-like structures. Spikelet primordia initiate normally on the abaxial surface of each new structure, but the adaxial surfaces lack spikelet primordia (Fig. 2 K–O).
In the early stage of Fas1-2 tassel (B73) development, primordia initiate from the apical surface, leading to a loss of growth of the tassel rachis as cells are recruited to new primordia that develop into branch-like structures with normal spikelets (Fig. 2 P–S). Fas1-R tassels in A188 are slightly different. At first, the IM enlarges while initiating normal branches, then the center of the apical meristem halts while the peripheral region fasciates to form a flat meristem with a deep central depression. The fasciated meristem then splits into many branched structures (Fig. 2 T–W).
In both alleles at this early stage, male and female inflorescences share a common phenotype. Growth of the central region is repressed and peripheral zone growth is promoted, forming inflorescences with deep branching. Fas1-2 and Fas1-R differ in that the bifurcations persist longer in Fas1-R ears and sometimes lead to secondary axes in which the adaxial side lacks spikelets.
Duplication of Both zmm8 and drl2 Cause Fascicled Inflorescences.
Fas1-R was first mapped on the long arm of chromosome 9 using the standard set of waxy reciprocal translocations (24). The final mapping region was narrowed to 167 kb located in bin 9.06 between IDP580 and custom marker SSR10 using 426 individuals (SI Appendix, Fig. S1). Fas1-2 was mapped in a B73 F2 population using bulked-segregant RNA sequencing with 30 1- to 2-cm ears of Fas1-2 and a similar pool of normal siblings, followed by fine mapping with 7,680 F2 individuals. The mapping region was narrowed to ∼160 kb between marker BMC5 and BMC6 on chromosome 9, which showed a consistent mapping region with Fas1-R (Fig. 3A and SI Appendix, Fig. S1). (Mapping primers are listed in SI Appendix, Table S3.) In B73, two genes reside in this interval: the MADS-box gene, zmm8 (Zm00001d048082), and the YABBY gene, drooping leaf2 (drl2, Zm00001d048083) (Fig. 3A).
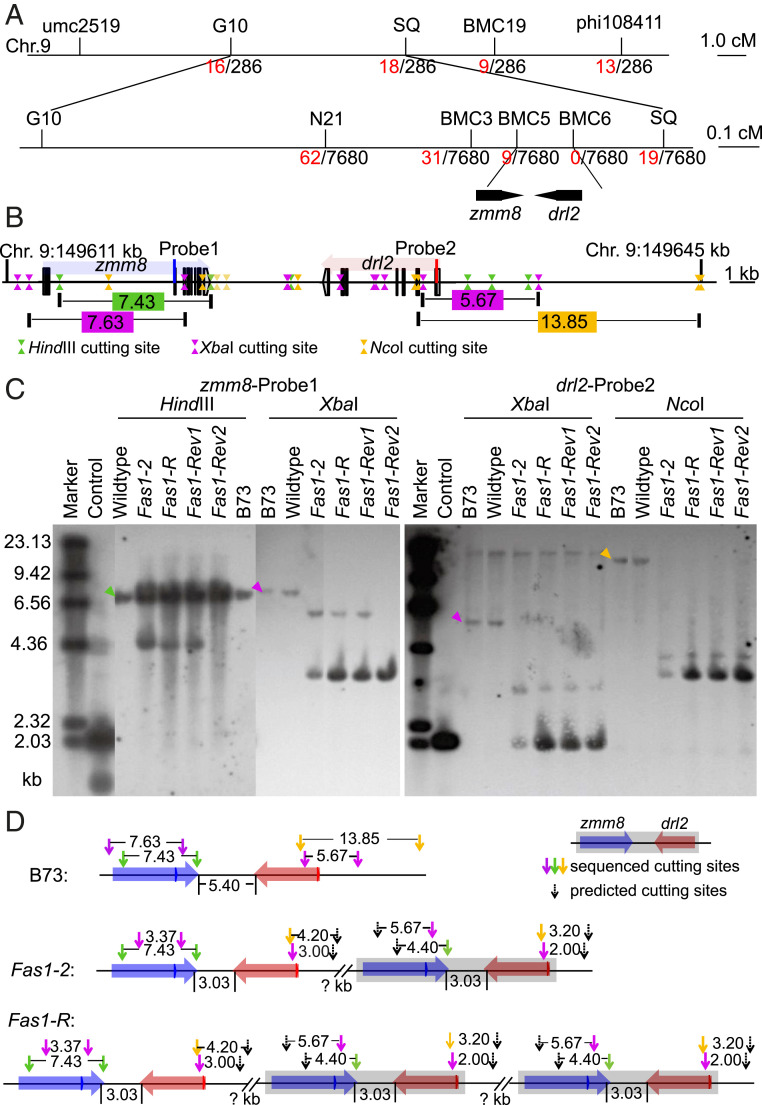
zmm8 and drl2 genes are responsible for the fascicled inflorescences. (A) Genetic mapping of Fas1-2 and Fas1-R. The black markers were used for genetic mapping in a Fas1-2 F2 population, the colored and black numbers show the numbers of recombinants and the population sizes. The Fas1 locus, flanked by BMC5 and BMC6, delineated a 160-Kb region on Chr9 (B73 RefGen_version 4) containing two annotated genes: zmm8 and drl2. (B) The genomic structure of zmm8 and drl2 in a 35-kb genomic region. The green, purple, and yellow triangles refer to the site of HindIII, XbaI, and NcoI restriction enzymes, respectively. The numbers in the boxes show the distance of the two closest cut sites of each enzyme (kb). The blue and red lines refer to zmm8 and drl2 specific probes. (C) A DNA blot with zmm8-specific probe1 and drl2-specific probe2 using digested genomic DNA of B73, wild-type Fas1-2, Fas1-R, Fas1-Rev1, and Fas1-Rev2. The triangles refer to the bands expected in the B73 RefGen_version 4 genome when digested by each enzyme. (D) A possible arrangement of zmm8 and drl2 in Fas1-2 and Fas1-R. The blue and red boxes refer to zmm8 and drl2. The arrows with pink, green, and yellow color refer to sequence-defined cutting sites, and the black arrows show the predicted cutting site. The numbers show the physical distance (kb).
We carried out a revertant screen of Fas1-R and identified 2 revertants in 3,378 plants. The high frequency of reversion suggested the possibility of gene duplication at the Fas1 locus. To determine if there was copy number variation at the Fas1 locus, we performed restriction fragment length analysis using DNA blots with specific probes located within zmm8 and drl2. Genomic DNA digested with HindIII followed by blotting with zmm8-specific Probe1, shows one specific band of 7,433 bp in B73 and the normal sibling, consistent with the B73 reference genome version 4 (Fig. 3 B and C). In Fas1-2 and Fas1-R, however, the 7,433-bp band has higher intensity, and there is an additional band of ∼4.4 kb. When digested with XbaI and hybridized with zmm8-specific Probe1, the normal individuals show the expected size band, but the mutants have two bands of different sizes (Fig. 3C). Sequence analysis confirmed that the difference in band number was not due to novel XbaI sites within the region of zmm8-specific Probe1 in Fas1 mutants. The lower band in Fas1-R is of higher intensity suggesting an increase in copy number (Fig. 3C). These results suggest that there could be two copies of zmm8 in Fas1-2, but two or more copies in Fas1-R. Consistent with the possibility of copy number variation, one of the revertants, Fas1-Rev2, lost one of the extra copies of zmm8.
With the drl2-specific Probe2, one band is identified in B73 and the normal sibling after digestion of genomic DNA with XbaI or NcoI. The sizes are consistent with B73 version 4 (5,674 bp for XbaI and 1,3847 bp for NcoI) (Fig. 3 B and C). Two different bands are found in Fas1-2 and Fas1-R for each enzyme, and the lower band is more intense only in Fas1-R and its derivative revertant alleles (Fig. 3C). These results support the hypothesis that there are two copies of drl2 in Fas1-2, but two or more copies in Fas1-R. However, unlike the results with the zmm8 probe, neither revertant showed any differences compared to the Fas1-R drl2-specific Probe2 banding pattern. Taken together, based on the copy number changes and available sequence, a possible arrangement of zmm8 and drl2 in Fas1-2 and Fas1-R is shown in Fig. 3D. Fas1-2 and Fas1-R are independent alleles, which contain two or more duplicated copies. The extra copies are tightly linked in both alleles.
In order to assess whether the duplication found in the Fas1 alleles was unique, we compared our contiguous genomic sequence from Fas1-2 containing zmm8, the intergenic region, and drl2 to the recently released maize genomes (https://nam-genomes.org) using basic local alignment search tool (BLAST) (25). The contiguous genomic sequence of Fas1-2 shares 99.92% sequence identity with the inbred B97. Remarkably, B97 has two copies of zmm8 (here referred to as zmm8-L and zmm8-R) and two copies of drl2 (referred to as drl2-L and drl2-R) at the syntenic region. Another inbred, Oh43, has one copy of drl2, but an extra truncated copy of zmm8 in addition to a full-length copy of zmm8 (SI Appendix, Fig. S2A).
To further study the gene duplications in B97 and Oh43, we compared the DNA blotting pattern between B73, Fas1-2, Fas1-R, B97, and Oh43 using zmm8-specific Probe1. Because of the similarity of zmm8 duplications in B97, two near-equal-length bands were produced when digested with EcoRI (11,712 bp and 11,706 bp) or HindIII (7,450 bp and 7,456 bp), and each was slightly bigger than in B73 (SI Appendix, Fig. S2 A and B). Using a HindIII digestion, Fas1-2 and Fas1-R produced an additional band of a different size (SI Appendix, Fig. S2 A and B). These results show that Fas1-2 and Fas1-R have more zmm8 copies than B97. Additional sequence variation exists between B97 and the Fas1 alleles in the introns and intergenic region (SI Appendix, Fig. S2C). Although copy number variation of zmm8 and drl2 exists in both B97 and Oh43, inflorescences of these two lines are normal, unlike Fas1 mutants.
zmm8 and drl2 Are Misexpressed at Meristematic Regions of Fas1 Immature Ear.
To understand how the extra gene copies might affect expression patterns, we carried out quantitative RNA studies of zmm8 and drl2 in immature inflorescences. Compared to the wild type, zmm8 was misexpressed in both 1- to 2-mm Fas1-2 ears and 2- to 3-mm Fas1-R ears. The expression level of drl2 was twofold higher in Fas1-2 compared to normal siblings in samples containing 1- to 2-mm ears surrounded by immature leaf primordia (Fig. 4 A and C). When all leaf tissues were dissected away from 2- to 3-mm Fas1-R ears, the elevated expression level of drl2 was more obvious (Fig. 4 B and D). Neither zmm8 nor drl2 were expressed in the 2- to 3-mm immature ears of Fas1-Rev1, Fas1-Rev2, B97, or Oh43 (Fig. 4B and SI Appendix, Fig. S3 F and G). Both genes were expressed in Fas1-Rev1 and Fas1-Rev2 at later ear developmental stages (5 to 8 mm and 8 to 12 mm) which contain FMs and floral organs (SI Appendix, Fig. S3 F and G).
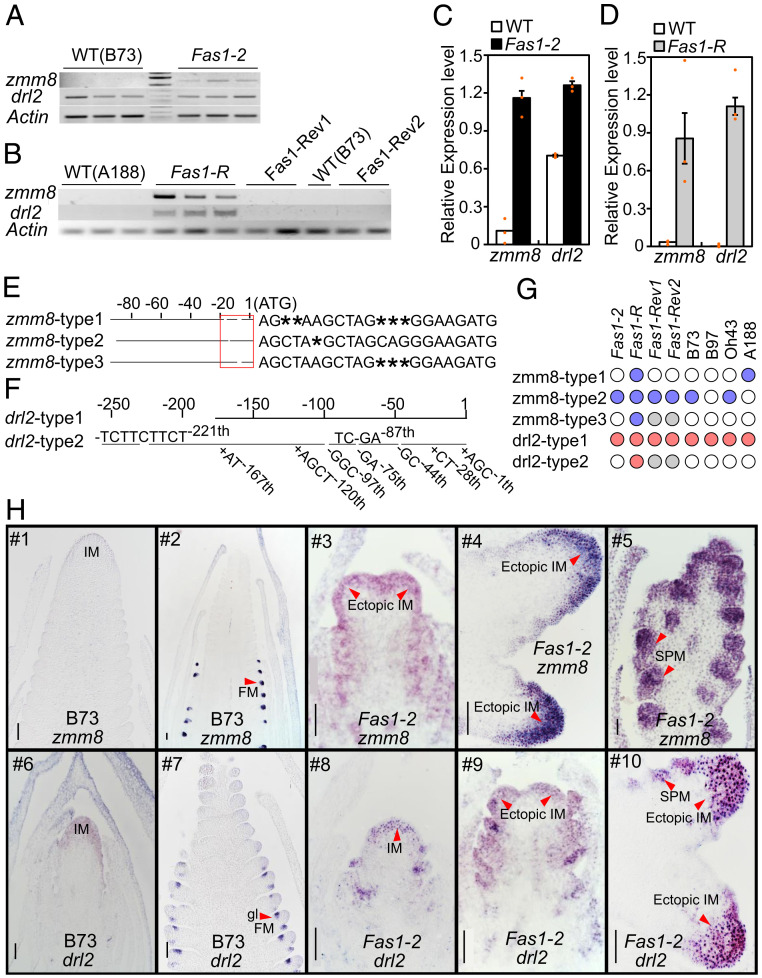
zmm8 and drl2 transcripts are misexpressed in Fas1. (A) An RNA expression analysis of zmm8 and drl2 in 1 to 2 mm ears of Fas1-2 by RT-PCR. (B) An RNA expression analysis of zmm8 and drl2 in 2 to 3 mm ears of Fas1-R, Fas1-Rev1, Fas1-Rev2, and the wild type (B73) by RT-PCR. (C, D) qRT-PCR of the same samples presented in A and B. The values are means ± SE (n = 3). (E) Three types (zmm8-type1, zmm8-type2, and zmm8-type3) of 5′−UTR sequence of zmm8 in Fas1-R heterozygotes identified by 5′ RACE. Sequence variations in the red box are located at 1 to −20, upstream of ATG. A star denotes the absence of base compared to other aligned sequences. (F) Two types (drl2-type1 and drl2-type2) of 5′−UTR sequence of drl2 in Fas1-R heterozygotes identified by 5′ RACE. Sequence variations in the red boxes are located at 1 to −250, upstream of the ATG. (G) Presence and expression of different types of zmm8 and drl2 transcripts. The blue and red circles stand for zmm8 and drl2 sequences that are present and expressed, respectively; the gray circles stand for sequences that are present but not expressed; and the empty circles represent sequences that were not present. (H) An mRNA in situ hybridization pattern of zmm8 and drl2 in B73 and Fas1-2 ear. The arrowheads in (#2) point to the FM, in (#3, #4, #9, #10) point to ectopic IMs, in (#5) to SPM, and in (#7) indicate glume primordia (gl). (Scale bar, 100 μm.)
In order to explore the possibility of different transcripts originating within the Fas1 locus, we carried out 5′ rapid amplification of cDNA ends (RACE) of zmm8 and drl2 to amplify all possible transcripts in immature ears of Fas1-R/+ (A188 background) and A188. After amplification with gene specific primers (GSP1 and GSP2) and subcloning, more than 20 clones were sequenced. Three types of zmm8 full-length cDNA have the same coding sequence but different 5′ untranslated region (UTR) in Fas1-R/+ (Fig. 4E). Genomic DNA amplifications with primers designed specifically for Fas1-R 5′ UTR showed zmm8-type2 and zmm8-type3 fragments existed in Fas1-R/+ only and not in A188 genomic DNA (Fig. 4G). When evaluating B73, B97, and Oh43, we found zmm8-type3 sequence to be unique to the Fas1 alleles (Fas1-R, Fas1-Rev1, and Fas1-Rev2) (Fig. 4 E and G and SI Appendix, Fig. S3 A–C). drl2-specific primers revealed two types of 5′-UTR sequences in Fas1-R/+, one in common with A188 and likely arising from the wild-type copy in the heterozygote. The other product had several indels and insertions located 1 to −250 bp upstream of the ATG start codon (Fig. 4F). The sequence in Fas1-R was also confirmed by genomic DNA (gDNA) amplifications. The drl2-type1 was present in other inbreds, but drl2-type2 was found only in the Fas1-R–related alleles (Fas1-R, Fas1-Rev1, and Fas1-Rev2) (Fig. 4G and SI Appendix, Fig. S3 D and E).
Although the Fas1 revertants contain the Fas1 unique 5′-UTR genomic sequence for zmm8 and drl2, these sequences are not expressed. Sequencing of the transcripts showed they were zmm8-type2 and drl2-type1. These findings suggest that it is not simply additional gene copies that lead to the early misexpression of these two transcription factors, and thus the Fas1 phenotype, but promoters or enhancers resulting from the gene rearrangements unique to Fas1-2 and Fas1-R.
To understand how misexpression of zmm8 and drl2 leads to a fascicled inflorescence phenotype, we carried out in situ hybridization using antisense probes. zmm8 is expressed only in the upper floret and not in the lower floret of each spikelet (26). drl2 is expressed in leaf primordia of the shoot apical meristem (SAM) and lateral floral primordia of the floret, controlling midrib patterning and FM determinacy (27, 28). Consistent with previous reports (26), we did not detect zmm8 expression in the earlier stage of normal ear development when SPMs are initiating but identified zmm8 expression in FM primordia (Fig. 4H #1 and #2). Intriguingly, in Fas1-2 ears, zmm8 signal was observed in the IMs and SPMs (Fig. 4H #3–#5), although it was not observed in the sunken domain of the inflorescence tip (Fig. 4H #4). In normal ears, drl2 mRNA was not visible in 1-mm ears (Fig. 4H #6) but was detected in lateral floral primordia of later stages (Fig. 4H #7). In the Fas1-2 ears, however, drl2 signals could be observed in the ectopic IMs and SPMs (Fig. 4H #8–#10). These results show that zmm8 and drl2 are misexpressed in the IMs of the Fas1 ear, which may suppress the meristematic activities of the central region and promote meristematic activities of the peripheral region, leading to highly branched “bundled” inflorescences.
In order to determine if simple overexpression of zmm8 could create the Fas1 phenotype, we overexpressed the gene behind the ubiquitin promoter in the inbred line ZZC01. The plants had a normal IM, although the expression levels were much higher than controls (SI Appendix, Fig. S4 A and B). We also knocked out zmm8 and its paralog zmm14 using CRISPR-Cas9. The loss of function phenotype is reminiscent of the triple mutant of drl1, drl2 and the maize AGAMOUS ortholog, zag1 (28). The FM shows failure of floral organ conversion and instead forms indeterminate branches with increased and elongated glumes (SI Appendix, Fig. S4 H–L). This result suggests that zmm8/14 and drl1/2 have the capacity to regulate similar floral developmental pathways. To test the possibility that ZMM8 and DRL2 interact with each other to cause the Fas1 phenotype, we carried out yeast two-hybrid and luciferase complementation image (LCI) assays. We found that ZMM8 physically interacted with DRL2 in yeast and in tobacco (SI Appendix, Fig. S4 F and G). Given that the normal expression patterns of drl2 and zmm8 do not overlap (Fig. 4H), it is possible that the misexpression of both zmm8 and drl2 in overlapping domains in Fas1 mutants allows for an interaction that does not occur when only zmm8 is misexpressed.
Based on the gene-copy number variations and the misexpression pattern, we hypothesize that the extra copies led to the formation of promoters or enhancers, which result in spatial and temporal misexpression of both zmm8 and drl2 in Fas1 IMs. One of the revertants, Fas1-Rev1, maintains the Fas1-R genomic structure but expresses zmm8-type2 and drl2-type1 transcripts with an additional truncated drl2, which could be the possible reason for the phenotype reversion. The other revertant, Fas1-Rev2, lost an extra copy of zmm8 and expresses zmm8-type2 and drl2-type1. Neither revertant expresses the Fas1-R specific transcripts (zmm8-type3 and drl2-type2), and the transcripts they do express only accumulate at the later floral development stage (Fig. 4G and SI Appendix, Fig. S3 F and G). We propose that the promoter(s) containing zmm8-type3– and/or drl2-type2–related sequences are required for the misexpression of zmm8 and drl2 in Fas1-R.
Central/Peripheral Zone Markers Are Misexpressed in Fas1 Inflorescence Meristems.
To better understand the biological processes that are altered in Fas1, 1- to 2-mm immature ears containing adjacent immature leaf primordia of Fas1-2 and normal siblings were collected for RNA sequencing. In total, we detected 3,227 up-regulation and 2,284 down-regulation genes (q < 0.05) (Fig. 5A). We found that zmm8 was not expressed in wild type but was expressed at a low level (0.35 fragments per kilo base per million on an average) in Fas1-2, the expression of drl2 was threefold higher in Fas1-2 (SI Appendix, Fig. S5A).
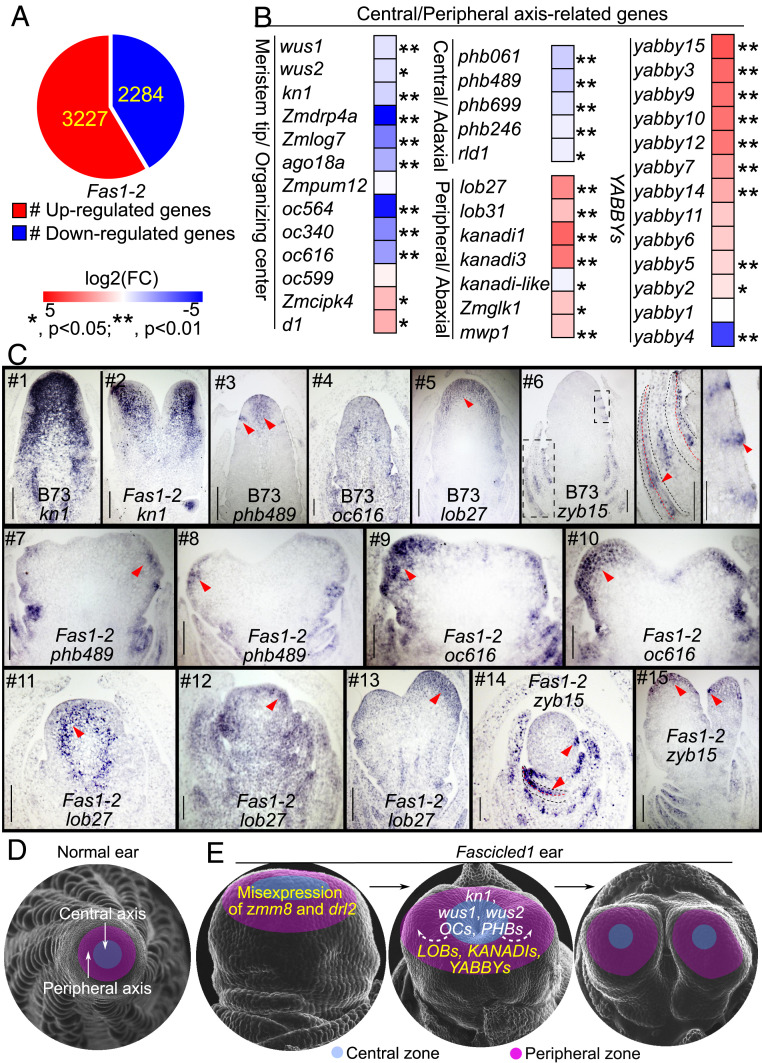
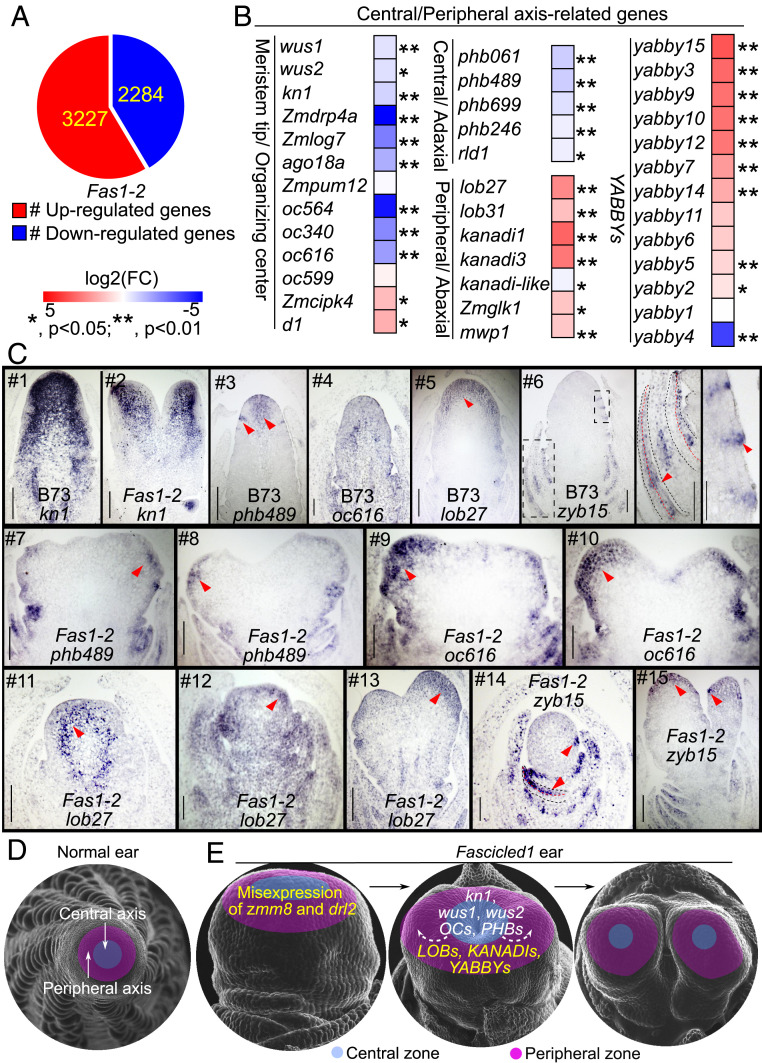
Central/peripheral cell fate of IM in Fas1. (A) DEGs detected by RNA sequencing in 1- to 2-mm ear of Fas1-2 allele with three biological replicates (P < 0.05). (B) DEGs that regulate central/peripheral cell fate in RNA sequencing data. *P < 0.05, **P < 0.01. (C) In situ hybridizations showed an expression pattern of kn1, phb489, oc616, lob27, and zyb15, in ear primordia of wild type and Fas1-2. The arrowheads point to the central and SPM boundary region of normal ear in #3, to apical meristem in #5, to adaxial side of young leaf in #6 (Middle) and #14 (lower arrow), to lateral organs (Right) in #6, #14 (upper arrow), and #15 (lower arrow), and to the peripheral region of ectopic IM in #7 to #10 and #12 and #13. (Scale bar, 100 μm.) (D, E) A model of zmm8 and drl2 misexpression causing a fascicled inflorescence. Maize inflorescences are radially symmetrical, containing a single central/peripheral axis. Duplications of zmm8 and drl2 in Fas1 cause their misexpression in the apical meristem of Fas1 inflorescences at the transition stage, down-regulating meristem central specific genes including kn1, wus, OCs, and PHBs, and up-regulating meristem peripheral specific genes including LOBs, KANADIs, and YABBYs (E), which leads to suppression of the meristematic activity of the central cells and promotion of the meristematic activity of the peripheral cells, resulting in repeatedly bifurcated inflorescences. The yellow and white color of the words in E mark the up- and down-regulated genes, and the arrowheads showed the signal moved from the middle to the peripheral region of the meristem.
We focused on expression of genes related with organ polarity and inflorescence architecture and found meristem marker genes, such as knotted1 (kn1), wuschel1 (wus1), and wus2, were dramatically down-regulated in Fas1-2. Several other genes which are specifically expressed in the SAM tip, including maize orthologs of DRP4A, LOG7, AGO18A, OC564, and OC340 (29) were also down-regulated significantly (Fig. 5B and SI Appendix, Table S4). Given that YABBY genes play a role in polarity in Arabidopsis, we examined other polarity markers: adaxial/central zone-identity genes PHABULOSA (PHB), PHAVOLUTA (PHV), and REVOLUTA (REV); and abaxial/peripheral zone-promoting genes YABBY2 and YABBY3, KANADI1, KANADI2, and KANADI3. The PHB (ath14) maize homologs phb061, phb489, phb699, and phb246 are all down-regulated in Fas1-2, while the AS2 homologous genes (lob27 and lob31) and KANADI homologous genes (kanadi1, kanadi3, kanadi-like, Zmglk1, and mwp1) are all up-regulated in Fas1-2. Intriguingly, 12/13 of the differentially expressed YABBYs (including Arabidopsis YABBY2, YABBY3, and YABBY5 homologous genes) were also up-regulated in Fas1-2 (Fig. 5B and SI Appendix, Table S4). Down-regulation of central zone genes and up-regulation of peripheral zone genes suggest that cell fate and organ polarity of Fas1-2 IMs are altered.
Expression levels of these genes were also analyzed in Fas1-R (A188) with 2- to 3-mm ears using qRT-PCR. The data revealed that markers for meristematic activity and central zone showed similar expression patterns to that in Fas1-2. In contrast, markers for peripheral promoting genes (lob27, kanadi1, Zmglk1, mwp1, and yabby9/10/14/15) had reduced expression, showing the opposite trends with that in Fas1-2 (SI Appendix, Fig. S3C). These differences could be attributed from differences in ear developmental stage (30), inbred background, or allele.
To further explore the cell fate changes in Fas1, five differentially expressed marker genes were selected for in situ hybridization. We found the meristem marker knotted1, strongly expressed in the normal ear, covering the entire region of the IM (Fig. 5C #1). In Fas1-2 with two ectopic IM, the expression domain was reduced (Fig. 5C #2). The transcripts of phb489, homologous to PHAB, are highly enriched in the central region of the tip of a normal ear and are also detected in the boundary region between the IM and SPM primordia (Fig. 5C #3). In Fas1-2, which has two independent growth axes, no phb489 signal was detected in the central region of this stage and only a weak signal in the peripheral region (Fig. 5C #7 and #8). oc616 has been reported as specifically expressed in the center of the SAM (29). We found the transcript signal distributed throughout the immature ear in B73 (Fig. 5C #6), but it was strong in the apical cells and very weak in the central region of Fas1-2 IM with two bifurcations (Fig. 5C #9 and #10). The repressed expression of these two central zone-marker genes suggests that growth of the central region in the Fas1 ear has been suppressed. lob27, the as2 homologous gene, is expressed in the apical meristem of B73 ear (Fig. 5C #5). In Fas1-2, it is expressed in the boundary region between peripheral and center when the ear width increased (Fig. 5C #11), then shows broad expression in the ear with increased mild bifurcations, and finally appeared only in the ectopic IM (Fig. 5C #12 and #13). yabby15 (zyb15) is a robust marker of lateral organs and is absent from meristems (313233–34). We found zyb15 mRNAs accumulate in the bracts and the adaxial side of the young leaf in B73 ear (Fig. 5C #6) as noted previously (31), however, in Fas1-2, it is stronger in the adaxial side of the lateral organs and also expressed in the apical cells of the IM with two bifurcations and the bracts (Fig. 5C #14 and #15). In summary, these marker genes accumulate in the peripheral region of ectopic IMs but weakly in the central domain of the Fas1 ear, a pattern consistent with zmm8 and drl2. These results suggest the misexpression of zmm8 and drl2 changes central/peripheral cell fate in Fas1 IMs by targeting these genes directly or indirectly.
Discussion
The two Fas1 alleles arose spontaneously with no available progenitors. In independent experiments, the alleles mapped to the same interval of chromosome 9, containing two genes that encode transcription factors, a MADS-box, zmm8 and a YABBY gene, drl2 (Fig. 3A). Both zmm8 and drl2 are normally expressed in floral organs, with zmm8 in the FM itself and drl2 in the lateral organs of the FM (Fig. 4H #1 and #2, #6 and #7). RNA analysis shows that in the Fas1 mutants, expression of both genes is increased and shifted earlier in development (Fig. 4 A–C). In situ hybridization in Fas1 shows expression in the IM rather than the floral organs (Fig. 4H #3–#5, #8–#10). DNA blots show extra copies of both genes in the Fas1 alleles (Fig. 3C). Given that the inbred B97 also has a duplication of this locus, but both zmm8 and drl2 maintain normal expression (SI Appendix, Figs. S2 A and B and S3 F and G), we hypothesize there is a new promoter or regulatory sequence that is responsible for the misexpression. Our RNA sequencing data suggest that the misexpression of the central/peripheral genes leads to the fascicled ear and tassel with deep branching (Fig. 5).
The Biological Function and Coregulation of zmm8 and drl2.
MADS-box genes have been classified into “ABCDE” clades, which are widely used as a framework in shaping floral organ identity (3536–37). In maize, zmm8 and its duplicate, zmm14, are AGAMOUS-LIKE6 E-class genes. In angiosperms, E-class proteins are necessary components of B- and C-class tetramers (38, 39). The E-class SEPALLATA (123–4) genes of Arabidopsis function redundantly and in combination with A-, B-, and C-class genes. Loss-of-function mutants lose determinacy of FMs and display homeotic conversions (40, 41). A mutant phenotype has been identified for the zmm8 ortholog in rice, leafy hull sterile1 (lsh1) encoding OsMADS1. Missense mutations in the MADS domain of lhs1 result in open flowers with leafy lemma, palea, and lodicule, reduced stamens, and additional carpels. Another rice mutant in the SEPALLATA subfamily is panicle phytomer2 (carrying a mutation in OsMADS34), which converts some spikelets to branches, and the flowers have elongated rudimentary glumes and sterile lemmas (42). These phenotypes suggest a function in specifying floral organ identity (43, 44). Indeed, we discovered that the double mutant of zmm8/zmm14 fails to initiate floral organs and is indeterminate (SI Appendix, Fig. S4). Thus, their expression in the FM is required to promote floral organ initiation and determinacy.
YABBY proteins function to promote laminar growth of lateral organs, thus distinguishing them from the radially symmetrical SAM (33). Four of six Arabidopsis YABBY genes including FILAMENTOUS FLOWER (FIL), YABBY2, YABBY3, and YABBY5 are expressed in the abaxial side of lateral organs and are responsible for abaxial cell fate identity. Loss of function of fil and yabby3 leads to narrow leaves with loss of laminar expansion (10, 11). In rice, mutation of the FIL-clade gene, TONGARI-BOUSHI1 (TOB1), results in a prematurely terminated SM and reduced growth of the lemma or palea in the floret (45), while the drooping leaf (dl) mutant in the CRC/DL-clade showed a homeotic transformation of carpels into stamens and lack of midrib (46, 47). In maize, drooping leaf1 (drl1) and drl2 mutants have unfused carpels in the female florets and extra stamens in the male florets (28), in addition to the loss of midrib (27). When combined with the loss of function of maize AGAMOUS (zag1) (28), the FMs are more indeterminate, similar to those of zmm8/zmm14 (SI Appendix, Fig. S4C). Thus, both zmm8 and drl2 affect a common process when expressed in their normal domain.
While the knockout mutant (zmm8/zmm14-KO) may not help solve the question of how misexpression of zmm8 in Fas1 leads to the fascicled phenotype, it does provide insight into the potential regulation or interaction between zmm8 and drl2 in Fas1. A gene regulatory network analysis in maize suggested that drl1/2 and MADS-box genes, including zmm8 and zmm14, are coregulated and coexpressed (28). Indeed, we found that ZMM8 and DRL2 physically interact with each other in heterologous systems (SI Appendix, Fig. S4 F and G). Given the fact that overexpressing zmm8 does not cause a Fas1 phenotype (SI Appendix, Fig. S4A), our results suggest that an interaction of ZMM8 and DRL2 proteins might produce the Fas1 phenotype, rather than it being due simply to an increase in the level of only zmm8. In Arabidopsis, ectopic expression of FIL or YABBY3 are sufficient to specify abaxial cell fate, and high levels lead to enlarged meristems that arrest (10, 11). Although it is still possible that overexpression of drl2 alone causes the Fas1 phenotype, Fas1-Rev2 lost an extra copy of zmm8 but not drl2.
In summary, both zmm8 and drl2 have roles in regulating FM determinacy but are not expressed in overlapping domains. In Fas1, the misexpression of zmm8 and drl2 overlaps in the immature IM, affecting cell fate in the central/peripheral zone and thereby activating an ectopic laminar program and disrupting signaling necessary for maintenance of radially symmetrical IMs.
Tandem Duplications that Cause Dominant Phenotypes.
A high reversion rate, as noted with Fas1-R, is also seen with one of the dominant alleles of Kn1, Kn1-O (48). The tandem duplication of 17 kb produced two promoters in Kn1-O, one with sequence similarity to wild type and the other in a novel position, juxtaposed to the 3′ end of the duplicated copy. The promoter is considered the cause of the mutant phenotype because transposon insertions into the promoter repressed the mutant phenotype (49). The codominant Tunicate locus which confers the pod corn phenotype, in which each kernel is surrounded by derepressed glumes, palea, and lemma, is a duplication of another MADS-box gene, zmm19 (5051–52), and the ectopic expression pattern of zmm19 was caused by the rearrangement of a 5′-regulatory region of zmm19 by an unknown mechanism.
We hypothesize that a similar event occurred at Fas1; regulatory sequences that initiate expression of both zmm8 and drl2 earlier than normal cause the phenotype. Given the Fas1-R specific 5′ UTR for both zmm8 and drl2, sequence variation may extend to the longer upstream region of the duplicated genes to produce promoters or regulatory regions. In fact, this region may harbor the variation in Fas1-Rev1, which is normal in phenotype yet retains the copy number variation of Fas1-R. Although there are duplicated copies of zmm8 and or drl2 in inbreds B97 and Oh43, the Fas1 mutants have additional copies, the Fas1-R specific 5′ UTRs of zmm8 and drl2 are not present in B97 and Oh43, and misexpression of zmm8 and drl2 is not detected at the ear transition stage in these inbreds. We hypothesize that in Fas1 alleles, promoters or enhancers formed as a result of the duplication events. Distal regulation of gene expression has been described for teosinte branched1 (53) and unbranched3 (54). The unknown regulatory sequences at Fas1, whether they are enhancer–promoter or promoter–promoter chromatin interactions, cause ectopic expression of zmm8 and drl2 at an earlier stage than in wild type. Finding the exact sequence of these promoters will require further study but could be used for re-engineering gene expression to improve maize ear architecture.
Establishment of Radial Symmetry of the Maize IM.
The maize IM is radially symmetrical, containing a central/peripheral axis (55). Gene expression patterns and mutant phenotypes connect the central/peripheral axis of symmetric organs to the adaxial/abaxial axis of laminar lateral organs (2, 8). The ectopic expression of drl2 and zmm8 was accompanied by the up-regulation of peripheral cell fate promoting genes like LOBs, KANADIs, and YABBYs, which may promote the meristematic activity of the peripheral cells in the IM. The meristem-activating genes kn1, wus, OCs, and central zone marker genes PHB are down-regulated in the apical meristem of the Fas1 ear, showing suppression of the meristem central zone (Fig. 5 D and E).
A similar phenotype of bifurcated IM has been described in rice for the double mutant of the TOPLESS (TPL)-like transcriptional corepressor encoding gene aberrant spikelet and panicle1 (asp1) and Arabidopsis CLV3 homolog floral organ number2 (fon2) (56). Similar to Fas1, the IM was enlarged and then bifurcated. The asp1;fon2 phenotype differs in that the bifurcated IMs retain radial symmetry. Interestingly, loss of the WUS ortholog, tillers absent1 (tab1), did not affect this phenotype (56). It would be interesting to know if YABBY genes are differentially expressed in this mutant. If not, it appears there is more than one mechanism to bifurcate the IM.
Although mutants in maize orthologs of the CLV genes have enlarged meristems similar to those of Fas1, we do not think these genes are central to this phenotype. The CLV2 ortholog, fasciated ear2 and fon2-like cle protein1 are down-regulated in Fas1-2. The mutants of these genes have loss of function phenotypes that cause fasciation by loss of spatial regulation of wus. However, both wus1 and wus2 were also down-regulated in Fas1-2. Other genes in the CLV-WUS pathway such as Wuschel Homeobox (WOX) genes, Clavata (clv3)/embryo surrounding region (ESR) (CLE) genes, G protein-encoding genes, and Barely any meristem genes were not consistently differentially expressed (SI Appendix, Fig. S5B). We hypothesize that rather than differential expression of genes in the CLV-WUS pathway, the Fas1 phenotype is due to differential expression of genes specifying central/peripheral zone cell fate which then activates a laminar program and disrupts maintenance of a radially symmetrical IM. We propose that the duplications and rearrangements of zmm8 and drl2 in Fas1 alleles cause their misexpression in the IM at the transition stage, leading to suppression of meristematic activity of central cells and promotion of meristematic activity of peripheral cells, resulting in the repeatedly bifurcated inflorescences of this neomorphic mutant.
Materials and Methods
Plant Materials and Phenotypic Characterization.
The dominant Fascicled ear1-Reference allele (Fas1-R) was obtained from the Maize Cooperative Center and was backcrossed to B73 nine times and to A188 six times. Phenotypic characterization was done in backcross progenies and F2 families. Paul Weatherwax first described the Fas1 mutant in 1954 (25), contrary to erroneous citations of an earlier description. Based on sequence similarity to B97, we purport that this allele is the “recent” allele described by Paul Weatherwax (23). Fas1-Rev1 and Fas1-Rev2 were isolated in two revertant screens with 1/1,878 and 1/1,500 revertants per total, respectively. Fas1-R (Orr) was a gift from Marshall Sundberg (Louisiana State University) after it had been maintained by Alan Orr in the original landrace in which it was found. This line was the progenitor for Fas1-Rev1.
Fas1-2 came from a Chinese breeding line. It was crossed to B73, then self-pollinated to construct F2 populations and backcrossed to B73 for observations. Fas1-2/B73 F1 individuals appear similar to Fas1-2 homozygous mutants with bifurcated inflorescences. In the F2 populations with 282 individuals, there were 207 mutants and 75 wild-type plants, consistent with the expected ratio of 3:1 (χ2 test, P < 0.001). Inflorescence traits were measured for both alleles (SI Appendix, Tables S1 and S2).
For the zmm8 overexpression line, the zmm8 coding sequence was cloned into the modified pCAMBIA3301 vector to create the recombinant vector pUbi::zmm8 + YFP. For the zmm8/14 CRISPR-Cas9 KO line, we designed two guide RNAs (gRNA) for each gene and ligated four gRNAs into vector CPB-ZmUbi-hspCas to produce recombinant vector ZmpU6::zmm8 (gRNA1) + OspU3::zmm8 (gRNA2) + ZmpU6::zmm14 (gRNA1) + OspU3::zmm14 (gRNA2). The gRNA sequences list is in SI Appendix, Table S3. All genetic transformation was performed by the China National Seed Group Corporation, and the transgenic genetic background is ZZC01. Phenotypic characterization was done on T0 plants. For the zmm8 overexpression line, two independent transgenic events of five and eight transgenic plants each were observed; and for the zmm8/14 CRISPR-Cas9 KO line, three independent events with three editing types for each gene were detected (SI Appendix, Table S5).
SEM Observation.
Immature ear and tassel from 1 to 5 mm of the Fas1-2 (in B73), Fas1-R (in A188) homozygotes or heterozygotes, and wild-type siblings (in B73) were collected and fixed in 50% FAA solution (50% ethanol, 3.7% formaldehyde, and 5% glacial acetic acid (V/V/V)), then dehydrated through a graded series of ethanol from 30 to 95%. Tissues were dried using a Tousimis autosamdri-815 critical point dryer, sputter coated with gold palladium for 30 to 60 s, and observed using a Jeol JSM-7900F SEM as previously described (57).
Fine Mapping of Fas1 Locus.
Fas1-R was first mapped between visible markers wx1 and Rld, on the long arm of chromosome 9 using waxy translocation lines (24). In addition, Fas1-R was fine mapped in 426 backcrossing individuals (A188 as recurrent parent) with molecular markers developed from the syntenic region of B73 using the IBM2 2005 Neighbors genetic map available at MaizeGDB (58). Primers used in the study are listed in SI Appendix, Table S3.
For the Fas1-2 mapping, 286 F2 individuals derived from Fas1-2 × B73 were constructed. For the bulked-segregant RNA sequencing mapping, 30/30 1- to 2-cm ears of the Fas1-2 and wild-type individuals were collected for RNA extraction (with TRIzol reagent (Life Technologies)). The two RNA libraries were sequenced on a NextSEq. 441 Illumina platform with PE75 (paired-end), and 18 Gb data were aligned to the maize genome B73 RefGen version 3 (AGP (A Golden Path) version 3.31) using Tophat2-PE [2.0.9 (59)]. Then, the initial mapping region was narrowed to ∼9 Mb, flanked by markers umc2159 and phi108411. Furthermore, to fine map the Fas1-2, 15 markers were developed (SI Appendix, Table S3), and a large mapping population with 7,680 F2 individuals was constructed.
DNA Blotting.
Approximately 1.5g B73 leaf tissue, wild-type Fas1-2, Fas1-R, Fas1-Rev1, and Fas-Rev2 were used for high quality (>1 μg ⋅ μL) genomic DNA extraction by hexadecyltrimethylammonium bromide methods. Approximately 30μg DNA sample was separately digested by restriction enzyme HindIII (50 μ with 10 × M buffer), XbaI (50 μ with 10 × M buffer), and NcoI (50 μ with 10 × K buffer) at 37 °C for 16 h. The digested gDNA products were separated by electrophoresis at 30 V for 16 h on 0.8% agarose gel with 1 × TAE buffer (40 mM Tris base, 20 mM acetic acid, 1 mM Ethylenediaminetetraacetic acid disodium salt dehydrate, pH 8.3), then transferred to a positively charged nylon membrane using a capillary method. Both zmm8 and drl2 gene-specific probes (SI Appendix, Table S3) were labeled by Digoxin, then blotted and detected using Roche Southern blot kit (DIG High Prime DNA Labeling and Detection Starter Kit II, Roche).
Gene Expression Analysis.
To analyze the expression pattern of zmm8 and drl2, 20 to 30 1- to 2-mm–length immature ears of the Fas1-2 (B73) and wild type (B73), six to eight 2- to 3-mm–length ears of the Fas1-R (A188) and wild type (A188) were collected, respectively. Total RNA was extracted using TRIzol reagent (Life Technologies) according to the manufacturer’s instructions, purified, and reverse transcribed using an EasyScript one-step gDNA removal and cDNA-synthesis Supermix Kit (Transgene). GSPs (SI Appendix, Table S3) combining 2 × GoTaq Green Master Mix (Promega) or SYBR Green PCR Master Mix (KAPA) were used for RT-PCR and qPCR, respectively. qPCR was performed with three biological and three technical replicates, the maize beta-actin (NM_001155179) gene was used as an internal control. RT-PCR and RNA sequencing were performed with three biological replicates.
Rapid-Amplification of cDNA Ends.
Gene 5ʹ RACE was performed with RNA extracted from 2- to 3-mm–length ears of Fas1-R heterozygotes (A188), wild type (A188), Fas1-2 homozygotes, and 5- to 8-mm–length ears of Fas1-Rev1, Fas1-Rev2 heterozygotes, and A188, B73 using GeneRacer Kit (Invitrogen, L1502–02). After PCR amplification with GeneRacer 5 primer and reverse GSP1 using 2 × GoTaq Green Master Mix (Promega), then followed by the second cycle PCR amplification with GeneRacer 5 primer and reverse GSP2. The PCR products were purified using QIAquick PCR purification kit (Qiagen), the purified PCR products were cloned into pGEM-Teasy vector (Promega). At least 20 positive clones were sequenced for each sample. The sequences were analyzed using CLC sequence viewer 7.6; gene models of zmm8 and drl2 in B73 RefGen_version 4 were used to guide annotation. GSPs for RACE assay were listed in SI Appendix, Table S3.
Transcriptome Profiling.
Approximately 1- to 2-mm–length immature ears of the Fas1-2 (B73) and wild type (B73) were collected for RNA extraction. After purification, RNAs were used for RNA sequencing by Beijing Genomics Institute on the Illumina system HiSeq2500 (Illumina Inc.). The FASTX-Toolkit was used to preprocess raw reads (60), followed by FASTQC program to assess the clean reads (61), then aligned to the B73 RefGen_version 4 using TOPHAT version 2.1.0 (59). CUFFLINKS version 2.1.1 (62) was used to normalize and estimate the gene expression level based on fragments per kilobase of transcript per million reads (63). The differentially expressed genes (DEGs) were also calculated by CUFFLINKS version 2.1.1 at a significance level of P < 0.05. The DEGs were listed in SI Appendix, Table S3.
mRNA In Situ Hybridization.
Immature ears of Fas1-2 (B73) and B73 1 to 5 mm in length were harvested and fixed in 4% PFA solution (4g paraformaldehyde (Sigma-Aldrich) dissolved in 100 mL 1 × PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 0.137 M NaCl, 2.7 mM KCl, pH 7.4), pH 6.5 to 7)), then dehydrated with an ethanol series and embedded in Paraplast Plus (Sigma) and sectioned to a thickness of 8 μm. Antisense RNA probes were amplified with GSPs (SI Appendix, Table S3) adding an SP6 sequence, then purified and synthesized using SP6 RNA polymerase with digoxigenin-UTP as a label (Roche), respectively. RNA hybridization and immunologic detection were performed as described previously (64).
Protein–Protein Interactions.
To analysis protein–protein interactions, firefly LCI and yeast one-hybrid assays (Y2H) were performed. In LCI, the full-length coding sequence of zmm8 and drl2 were cloned into JW771 (NLUC) and JW772 (CLUC) to produce ZMM8–NLUC and DRL2–CLUC vectors, respectively. Agrobacterium tumefaciens GV3101 cells were used for transformation, and the Nicotiana benthamiana was used for transient expression with three replicates. After 48 h of growth under a 16 h light/8 h dark cycle, 1 mM luciferin (Promega) was applied to the abaxial epidermis of each leaf, and the luciferin signal were detected and captured as described in (54).
For Y2H, the full-length CDS of zmm8 and drl2 was cloned into pGBKT7 and pGADT7 (Clontech) to generate pGBKT7–ZMM8 and pGADT7–DRL2, respectively. The constructs were transformed into yeast strain AH109 as described in (65), and yeast two-hybrid assays were performed according to the description of Matchmaker Gold Two-Hybrid System user manual (66). Primers used for vector constructions were listed in SI Appendix, Table S3.
Acknowledgements
We thank Joshua Strable, Sam Leiboff, Zhaobin Dong, and Lei Liu for reviewing the manuscript; Zengdong Tan and Manfei Li for the RNA sequencing analysis; Katarina Makmuri for assistance with mapping Fas1-R; Margaret Woodhouse of the maize genome database for assistance with the NAM genomes; and George Chuck and Nathalie Bolduc for technical advice. We also thank De Wood for assistance with SEM at the US Department of Agriculture (Albany, CA). This work was supported by the National Natural Science Foundation of China (91935305), the National Key Research and Development Program of China (2016YFD0100404), and the US NSF (IOS-1733606 and IOS-1755141).
Data Availability.
RNA-seq data are deposted at NCBI (National Center for Biology Information), accession number: PRJNA698219.
References
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
56
57
58
59
60
61
62
63
64
65
66