
 ,
Jonathan P. Davies,
Samantha M. Lisy,
Reyhaneh Tirgar,
Sirena C. Tran,
Lars Plate
,
Jonathan P. Davies,
Samantha M. Lisy,
Reyhaneh Tirgar,
Sirena C. Tran,
Lars Plate
Edited by Gaya K. Amarasinghe, Washington University School of Medicine, St. Louis, MO, and accepted by Editorial Board Member Michael F. Summers December 7, 2020 (received for review June 12, 2020)
Author contributions: K.M.A. and L.P. designed research; K.M.A., J.P.D., S.M.L., R.T., S.C.T., and L.P. performed research; K.M.A., J.P.D., and L.P. analyzed data; and K.M.A., J.P.D., and L.P. wrote the paper.
Viral infections continue to pose a grave global health threat, necessitating the development of new broadly applicable therapeutic strategies, including the targeting of conserved host processes that are exploited during viral infections. Flaviviruses, such as dengue and Zika, depend on extensive engagement of the endoplasmic reticulum (ER) proteostasis network to aid with replication, production, and secretion of virions. Here, we identify a host-centered antiviral approach by targeting these conserved ER protein quality-control processes that are required for viral infection. We find that the small-molecule regulator of ER proteostasis 147 can broadly and safely inhibit dengue and Zika infection without inducing toxicity to the host cells.
Flaviviruses, including dengue and Zika, are widespread human pathogens; however, no broadly active therapeutics exist to fight infection. Recently, remodeling of endoplasmic reticulum (ER) proteostasis by pharmacologic regulators, such as compound 147, was shown to correct pathologic ER imbalances associated with protein misfolding diseases. Here, we establish an additional activity of compound 147 as an effective host-centered antiviral agent against flaviviruses. Compound 147 reduces infection by attenuating the infectivity of secreted virions without causing toxicity in host cells. Compound 147 is a preferential activator of the ATF6 pathway of the ER unfolded protein response, which requires targeting of cysteine residues primarily on protein disulfide isomerases (PDIs). We find that the antiviral activity of 147 is independent of ATF6 induction but does require modification of reactive thiols on protein targets. Targeting PDIs and additional non-PDI targets using RNAi and other small-molecule inhibitors was unable to recapitulate the antiviral effects, suggesting a unique polypharmacology may mediate the activity. Importantly, 147 can impair infection of multiple strains of dengue and Zika virus, indicating that it is suitable as a broad-spectrum antiviral agent.
Flaviviruses are significant human pathogens that cause widespread mortality around the world. The genus encompasses several arthropod-borne viruses, including dengue (DENV), yellow fever, and Zika (ZIKV) (12–3). These viruses pose serious public health threats, particularly as increased global travel and climate change have caused an increase in the spread of both the virus and their mosquito vectors (Aedes albopictus and Aedes aegypti) (4, 5). It is estimated that 53% of the world population lives in areas suitable for dengue transmission, resulting in 50 to 100 million dengue infections per year, of which 500,000 proceed to dengue hemorrhagic fever and 22,000 are fatal (1, 6). ZIKV infection has been linked to serious neurological defects, including Guillain–Barré syndrome and microcephaly in newborns (7). While ZIKV diagnoses were relatively rare until the early 2000s, the 2015–2016 pandemic in the Americas highlighted the potential for rapid spread of the disease vectors (3).
Vaccines currently exist for only a limited range of flaviviruses (8). The first DENV vaccine, Dengvaxia, was approved by the Food and Drug Administration in early 2019 (9). However, its use remains limited to children with confirmed prior infection. In addition, no postexposure therapeutic options are available for patients infected with these viruses; current treatments only attempt to alleviate the symptoms (1011–12). Antiviral strategies often focus on directly targeting viral proteins. For example, while molecules inhibiting the flavivirus RNA-dependent RNA polymerase NS5 or the protease NS3 have been identified, the high mutation rate of the virus allows for resistance to be developed quickly (10, 13, 14). One increasing area of exploration for alternative therapeutic approaches is the targeting of host factors that are critical for virus propagation (15). As these proteins are not under genetic control of the virus, development of resistance is much less likely (1617–18). In addition, host-targeted therapeutics should be effective as broad-spectrum antivirals instead of targeting a single virus (1920–21).
Flaviviruses contain a genome of positive-sense, single-stranded RNA (+ssRNA) ∼11 kb in size. The viruses replicate and assemble around the endoplasmic reticulum (ER) membrane (22). As the genome is translated after entry into the cell, the single polyprotein is inserted into the ER membrane and is co- and posttranslationally processed into three structural (capsid, premembrane, and envelope) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) proteins (23). The host proteostasis network has been implicated in the maintenance of individual viral proteins and virus evolution (20, 2425–26). The proteostasis network is comprised of chaperones, cochaperones, and other protein quality-control factors that control protein folding, assembly, posttranslational modification, trafficking, and degradation. Genetic screens have identified many proteostasis factors as essential for DENV replication, including several components of the oligosaccharyl transferase (OST) complex, as well as the ER membrane protein complex (272829–30). Aside from promoting biogenesis of viral proteins, these components also have additional roles in organizing viral replication centers at the ER membrane (28, 29, 31). Additionally, diverse cellular chaperone and cochaperone systems are required for all stages of the viral life cycle, including virus entry and disassembly, folding of individual viral proteins, as well as assembly and egress of new virions (21, 32, 33). Pharmacologic inhibition of proteostasis factors, including the OST as well as cytosolic Hsp70 and Hsp90 chaperones, has been shown to be an effective strategy to reduce flavivirus infection in cell models (19, 21, 333435–36).
As flavivirus replication and polyprotein translation occurs at the ER membrane, infection results in expansion of the ER and remodeling of the ER proteostasis environment (31, 37, 38). DENV infection leads to modulation of the unfolded protein response (UPR), the adaptive stress response that remodels the ER proteostasis environment to counter stress caused by accumulation of misfolded proteins (39, 40). The UPR consists of three overlapping but distinct signaling branches downstream of the stress-sensing receptors IRE1 (inositol-requiring enzyme 1), ATF6 (activating transcription factor 6), and PERK (protein kinase R-like ER kinase) (41, 42). The first two branches primarily control the up-regulation of chaperones and other proteostasis factors to expand the protein-folding capacity of the ER. The PERK branch is responsible for translational attenuation via phosphorylation of eukaryotic initiation factor 2α (eIF2α) and subsequent activation of the integrated stress response. DENV has been shown to up-regulate the IRE1/XBP1s and ATF6 branches of the UPR, while suppressing activation of the PERK branch (39).
Given the dependencies of DENV and other flaviviruses on UPR modulation and an enhanced ER proteostasis environment in the host cells during infection, we sought to explore whether these dependencies can be perturbed pharmacologically to impair viral infection. Pharmacologic remodeling of ER proteostasis pathways has become an attractive strategy at correcting imbalances associated with diverse phenotypes related to protein stress and misfolding without affecting endogenous protein maturation or causing toxicity (43444546–47). In particular, compound 147, which was developed as a preferential activator of the ATF6 pathway, has broadly beneficial effects at reducing secretion of amyloidogenic proteins and protecting against oxidative organ damage from ischemia/reperfusion (43, 46).
Here, we demonstrate that ER proteostasis remodeling by compound 147 serves as an effective strategy to reduce flavivirus infection. We determine that 147 reduces viral replication but particularly lowers the infectivity of secreted virions. The replication and assembly defect is surprisingly not mediated through activation of ATF6. Instead, the activity is mediated by upstream covalent modifications of protein targets by 147. Prior work identified protein disulfide isomerases (PDIs) as critical targets for the 147-dependent ATF6 activation (48); however, our data suggest that PDIs are not fully responsible for the antiviral activity. Finally, we show that 147 treatment can reduce proliferation of multiple DENV serotypes and several ZIKV strains, demonstrating that the pharmacologic agent could be a broadly effective strategy against flaviviruses and other viruses that depend on ER proteostasis processes.
DENV is known to activate the UPR in infected host cells and was previously found to specifically up-regulate the ATF6 and IRE1/XBP1s signaling arms, which transcriptionally up-regulate ER protein quality-control factors and adjust proteostasis capacity (Fig. 1A) (39, 42). To confirm the activation of ER proteostasis pathways by DENV, we performed an infection time-course with DENV serotype 2 (DENV2) in Huh7 liver carcinoma cells and measured transcript levels of an ATF6-regulated gene (PDIA4) and an XBP1s-regulated gene (ERDJ4) by qRT-PCR. Both PDIA4 and ERDJ4 transcripts exhibited a time-dependent induction in response to DENV2 infection (Fig. 1B). The late onset of UPR activation and extent of transcript and protein induction was consistent with prior studies in other cell lines (39). While a similar time-dependent up-regulation of ATF6 and XBP1s-regulated protein products could be observed by western blot, the up-regulation of individual proteins was variable (SI Appendix, Fig. S1 A and B). To more broadly understand how DENV regulates the proteostasis network in Huh7 cells, we performed quantitative proteomics analysis at 24 and 36 h postinfection (hpi) with DENV2 to quantify global changes in protein expression (SI Appendix, Fig. S1 C and D and Dataset S1). We found that DENV2 activated specific factors involved in ER proteostasis maintenance, such as DNAJC10, BiP, SRPRB, and HYOU1. We also filtered previously defined gene sets consisting of ∼20 specific genes for each of the UPR signaling branches to quantify the cumulative activation of ER proteostasis pathways (Fig. 1C) (49). This analysis indicated a mild stimulation of ATF6- and XBP1s-regulated proteins, while PERK-regulated proteins remained largely unchanged. In contrast, we did not observe up-regulation of cytosolic proteostasis factors that are under control of the heat shock response (HSR) (50). Overall, our results confirm that DENV2 preferentially remodels ER proteostasis pathways through ATF6 and IRE1/XBP1s-dependent up-regulation of specific chaperones and protein-folding factors.

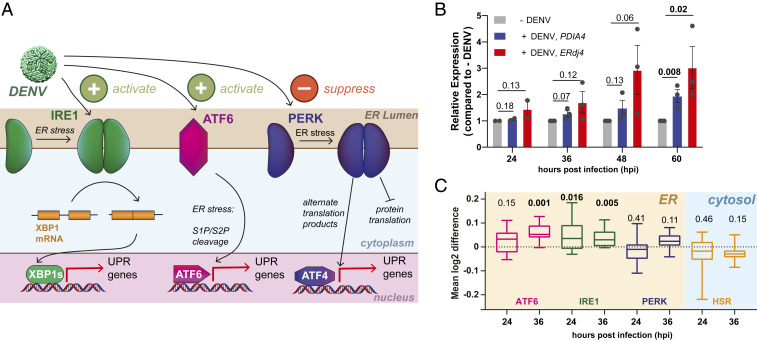
DENV infections activates the ER UPR. (A) Schematic overview of the UPR showing the three signaling branches: IRE1/XBP1s, PERK, and ATF6. ER protein misfolding stress induces dimerization of IRE1 and PERK, followed by autophosphorylation and activation of downstream signaling pathways. ER stress activation of ATF6 results in trafficking to the Golgi and proteolytic cleavage by S1/S2 proteases. Prior work showed that DENV increased activity of the IRE1/XBP1s and ATF6 branches, while simultaneously preventing activation of the PERK branch (as indicated at the top) (39). (B) Time course of qPCR data showing transcriptional up-regulation of an IRE1/XBP1s target (ERdj4) and an ATF6 target (PDIA4) in response to DENV activation (MOI 3) in Huh7 cells. Error bars shows SEM from two to three biological replicates and P values from two-tailed unpaired Student t tests are displayed. (C) Box plots of proteomics data showing the aggregate up-regulation of IRE1/XBP1s and ATF6 protein targets at 24 and 36 hpi. PERK and cytosolic HSR targets are not affected. Huh7 cells were infected with DENV (MOI 3) for 24 h or 36 h. Gene sets for UPR and HSR pathways were defined based on prior transcriptional profiles (49). P values from two-tailed Wilcoxon signed rank tests are indicated. Cell-wide proteomics data comparing DENV infected to noninfected Huh7 cells is shown in SI Appendix, Fig. S1 C and D.
Considering the dependencies of DENV and other flaviviruses on UPR activation, we determined the impact of the proteostasis regulators 147 (a preferential activator of the ATF6 branch), and Ceapin-A7 (Cp-A7; a selective inhibitor of the ATF6 branch) on DENV infection (Fig. 2A). We pretreated Huh7 cells with the respective compounds 16 h prior to infection by DENV2 to induce or inhibit UPR-dependent proteostasis remodeling, and we retreated immediately after infection. Quantification of DENV viral titers by a focus-forming assay demonstrated a significant reduction in infection with compound 147 at 24 and 36 hpi (Fig. 2B). In contrast, Cp-A7 treatment only resulted in a small reduction in viral titers at 24 hpi but the infection recovered at 36 hpi. Consistent with experiments in other cell lines, treatment with Cp-A7 did not induce a large amount of cell toxicity. Compound 147 at 10 µM caused a small reduction (approximate 20%) in cell proliferation when quantifying ATP levels by CellTiter-Glo (SI Appendix, Fig. S2A) (46). We conducted dose–response studies with 147 demonstrating that the compound was effective at reducing DENV proteins at an IC50 of ∼1 µM (SI Appendix, Fig. S2B). Cell viability was maintained above 90% at this concentration (SI Appendix, Fig. S2C). To determine if this viability change was a result of active cell death, we measured the increase in caspase activity induced by DENV, 147, or the combination of both to quantify apoptosis induction (SI Appendix, Fig. S2D). On their own, DENV and 147 did not stimulate an increase in caspase activity, while the combined DENV infection with 147 treatment (above 3 µM) led to a very mild 1.5-fold increase in caspase activity. However, this induction was much lower than with short treatment of the known caspase activator staurosporin (SI Appendix, Fig. S2D), indicating that any change in ATP levels is likely due to cytostatic activity, rather than a cell-death–mediated pathway. These results demonstrate that modulation of the ER proteostasis network with the preferential ATF6 activator 147 could represent an effective strategy to impair DENV2 infection.
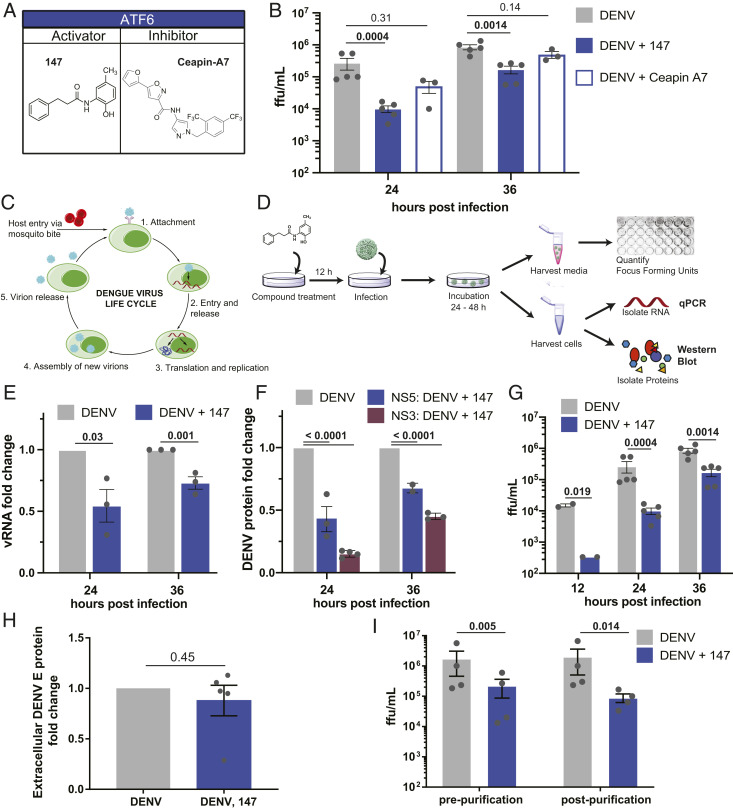
Treatment with small molecule 147 reduces vRNA, protein, and titer levels. (A) Chemical structures of small-molecule ATF6 activator 147 and ATF6 inhibitor Cp-A7. (B) Focus forming assay to quantify changes in dengue infection in response to treatment with ATF6 modulator compounds. Huh7 cells were treated with compounds, and 16 h later infected with DENV2 BID-V533 at a MOI of 3 for 3 h. Media and treatments were replaced, and cells and media were harvested at indicated time points and virus focus forming units (ffu/mL) quantified. Error bars show SEM and P values from ratio paired t tests are indicated. (C) Schematic of the flavivirus life cycle. After attachment of the virus to a cellular receptor and clathrin-mediated endocytosis, fusion of the envelope protein with the endosome causes release of the capsid and RNA genome. The positive-sense genome is then translated, providing the machinery to begin forming replication pockets at the ER membrane. Once sufficient proteins are available, new virions are assembled, trafficked through the Golgi network, and released into the extracellular space. (D) Experimental workflow for quantifying vRNA, viral protein, and titer levels. After pretreatment with compound 147, cells are infected, retreated with 147, and left for harvesting at later time points. Media samples are taken to quantify ffu; cellular samples taken to quantify vRNA by qPCR or viral proteins by western blot/proteomics analysis. (E) Bar graph showing reduction in vRNA in response to 147 treatment as outlined in D 24 and 36 hpi. Error bars correspond to SEM of three biological replicates and P values from unpaired t tests are shown. (F) Bar graph showing reduction in NS3 and NS5 viral protein levels in response to 147 treatment as outlined in D 24 and 36 hpi. Error bars correspond to SEM from two to three biological replicates per protein and P values from unpaired t tests are shown. Representative western blots are shown in SI Appendix, Fig. S2E. (G) Bar graph showing reduction of DENV viral titers in response to 147 treatment as outlined in D 24 and 36 hpi. Error bars correspond to SEM from two to five biological replicates and P values from ratio paired t tests are shown. (H) Quantification of envelope (“E”) protein from extracellular virus showing that 147 does not reduce viral structural proteins levels. Secreted virions (24 hpi) were purified and concentrated from the media by ultracentrifugation on a sucrose cushion. The same volume of sample was analyzed by SDS/PAGE and western blots (representative blot of E and prM proteins shown in SI Appendix, Fig. S2K). Error bars correspond to SEM from five biological replicates and P value from unpaired t tests are shown. (I) Bar graph showing reduction of DENV viral titers in response to 147 treatment 24 hpi as pre- and postpurification, as described in H. Error bars correspond to SEM from four biological replicates and P values from ratio paired t tests are shown.
The ER plays critical roles in several stages of the viral life cycle, including replication of viral RNA (vRNA) at replication centers on the cytosolic side of the ER membrane, translation and proteolytic processing of the viral polyprotein in the ER membrane, as well as the folding, assembly, and secretion of new virions (51, 52). To determine at what stage in the viral life cycle the compound treatment impaired viral propagation, we investigated the impact of compound 147 on vRNA and protein levels at different time points postinfection (Fig. 2 C and D). The replicative cycle of DENV lasts ∼24 h; therefore the 12- and 24-hpi time points corresponds to initial infection, while the 36-hpi time point includes reinfection. Quantification of vRNA by qPCR indicated a modest but significant reduction of 50% at 24 hpi, which was attenuated to 30% reduction at 36 hpi (Fig. 2E). Western blot quantification of NS3 and NS5 viral protein in Huh7 cell lysates from DENV2-infected cells showed a 35 to 80% reduction in viral nonstructural proteins in response to 147 treatment (Fig. 2F and SI Appendix, Fig. S2E). This highlighted that the ER proteostasis regulator exerted a more pronounced effect on viral protein production relative to replication of vRNA. In contrast, infectious viral titers showed a far greater reduction reaching 98% at 12 hpi and sustaining 80% reduction at 36 hpi (Fig. 2G). We confirmed that reduction in viral proteins are sustained 48 hpi and that renewed treatment with 147 24 hpi does not further reduce viral propagation (SI Appendix, Fig. S2F).
The intensified effect of 147 on viral titers compared to vRNA and protein levels suggests that the compound predominantly acts on a stage subsequent to translation of viral proteins, which would be consistent with disruption of viral maturation or secretion pathways through modulation of ER proteostasis. We confirmed that 147 did not impair viral entry. Omission of the pretreatment prior to infection and treatment of Huh7 cells with the compound only after infection still resulted in a comparable reduction in viral protein and titers, confirming that the compound must act at a postentry stage (SI Appendix, Fig. S2 G–J).
To further disentangle the mechanism by which 147 may be impairing viral assembly and egress, we compared the reduction in intracellular vs. extracellular viral protein levels by western blot. Extracellular virus secreted into the media was concentrated 24 hpi by ultracentrifugation. While the reduction of nonstructural protein levels intracellularly was up to 80% (Fig. 2F and SI Appendix, Fig. S2E), matching the decrease seen with nonstructural proteins, no decrease was observed in the extracellular structural protein levels (E and prM) (Fig. 2H and SI Appendix, Fig. S2K). Importantly, the reduction in secreted structural protein levels is markedly less than the observed ∼95% reduction in infectious titer levels observed from the same samples (Fig. 2I). These combined findings suggest that 147 is affecting DENV assembly and secretion in a way that may lead to secretion of noninfectious virions. To support this mechanism, we performed qPCR on the purified extracellular virus samples to measure if there was a change in RNA similar to the change in titer. In support of the assembly defect, this reduction in extracellular vRNA was much smaller than the reduction in titers and not statistically significant (SI Appendix, Fig. S2L). These data suggest that the reduction in titer is not simply due to the presence of empty virions in the supernatant.
Given that flavivirus infection activates the ATF6 pathway, it seemed surprising that preactivation could reduce viral propagation. We therefore sought to investigate whether ATF6 activation and induction of ATF6-targeted proteostasis factors was required for the 147-dependent inhibition of DENV infection. We first confirmed by qPCR and quantitative western blot analysis that treatment with 147 activated the ATF6 regulated gene HSPA5 (BiP) in Huh7 cells (SI Appendix, Fig. S3A). To probe the impact of ATF6 activation on the compound activity, we took advantage of the ATF6 inhibitor Cp-A7, which inhibits ATF6 (53, 54). Cp-A7 mediates a neomorphic interaction between ATF6 and the peroxisomal membrane protein ABCD3, keeping ATF6 in a trafficking-incompetent oligomeric state, which cannot be activated by 147 (Fig. 3A) (55). We confirmed by qPCR and western blot that cotreatment with 147 and Cp-A7 could attenuate the 147-dependent induction of ATF6 target genes in Huh7 cells (SI Appendix, Fig. S3 A and B). Furthermore, quantitative proteomics data of cell-wide expression changes confirmed the reduced induction of larger set of ATF6 target genes (SI Appendix, Fig. S3C and Dataset S2). We then investigated the addition of Cp-A7 on the 147-mediated reduction of DENV propagation. Cotreatment of Cp-A7 did not diminish the reduction in vRNA or NS3 protein (Fig. 3 B and C). Furthermore, the addition of Cp-A7 only partially recovered the DENV2 viral titers at 24 hpi (Fig. 3D). These results highlight that the reduction in viral titers could not be fully attributed to the 147-mediated induction of ATF6 target genes.
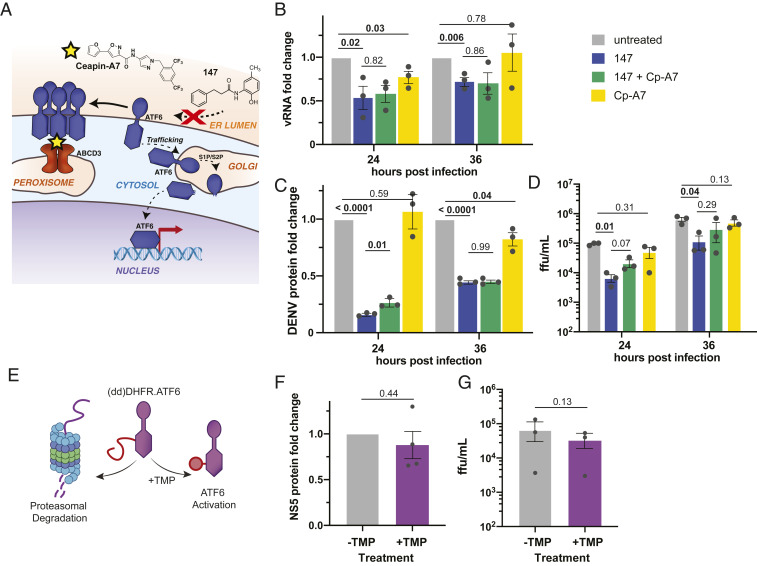
ATF6 inhibition does not attenuate the 147-mediated reduction in DENV replication. (A) Cp-A7 acts as an ATF6 inhibitor downstream of 147 by tethering inactive ATF6 to the peroxisomal membrane protein ABCD3 preventing ATF6 trafficking to the Golgi and activation through S1/S2 cleavage. (B) Bar graph showing the reduction in vRNA levels in response to 147 and Cp-A7 treatment measured by qRT-PCR in Huh7 cells infected with DENV as outlined in Fig. 2D. Cp-A7 does not attenuate the 147-mediate reduction. Error bars correspond to SEM from three biological replicates and P values from unpaired t tests are shown. (C) Bar graph showing the reduction in NS3 viral protein levels in response to 147 and Cp-A7 treatment measured by western blot in Huh7 cells infected with DENV as outlined in Fig. 2D. Cp-A7 does not attenuate the 147-mediate reduction. Error bars correspond to SEM from three biological replicates and P values from unpaired t tests are shown. Representative western blots are shown in SI Appendix, Fig. S2E. (D) Bar graph showing the reduction in DENV viral titers in response to 147 and Cp-A7 treatment in Huh7 cells infected with DENV as outlined in Fig. 2D. Cp-A7 only minimally attenuates the 147-mediate reduction at 24 hpi. Error bars correspond to SEM from three biological replicates and P values from paired ratio t tests are shown. (E) Schematic of the ddDHFR.ATF6 construct, which is constitutively degraded in the absence of the stabilizing ligand TMP. TMP addition leads to accumulation of ATF6 and transcriptional activation of ATF6 target genes. (F) Graph showing NS5 viral protein levels in DENV infected Huh7 cells that were transiently transfected with ddDHFR.ATF6. ATF6 was activated through addition of TMP 16 h prior to DENV infection (MOI 3). Data points were collected 24 hpi. Error bars correspond to SEM from three biological replicates and P value from unpaired t test is shown. Representative western blots are shown in SI Appendix, Fig. S3G. (G) Graph showing DENV viral titers in infected Huh7 cells that were transiently transfected with ddDHFR.ATF6. ATF6 was activated through addition of TMP 16 h prior to DENV infection (MOI 3) and DENV ffu were quantified 24 hpi. Error bars correspond to SEM from three biological replicates and P value from ratio paired t test is shown.
To further probe whether the reduced viral propagation could be ascribed to activation of the ATF6 pathway, we took advantage of an orthogonal chemical genetic approach to selectively induce the ATF6 pathway independent of global ER stress. We transiently transfected a destabilized domain (dd)DHFR.ATF6 construct into Huh7 cells. This construct is constitutively degraded in the absence of a small-molecule ligand, but can be stabilized through addition of trimethoprim (TMP), leading to accumulation of DHFR.ATF6 and selective induction of ATF6-regulated genes (Fig. 3E) (42). We confirmed TMP-dependent up-regulation of ATF6-regulated targets BiP, PDIA4, and GRP94 in Huh7 cells (SI Appendix, Fig. S3 D–F). Next, we pretreated Huh7 cells with TMP to stabilize ddDHFR.ATF6, infected cells with DENV2, and quantified propagation of the virus by monitoring viral protein levels by western blot and measuring infectious titers. Chemical-genetic ATF6 activation did not lead to a measurable reduction in viral protein levels (Fig. 3F and SI Appendix, Fig. S3G). At the same time, a moderate decrease in viral titers could be observed, but this reduction in viral propagation was much lower than seen with 147 treatment (Fig. 3G). Together, the results from ATF6 inhibition and chemical-genetic ATF6 activation indicate that induction of ATF6-regulated ER proteostasis factors does not impact vRNA replication or production of viral proteins, and only partially accounts for the reduction in DENV2 viral titers.
Considering that reduced viral propagation in response to 147 was only partially mediated by ATF6 activation, we sought to explore other mechanisms of how the molecule could impair the virus. Previous studies showed that 147 is a prodrug and requires metabolic activation in cells to generate a reactive p-quinone methide that can then form protein adducts with reactive cysteine residues (Fig. 4A) (46, 48, 56). In several cell types, ER-resident PDIs were identified as the common protein targets of 147, and this modulation of PDIs was linked to the activation of ATF6 (48). To explore whether covalent targeting of reactive thiols by 147 is required for the reduction in DENV propagation, we blocked the covalent modifications through addition of an excess of the small-molecule thiol 2-mercaptoethanol (BME) to the cells treated with 147 (Fig. 4A). We confirmed that this addition did not impair cell viability (SI Appendix, Fig. S4B), and that BME alone only had a minimal effect on NS3 viral protein levels (SI Appendix, Fig. S4A). When BME was added to DENV-infected cells that were treated with 147, this resulted in a partial to complete recovery of viral proteins at 24 and 36 hpi, respectively (Fig. 4B and SI Appendix, Fig. S4C). Similarly, the reduction in DENV2 viral titers was attenuated by the addition of BME (Fig. 4C). These results indicate that the targeting of cellular thiol groups is required for the inhibition of virus propagation.
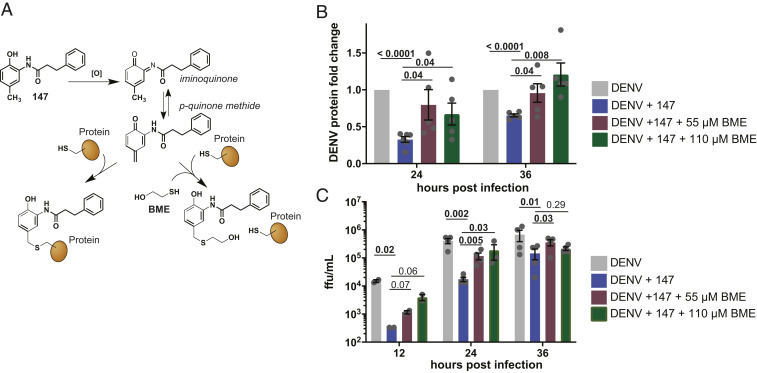
147-mediated reduction in DENV infection is sensitive to small-molecule thiols. (A) Schematic outlining the metabolic activation mechanisms of 147. After oxidation by P450 enzymes, the generated p-quinone methide can react with thiol nucleophiles such as cysteine residues on cellular protein targets. Addition of exogenous free thiols, such as BME, can quench the active form of 147 before it reacts with protein targets. (B) Graph showing reduction in DENV NS3, NS5, or E protein levels in response to 147 treatment and addition of BME. Huh7 cells were pretreated with 147 and indicated concentrations of BME 16 h prior to DENV infection (MOI 3) and immediately after infection. Protein levels were quantified by western blot. Error bars show SEM and P values from unpaired t tests are shown. Representative western blot is shown in SI Appendix, Fig. S4C. (C) Bar graph showing DENV viral titer levels in response to 147 treatment and rescue through addition of BME. Huh7 cells were pretreated with 147 and indicated concentrations of BME 16 h prior to DENV infection (MOI 3) and immediately after infection, and viral ffu were quantified by a focus forming assay. Error bars show SEM and P values from ratio paired t tests are shown.
We next explored whether targeting of specific PDI proteins by 147 was required for the reduced viral replication. Previous studies determined the covalent targets of 147 in HEK293T, HepG2, and ALLC plasma cells (48). However, it was conceivable that the metabolic activation mechanism differs in cell types and could result in alternative targets. We therefore determined whether 147 could similarly target PDIs in Huh7 cells. We took advantage of the active analog 147-20, which contains an alkyne handle that enables further click chemistry derivatization with desthiobiotin after protein labeling, followed by isolation of the targeted proteins on streptavidin resin (Fig. 5A). The desthiobiotin probe also contained a TAMRA fluorophore that allowed gel-based visualization of the targeted proteins (Fig. 5B). We confirmed that 147-20 retained activity reducing DENV titer levels and NS3 protein levels in infected cells (SI Appendix, Fig. S5 A–C). In contrast, treatment with 147-4, an inactive analog where generation of the quinone methide is blocked by the trifluoromethyl group, did not reduce infection. We probed for the presence of specific PDIs (PDIA4, PDIA1, PDIA6), which were identified as 147 targets in other cell lines. These proteins were clearly detectable at molecular weights of prominent labeled bands that were competed by 147, confirming them as targets in Huh7 cells (Fig. 5B and SI Appendix, Fig. S5D) (48). In DENV-infected cells that were treated with 147-20, no additional labeled protein bands were observed, indicating that viral proteins are not directly targeted by the compound (SI Appendix, Fig. S5E).
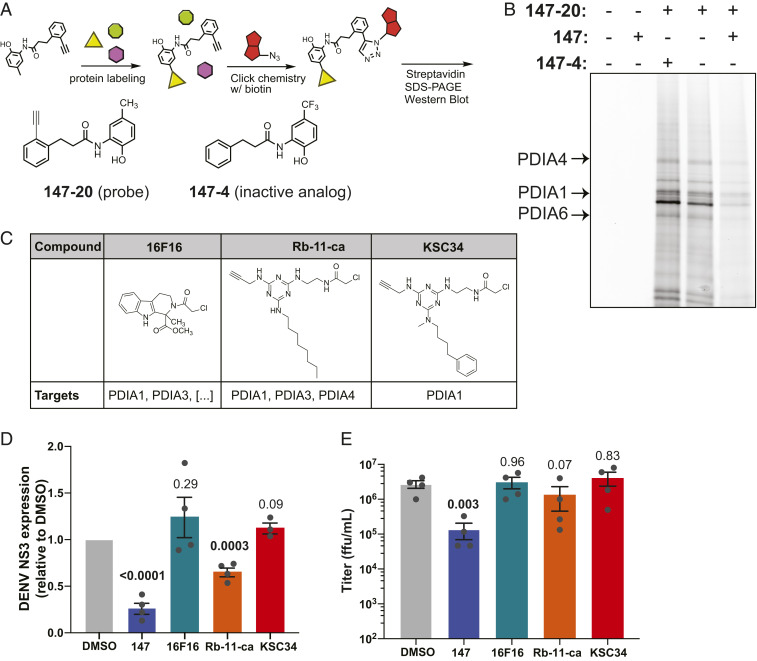
Modification of individual protein disulfide isomerases by 147 is not sufficient to reduce viral infection. (A) Illustration of the chemoproteomic workflow for target identification using chemical derivatives of 147. Cells are treated with 147-20, an alkyne analog of 147 that retains activity and can covalently label proteins as outlined in Fig. 4A. The alkyne handle enables derivatization with either a fluorophore or biotin azide for detection or affinity purification of protein targets. 147-4 is an inactive analog of 147 used to determine specificity in competition experiments. (B) Fluorescence SDS/PAGE image identifying proteins labeled by 147-20 in Huh7 cells. Huh7 cells were treated with 147-20 (3 µM) and threefold excess competitor (active 147, or inactive 147-4) for 18 h. Labeled proteins in cell lysates were derivatized with a TAMRA-desthiobiotin azide, proteins resolved on SDS/PAGE and a fluorescence image of the gel is shown. The pattern of labeled proteins reveals similar targets as seen in other cell lines (48). The location of PDIA4, PDIA6, and PDIA1/P4HB is indicated. SI Appendix, Fig. S5D shows images of western blot overlays probing for individual PDI targets. (C) Table showing chemical structures of PDI inhibitors 16F16, Rb-11-ca, and KSC-34 and their target selectivity for PDI isoforms. (D) Graph quantifying DENV NS3 protein levels in Huh7 cells in response to treatment with small-molecule PDI inhibitors. Huh7 cells were treated with 147 or PDI inhibitors 16 h prior to DENV infection (MOI 3) and NS3 protein levels were quantified by western blot 24 hpi. Only 147 and Rb-11-ca treatment lead to a significant reduction in viral protein. Error bars show SEM and P values from unpaired t tests are shown. Representative western blots shown in SI Appendix, Fig. S5J. (E) Graph showing DENV viral titers in Huh7 cells treated with 147 or PDI inhibitors. Cells were treated with the corresponding molecules for 16 h prior to DENV infection (MOI 3). Viral ffu were quantified 24 hpi by focus forming assay. Only 147 treatment reduces viral titers. Error bars show SEM and P values from ratio paired t tests are shown.
Considering the targeting of multiple PDI enzymes, we were interested in whether the inhibition of PDIs could have an important role in the reduction of viral propagation. We created stable knockdown cell lines of several PDIs using short-hairpin RNAs (shRNAs) (SI Appendix, Fig. S5F). These cell lines were then infected with DENV2 and treated with 147 to assess the effect of PDI knockdown on propagation of the virus. Individual knockdown of PDIA1 (P4HB), PDIA4, or PDIA6 did not result in a decrease in viral titers, indicating that none of these individual PDIs are essential for viral propagation (SI Appendix, Fig. S5G). Only knockdown of PDIA3 resulted in a small reduction in viral titers. While knockdown for some of the PDIs was mild, 147 has been shown to incompletely target these proteins with ∼25% of PDIA4 being labeled (48). Thus, even mild knockdown of the proteins should be sufficient to mimic the incomplete targeting by 147. Next, we measured viral infections in the knockdown cell lines in the presence of 147 to determine whether particular PDIs are required for the 147-mediated virus inhibition. We observed no measurable attenuation in the reduced viral titers with any single PDI knockdown, including the PDIA3 knockdown, suggesting that no individual PDIs are responsible for the 147-mediated effect (SI Appendix, Fig. S5G).
To further explore whether PDI inhibition could be responsible for the reduction in DENV infection, we took advantage of several published small-molecule inhibitors of PDIA1 (Fig. 5C) (5758–59). Importantly, these molecules display different degrees of specificity toward PDIA1 relative to other PDIs, mirroring the polypharmacology observed with 147. We profiled the specific PDIs targeted by the compounds in Huh7 cells by modifying the alkyne handles on the molecule with a fluorophore. KSC-34 displayed the greatest preference for PDIA1, while Rb-11-ca also labeled PDIA4, PDIA6, and several other proteins (SI Appendix, Fig. S5H). This is consistent with observations in other cell types and the development of KSC-34 as a more specific derivative of Rb-11-ca (58); 16F16 does not contain an alkyne handle but previous studies show even broader reactivity toward PDIs than Rb-11-ca (59). We then tested the effect of the compounds on DENV2 infection in Huh7 cells using the same treatment regime as for 147. KSC-34 and 16F16 were unable to reduce viral NS3 protein levels (Fig. 5D and SI Appendix, Fig. S5J). Rb-11-ca was able to lower viral protein production by 40%; however, this reduction did not translate into lower viral titers (Fig. 5E). Reduction in viral protein levels with Rb-11-ca could be attributed to compound toxicity, as we observed a 40% reduction in cell proliferation (SI Appendix, Fig. S5I). These results indicate that targeting of individual PDIs is not solely responsible for the reduced viral titers by 147 and that these existing PDI inhibitors are not effective at inhibiting DENV replication.
To identify additional mechanisms that could contribute to the 147-mediated reduction in DENV viral propagation, we turned to untargeted quantitative proteomics of the affinity-enriched samples to identify additional protein targets. We used tandem mass tags for relative quantification of proteins in the 147-20–enriched samples relative to the competition samples (147-20 treated with threefold excess 147) (Fig. 6A). This analysis confirmed that PDIs were the most highly enriched proteins that were covalently targeted by 147-20 (Fig. 6B, SI Appendix, Fig. S6A, and Dataset S3). While labeling of PDIA4, PDIA6, and PDIA1 was confirmed, we identified additional targeted PDIs (TXNDC5, PDIA3, TMX1) and two other highly enriched protein targets (GSTO1 and ALDH1A1). We compared these targets to previous datasets of 147-20 targets in HEK293T, HepG2, and ALMC-2 plasma cells (Fig. 6C and SI Appendix, Fig. S6B) (48). The additional proteins were targets specific to Huh7 cells and were not observed in other cell types, suggesting that they could have a unique role in the reduction in virus propagation in the Huh7 cells. To determine if targeting of any of these novel proteins could be responsible for the effects of 147, we similarly created stable knockdown cell lines of GSTO1, ALDH1A1, TXNDC5, and TMX1 using lentiviral delivery of shRNA. We validated knockdown by western blot (SI Appendix, Fig. S6C) and subsequently repeated the infection experiments to determine if knockdown of any single protein attenuated viral replication. No individual knockdown of these protein was sufficient to attenuate the 147-mediate reduction in DENV virus proteins (Fig. 6D). Furthermore, all knockdowns produced similar DENV titers (SI Appendix, Fig. S6D) as the mock-transduced control. All cell lines retained sensitivity to 147 treatment to reduce viral propagation. Overall, these results showed that the 147-mediated DENV inhibition is unlikely to be mediated by a single protein target.
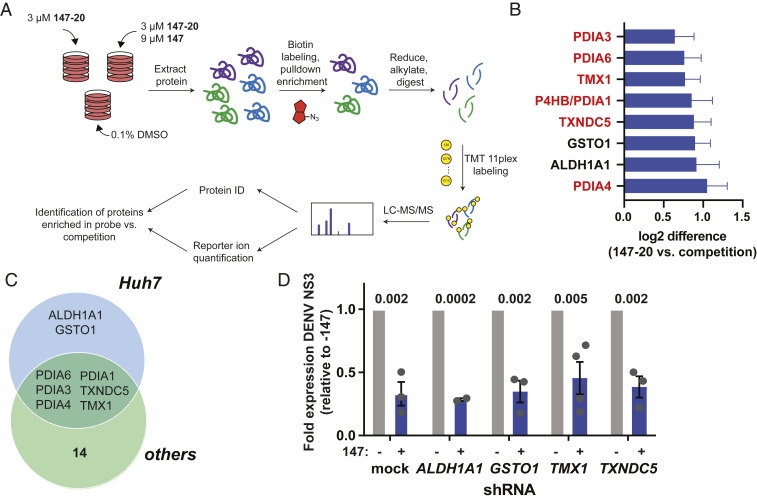
Identification of covalent protein targets of 147 in Huh7 cells. (A) Illustration of the workflow for the chemoproteomic target identification. Huh7 cells were treated with 3 µM 147-20 alone or in competition with 9 µM 147 for 18 h. Cell lysates were labeled with a TAMRA-desthiobiotin probe, labeled proteins were isolated on streptavidin beads, eluted, and digested with trypsin. Individual samples were then labeled with tandem mass tags, pooled, and subjected to LC-MS/MS for identification of proteins and quantitative comparison of proteins in the 147-20 treated compared to the 147-20/147 competition. (B) Graph showing the most highly enriched proteins targets in the 147-20 treated samples compared to the competition with 147. The full data are shown in SI Appendix, Fig. S6A. High-confidence targets were filtered that displayed a log2 enrichment ratio greater than 2 SDs of the distribution of enrichment ratios across four biological replicates. (C) Venn diagram showing the comparison of high-confidence protein targets of 147-20 in Huh7 cells and other cells lines (HEK293T, HepG2, and ALMC-2) identified in a prior study (48). SI Appendix, Fig. S6B shows the overlapping targets with individual cell lines. (D) Bar graph showing reduction in DENV NS3 protein levels in response to 147 treatment in Huh7 knockdown cell cells ALDH1A1, GSTO1, TNXDC5, and TMX1. Stable shRNA knockdown cell lines were generated by lentivirus transduced with a pool of 2 to 3 shRNAs for each target. Knockdown was validated by western blot (SI Appendix, Fig. S6C). Representative western blot is shown in SI Appendix, Fig. S6E.
Studies until this point were carried out with DENV2 BID-V533 (isolated in 2005 in Nicaragua) in Huh7 cells. To determine the scope of 147 antiviral activity, we tested the effect against DENV2 propagation in HepG2 cells, another liver carcinoma cell line commonly used as an infection model. Prior data also showed that 147 treatment is nontoxic in HepG2 cells and can reduce amyloidogenic protein secretion (46). When testing the impact on DENV titers in HepG2 cells, 147 treatment resulted in a significant reduction in DENV propagation 48 hpi (Fig. 7A). A similar decrease in protein levels was also detected (SI Appendix, Fig. S7A). Next, we proceeded to test whether the compound could be more broadly effective against other DENV strains. We tested 147 against a different DENV2 strain (16681, isolated in 1984 in Thailand) as well as against strains from DENV serotypes 3 and 4 (60). Treatment with compound 147 was able to reduce viral titers consistently greater than 95% (Fig. 7 B–D) and protein levels were similarly reduced (SI Appendix, Fig. S7B). Finally, we sought to test whether compound 147 could be useful to treat other flaviviruses. We measured the effect against two different strains of ZIKV: MR766 (isolated from Eastern Africa in the 1950s) and PRVABC59 (isolated from Puerto Rico in 2015) (61, 62). The molecule displayed similar activity at reducing ZIKV titers and protein levels for both strains (Fig. 7 E and F and SI Appendix, Fig. S7 C and D). Similar to earlier effects with DENV2 strain BID-V533, a small reduction in cell proliferation was seen with 10 µM 147 treatment during virus infection (SI Appendix, Fig. S7E). To validate the observed antiviral effects were independent of the viability reduction, we repeated the experiments at 1 µM 147, the approximate IC50 observed in SI Appendix, Fig. S2B. At this concentration, we observed cell proliferation above 90% with 147 treatment in the context of infection with each virus. As expected, the lower 1 µM dose of 147 resulted in a muted antiviral effect, but ZIKV MR766 still showed significant inhibition on treatment with 147 (SI Appendix, Fig. S7 F–J). These results provide evidence that compound 147 is broadly active against multiple flaviviruses in multiple cell lines, thus facilitating its potential application as an extensive host-centered antiviral agent.
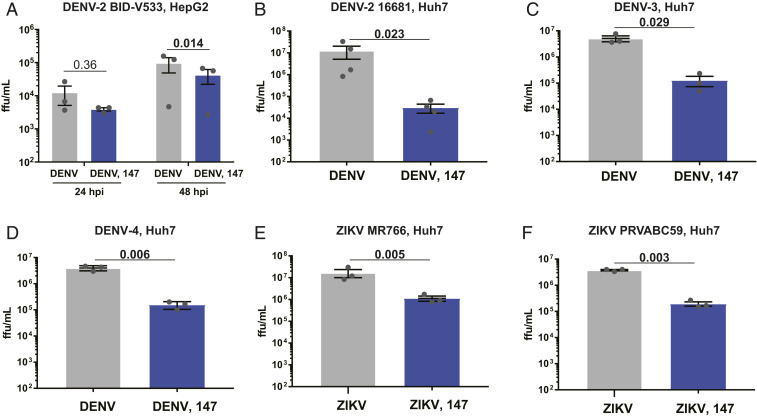
Compound 147 can reduce infection of multiple DENV strains, serotypes, as well as ZIKV. (A) Reduction of DENV2 infection in response to 147 in HepG2 liver carcinoma cells. HepG2 cells were pretreated with 147 (10 µM) for 16 h prior to infection with DENV2 BID-V533 (MOI 3). Viral titers were determined 24 and 48 hpi by focus forming assays. (B) Graph showing reduction in infection with DENV2 strain 16681 (isolated in Thailand in 1984) in response to treatment with compound 147. Prior studies were carried out with DENV2 BID-V533 (Nicaragua). Huh7 cells were pretreated with compound 147 (10 µM) 16 h prior to DENV infection (MOI 3) and viral titers were determined 24 hpi. (C and D) Treatment with 147 reduces infection of DENV serotype 3 and 4. Huh7 were pretreated with 147 (10 µM) for 16 h prior to infection with DENV3 Philippines/H87/1956 (C) or DENV4 H241 (D) (MOI 3). Viral titers were determined 24 hpi by focus forming assays. (E and F) Compound 147 is similarly active at reducing infection of Zika virus. Huh7 cells were pretreated with 147 (10 µM) for 16 h prior to infection with ZIKV strain MR766 (E) or ZIKV strain PRVABC59 (F). Viral titers were determined 24 hpi by focus forming assay. All error bars show SEM from three biological replicates and P values from ratio paired t tests are shown.
The continued health burden from arbovirus infections, such as dengue and Zika, combined with the lack of effective vaccines and therapeutics against these viruses, highlights a need for the development of new antiviral strategies. Therapeutic methods that target host factors required for viral propagation provide a path unlikely to elicit drug resistance and may act broadly across several virus species (17, 18, 20). However, a challenge remains to target host factors that selectively impair viral replication without causing toxicity to the host cell and infected organism. Prior studies with 147 have already indicated that the compound has broad potential to safely ameliorate proteostasis imbalances that are associated a variety of disease conditions (43, 46). The compound is effective at reducing the secretion and aggregation of amyloidogenic proteins, such as immunoglobulin light chains and transthyretin (46). In addition, selective remodeling of ER proteostasis pathways through an ATF6-dependent mechanism by 147 was shown to prevent oxidative organ damage in mouse models of ischemia/reperfusion (43). The same study also showed that 147 could be safely administered to mice through intravenous injections and was able to activate ER proteostasis pathways in multiple tissues, including liver, kidney, and heart (43). Here, we expand the therapeutic utility of compound 147 by establishing that the small molecule serves as a promising strategy to target multiple flaviviruses without causing significant toxicity in the host cell.
Treatment with the small-molecule proteostasis regulator 147 had the most profound effect on viral titer levels, indicating that the compound was most active at preventing the assembly and secretion of mature infectious virions. This is consistent with the compound targeting critical proteostasis processes that could be required for production of infectious virions and are thus crucial to the viral life cycle (32, 33). To support this mechanism, we found little to no reduction in extracellular virus structural protein levels, in contrast to intracellular protein levels that were reduced by up to 80% and extracellular viral titers which were reduced by ∼95%. A possible explanation for this observation is that while similar numbers of virions are being secreted from the cell, these virions possess some structural defect that leave them unable to infect cells. Compound 147 had mild cytostatic effects in Huh7 cells at the higher doses, but other antiviral effects persisted at lower doses where cell proliferation was not impacted. Importantly, prior studies showed that 147 is generally nontoxic in multiple cell lines and well tolerated in mice (43, 46).
While 147 was designed as a preferential activator of the ATF6 branch of the UPR, we found that ATF6 activation did not mediate the antiviral effect. The ATF6 inhibitor Cp-A7 on its own also did not reduce virus infection, which is consistent with prior knockout of ATF6 not affecting DENV propagation (39). These results prompted us to examine alternative mechanisms for the antiviral effects by investigating the specific protein targets of 147. Prior work showed that 147 is a prodrug that is metabolically oxidized to an iminoquinone or quinone methide, which covalently targets nucleophilic cysteine residues on cellular proteins (46, 48, 56). We showed that the metabolic activation mechanism and covalent targeting of reactive thiols is similarly required to reduce virus infection. Furthermore, chemoproteomic target identification in Huh7 cells identified a similar set of PDIs—including PDIA1, PDIA4, and PDIA6—as 147 targets in this cell line. However, knockdown of individual PDIs was not sufficient to reduce viral titers suggesting that these individual PDIs are not responsible for the inhibition of viral infection.
We addressed whether inhibition of multiple PDIs could be required by testing the effect of existing PDI inhibitors that display varying selectivity toward PDIA1/P4HB and additional PDIs also modified by 147. None of the PDI inhibitor compounds significantly reduced DENV viral titers, suggesting that 147 elicits its antiviral activity through an alternative mechanism of action. Importantly, one of the compounds, Rb-11-ca, exhibited considerably more toxicity in Huh7 cells, even at the highest concentration that we tested for 147. Toxicity has also been observed for other PDI inhibitors (63). This further highlights the unique properties of compound 147. The low toxicity could be explained by previous observations showing that less than 25% of the PDIA4 pool is labeled by the compound, indicating that PDIs are not globally inhibited by the molecule (48). Furthermore, endogenous protein secretion and formation of disulfide bonds in secreted proteins, including fully assembled immunoglobulins, are not affected by 147 (46). This lack of perturbation to normal proteostasis processes and selective inhibition of viral propagation makes 147 an ideal candidate for a host-centered antiviral strategy.
To address whether alternative protein targets besides the most prominent PDIs could be responsible for reduction in virus propagation, we compared our identified targets in Huh7 cells to the prior list of covalently modified proteins in other cell lines (48). In addition to the PDIs studied above, additional members of the PDI family were identified (TXNDC5, PDIA3, TMX1), as well as two unique proteins that were identified only in Huh7 cells: ALDH1A1 and GSTO1. However, knockdown of these proteins was also insufficient to recapitulate the reduction in viral replication induced by 147. These studies highlight the unique polypharmacology needed for 147’s antiviral effects, which likely requires a combination of protein targets.
The recent COVID-19 pandemic has refocused attention on the need for broad-spectrum antiviral agents that could inhibit future virus threats (13). Targeting essential host pathways that are commonly hijacked by virus infection without causing significant host toxicity is recognized as one important strategy to combat future infection with unknown pathogens. We determined that compound 147 has broad antiviral activity against multiple DENV strains and serotypes and that the compound is also effective against multiple ZIKV strains. These results highlight broad utility of the proteostasis regulator compound to reduce flavivirus infection by targeting conserved host cell processes that are commonly exploited by the virus. Our results here now provide an additional therapeutic application of 147 as a broad antiviral agent.
Cells were maintained in DMEM with high glucose and supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% glutamine. All cell lines except C6/36 were kept at 37 °C, 5% CO2. C6/36 cells were kept at 28 °C, 5% CO2.
For experimental infections of cell lines, media was removed from cells and virus was added at a multiplicity of infection (MOI) of 3 (DENV) for 3 h or MOI of 0.5 for 1 h (ZIKV) (unless otherwise noted). Inoculum was removed, cells were washed, and media containing chemical compounds or DMSO was added for the remainder of the experiment.
Confluent Vero cells in 96-well plates were inoculated with 10-fold serial dilutions of DENV or ZIKV in BA-1 diluent (1× M199 media, 5% BSA, 1× l-glutamine, 1× penicillin/streptomycin, 0.04% sodium bicarbonate, 50 mM Tris) for 2 h. The cells were overlaid with a 1:1 mixture of 2× nutrient overlay (2× Earle’s Balanced Salt Solution, 2× Ye-Lah Medium, 4% FBS, 0.4% sodium bicarbonate, 0.1 mg/mL gentamycin, 0.5 mg/mL amphotericin B) + 2.4% methylcellulose in water. After 2 d (ZIKV, DENV3, DENV4) or 3 d (DENV2), overlay was removed, and cells were fixed in ice cold 85% acetone for 30 min. Infectious foci were stained with a primary pan-flavivirus 4G2 antibody (1:1,000 in 5% BSA, TBST) and secondary HRP antibody (1:1,000 in 5% milk, TBST), then visualized using 400 µL 8 mg/mL 3-amino-9-ethylcarbazole (Sigma) in 10 mL 50 mM sodium acetate (Fisher) + 4 µL 30% H2O2 (Fisher) and exposed for 15 to 45 min until foci were visible.
Cell pellets were lysed in RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate) + Roche cOmplete protease inhibitor. Cells were left on ice for at least 10 min and lysate was cleared at 17,000 × g for 10 min. Cleared lysate concentrations were normalized and gel samples were separated by SDS/PAGE, transferred to PVDF membranes and immunoblotted with respective antibodies.
RNA was prepared from cell pellets using the Zymo Quick-RNA miniprep kit. cDNA was synthesized from 500 ng total cellular RNA using random primers (IDT), oligo-dT primers (IDT), and Promega M-MLV reverse transcriptase. qPCR analysis was performed using Bio-Rad iTaq Universal SYBR Green Supermix combined with primers for genes of interest (listed in SI Appendix, Table S1) and reaction were run in 96-well plates on a Bio-Rad CFX qPCR instrument.
Cell viability was determined using the CellTiter-Glo reagent (Promega). Huh7 cells were seeded into 96-well plates and treated with 100× dilutions of compounds in culture media. Sixteen hours after treatment, media was removed and cells were infected as described above (for −DENV samples, no virus was added). Media was replaced 24 h after infection and 100 µL Cell Titer Glo reagent was added and luminescence was measured for each well on a Synergy HT plate reader. Caspase activity was determined using the EnzCheck Caspase 3 Assay Kit #2 (Molecular Probes). Huh7 cells were seeded into 96-well plates and treated with 100× dilutions of compound in culture media. Sixteen hours after treatment, media was removed and cells were infected as described above (for −DENV samples, no virus was added). Virus was removed and cells were retreated for 24 h (or 12 h for staurosporin). Cells were lysed on ice and DMSO or 25 µM Z DEVD CHO inhibitor and Z DEVD-R110 substrate solutions were added. Fluorescence was measured using a Synergy HT plate reader (excitation/emission 496/520 nm).
Huh7 cells were treated with 3 µM 147-20 or a combination of 3 µM 147-20 and 9 µM 147. Cells were harvested 16 to 18 h posttreatment and lysed as described above. Click chemistry using 100 µM Cy5 azide or TAMRA desthiobiotin azide was performed and samples from Cy5 reactions were run directly on SDS/PAGE gels for analysis. For enrichment of proteins, samples were cleaned via methanol/chloroform precipitation, resolubilized in urea, and subsequently enriched on streptavidin agarose beads (Thermo Scientific).
Virus was purified using a modified version of a previously published protocol (64). In brief, virus-containing media (9 mL) was layered on top of 3 mL of sterile-filtered 20% (wt/vol) sucrose and spun at 167,000 × g (Sorvall Surespin 630) for 3 h at 4 °C to pellet virus. Media supernatant and sucrose were carefully removed and virus pellet was briefly air-dried for 10 min before resuspension in 100 μL ice-cold 5 mM HEPES, pH 7.9
We thank Dr. Tom Voss (Vanderbilt University Medical Center), Dr. Claire Huang (Center for Disease Control), and Dr. Eranthie Weerapana (Boston College) for providing critical reagents and material; Dr. Renã Robinson (Vanderbilt University) and members of her group for access to mass spectrometry instrumentation; and members of the L.P. group for critical reading of the manuscript. Work was funded by R35GM133552 (National Institute of General Medical Sciences). K.M.A. and S.C.T. were supported by T32 AI112541 (National Institute of Allergy and Infectious Diseases). K.M.A. was supported by the National Science Foundation Graduate Research Fellowships Program (1937963). J.P.D. and S.M.L. were supported by T32 GM008554 (National Institute of General Medical Sciences). We thank Vanderbilt University for providing critical start-up funds.
Processed proteomics data are included in Datasets S1–S3. The full mass spectrometry proteomics datasets have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD022453. Extended materials and methods can be found in SI Appendix.
2
3
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
 Small-molecule endoplasmic reticulum proteostasis regulator acts as a broad-spectrum inhibitor of dengue and Zika virus infections
Small-molecule endoplasmic reticulum proteostasis regulator acts as a broad-spectrum inhibitor of dengue and Zika virus infections
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
