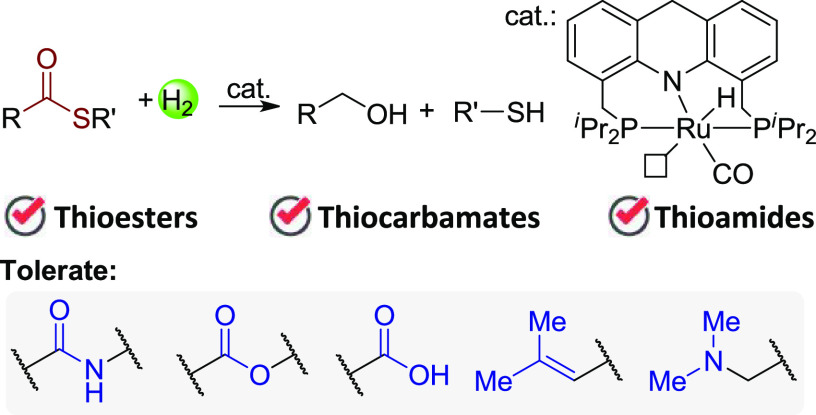
Direct hydrogenation of thioesters with H2 provides a facile and waste-free method to access alcohols and thiols. However, no report of this reaction is documented, possibly because of the incompatibility of the generated thiol with typical hydrogenation catalysts. Here, we report an efficient and selective hydrogenation of thioesters. The reaction is catalyzed by an acridine-based ruthenium complex without additives. Various thioesters were fully hydrogenated to the corresponding alcohols and thiols with excellent tolerance for amide, ester, and carboxylic acid groups. Thiocarbamates and thioamides also undergo hydrogenation under similar conditions, substantially extending the application of hydrogenation of organosulfur compounds.
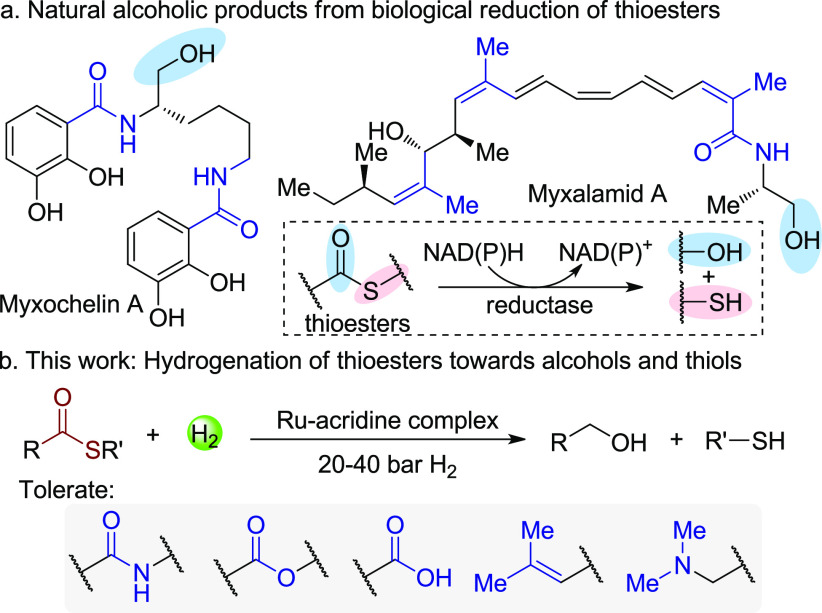
The four-electron reduction of thioesters by NAD(P)H to thiols and alcohols is well-known as the terminating step of non-ribosomal peptide synthetase and polyketide synthase involved biosynthetic processes.1 Due to the mild biological reducing conditions, multifunctionalized natural alcoholic products such as myxochelin A and myxalamid A are directly accessed (Scheme 1a).1a In contrast, current synthetic methods for the reduction of thioesters to alcohols and thiols still largely rely on the use of stoichiometric amounts of hydride reagents,2 which not only generate significant waste, but also limit application in synthesis because of possible over-reduction of unsaturated functional groups. Inspired by biological reduction of thioesters and also considering the ubiquity of thioesters in foods, cosmetics, antibiotics, and natural products,3 developing a selective and green reducing method for thioesters can access various alcohols and widely used mercaptans4.

Hydrogenation of Thioesters toward Thiols and Alcohols
In the context of producing high-value chemicals from bio-based feedstocks through more sustainable methods, hydrogenation of carboxylic acids and their derivatives is of great importance for generating versatile organic building blocks in a waste-free fashion, as compared to traditional reducing methods using hydride reagents.5 Recent years have witnessed great progress in this area using H2 in transforming carboxylic acids,6 esters,7 amides,8 and even carbonates, carbamates, and ureas9 to a vast number of alcohols and amines. Nonetheless, to the best of our knowledge, the hydrogenation of thioesters is yet to be reported,10 possibly due to the generation of thiols which might retard typical metal-catalyzed hydrogenation systems. Here we report our discovery of the hydrogenation of thioesters directly toward thiols and alcohols catalyzed by a Ru-acridine complex. Functional groups including amide, ester, carboxylic acid, and trisubstituted double bonds are tolerated (Scheme 1b). In addition, this system also enables hydrogenation of thiocarbamates and thioamides, demonstrating its efficiency and versatility.
Very recently, our group developed a Ru-acridine complex (Scheme 2a, Ru-1)-catalyzed synthesis of thioesters by dehydrogenative coupling of thiols and alcohols, evolving H2 as the only byproduct.11a In studying this reaction, a Ru-thiolate acridine complex (Ru-3) was isolated from the reaction of Ru-1 and hexanethiol, which was also a catalytically active species. Interestingly, Ru-3 was demonstrated to heterolytically split H2 at 1 bar to generate a hydrido-thiol complex (Ru-2) at room temperature. In accordance with the ease of regenerating the hydrido-thiol ruthenium complex, we wondered whether this system can hydrogenate thioesters under H2 pressure, despite the accumulation of free thiol during the reaction.
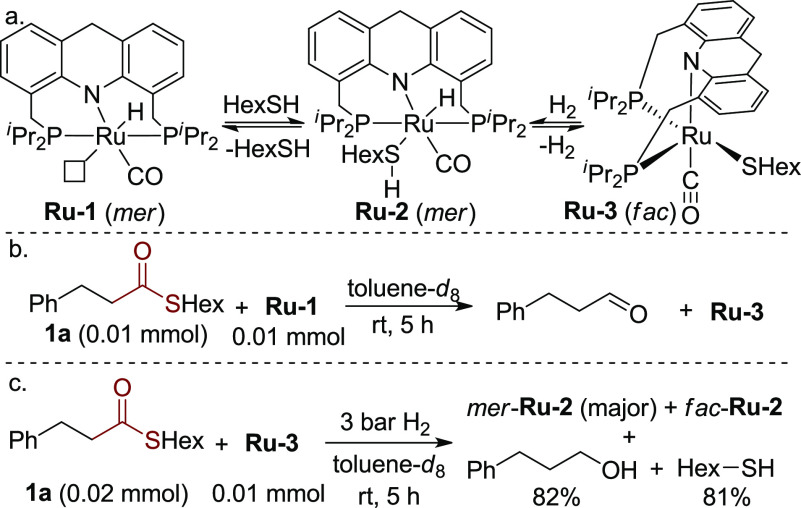
Stoichiometric Experiments toward Hydrogenation of Thioesters
Toward this end, a preliminary stoichiometric experiment was carried out in which equivalent amounts of thioester 1a and Ru-1 were mixed in toluene at room temperature. Interestingly, all of Ru-1 transformed into Ru-3 with the generation of the corresponding aldehyde in a few hours, indicating the capability of Ru-1 to reduce the thioester (Scheme 2b).12 However, in a catalytic reaction in which excess thiol is present, it is important that Ru-3 would still be capable of catalyzing the hydrogenation of thioesters under H2. Therefore, the Ru-thiolate complex (Ru-3) was tested in hydrogenation of 2 equiv of thioester 1a under 3 bar H2 in toluene at room temperature (Scheme 2c). Encouragingly, after 5 h, nearly no thioester 1a was observed by NMR, and alcohol and thiol were generated, while Ru-3 converted to Ru-hydrido-thiol isomers (mer- and fac-Ru-2).
Based on this promising result, catalytic experiments were further explored directly using Ru-1 as the catalyst (Table 1). While low conversion was observed using toluene as solvent (entry 1, 23%), 50% of thioester 1a was successfully hydrogenated to the alcohol and hexanethiol in dioxane under 10 bar H2 at room temperature after 36 h (46% and 48% yields, respectively, entry 2). However, the conversion was still incomplete even upon prolonging the reaction to 5 days (entry 3, 70%). We propose that under these conditions, the thiol can still inhibit the hydrogenation upon accumulation during the reaction. To further verify this point, a control experiment was performed by adding 1 equiv of hexanethiol before the reaction. As expected, only 17% conversion of the thioester was observed under the same conditions (entry 4). Notably, employing 3-phenylpropionaldehyde as the substrate (instead of thioester 1a) in the presence of 1 equiv of hexanethiol, nearly full conversion of the aldehyde was observed (entry 5). These experiments, taken together (entries 4 and 5), indicate that the presence of thiol slows down only the step of thioester conversion to aldehyde. In addition, in all of the incomplete reactions, only trace aldehyde was detected after the reaction, supporting that the step from aldehyde to alcohol is not rate-determining.

| entry | T (°C) | H2 pressure (bar) | conversion of 1a (%)b | yields of 2a/3a (%)b |
|---|---|---|---|---|
| 1c | r.t. | 10 | 23 | 19/22 |
| 2 | r.t. | 10 | 50 | 46/48 |
| 3d | r.t. | 10 | 70 | 64/68 |
| 4e | r.t. | 10 | 17 | 11/14 |
| 5f | r.t. | 10 | 92 | 81/– |
| 6 | r.t. | 40 | 58 | 52/56 |
| 7 | 135 | 20 | >99 | 94(92)/96 |
a Conditions: 1a (0.5 mmol), catalyst Ru-1 (1.0 mol%), dioxane (1 mL), 36 h.
b Conversions/yields were determined by GC using benzyl benzoate as internal standard; isolated yields in parentheses.
c Toluene (1 mL) as solvent.
d 5 days.
e HexSH (1 equiv) was added before the reaction.
f 3-Phenylpropionaldehyde (0.5 mmol) was used as substrate in the presence of HexSH (1 equiv); 4% ester was generated.
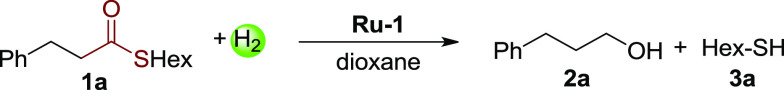
To promote the reaction, we then evaluated the effect of hydrogen pressure and temperature. Increasing H2 pressure to 40 bar, the conversion was only slightly improved, indicating that H2 pressure is not the key factor of the hydrogenation (Table 1, entry 6, 58% conversion). Interestingly, heating the reaction to 135 °C under 20 bar H2 resulted in full conversion of the thioester with no side reactions, affording the alcohol and the thiol in 94% and 96% yields, respectively (entry 7). In consideration of the former results (entries 2–4), heating appears to facilitate the dissociation of thiol from the ruthenium center (Scheme 3, i), possibly suggesting that Ru-1 is the active species in the initial conversion of the thioester to aldehyde. Prior mechanistic insights on related systems indicate that Ru-1 likely first isomerizes to fac-Ru-1 with a vacant site cis to the hydride,11b,11c which might facilitate the insertion of the coordinated thioester into the Ru-hydride bond (Scheme 3, ii, I to II). In contrast, based on the result that the presence of thiol does not affect the hydrogenation of aldehyde to alcohol (entry 5), an outer-sphere transition state III(11b) is proposed for aldehyde hydrogenation, in which Ru-2 is the active species. In the whole pathway, the presence of H2 ensures the regeneration of the Ru-H species from Ru-3 (Scheme 3, iii), thereby driving the hydrogenation reaction.
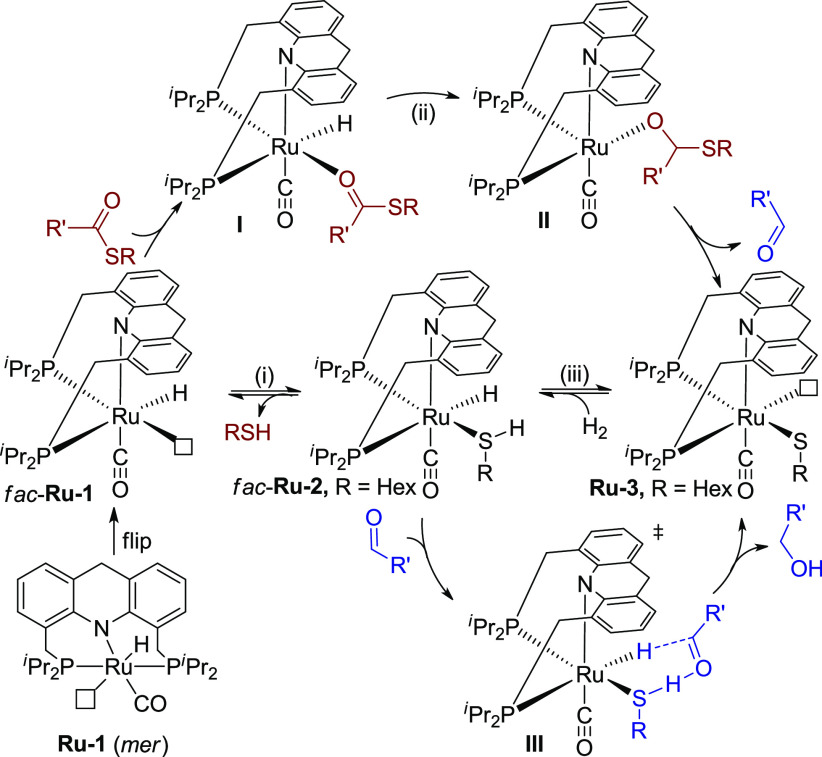
Proposed Mechanism
To further prove the high efficiency of the developed system,13 a scaled-up reaction was carried out under 30 bar H2 at 150 °C. Notably, 5 mmol thioester 1a was fully converted to the products with 0.2 mol% catalyst in only 2 h (Scheme 4, 1a, 95% yield of the alcohol, and 99% yield of hexanethiol). Subsequently, S-hexyl thioesters based on different acyl chains were studied at 0.5 mmol scale under 20 bar H2 at 135 °C (Scheme 4). First, a bi-thioester compound 1b was successfully hydrogenated to 2 equiv of hexanethiol and hexanediol in 93% and 94% yields, respectively. Additionally, a thioester with a tertiary amine group was also transformed to the corresponding products in excellent yields (1c). To our surprise, both ester and trisubstituted alkene groups remained untouched under the hydrogenation conditions, demonstrating the excellent selectivity of the system toward hydrogenation of thioester group (1d and 1e). In these two cases, 2% hexanethiol was added before the reaction to avoid the possible hydrogenation of the ester group or double bond by the first turnover of Ru-1.7e However, functional groups more prone to hydrogenation, terminal alkenes and ketones for example, were hydrogenated simultaneously in the corresponding products (1f and 1g). Even in the presence of 1 equiv of hexanethiol, the ketone group in 1g was fully hydrogenated. The reaction system appears sensitive to steric hindrance about the carbonyl group. For example, when thioester 1h with substantial steric hindrance was used as substrate, only 66% conversion of the thioester was achieved under 40 bar H2 at 150 °C. Lastly, employing benzothioate 1i, excellent yields of benzyl alcohol and hexanethiol were obtained under 40 bar H2 at 150 °C (97% for both). Interestingly, upon addition of a catalytic amount of the Lewis acid In(OTf)3, hydrogenative deoxygenation of thioester 1i was achieved, resulting in formation of the corresponding sulfide in 90% yield (see Supporting Information).14
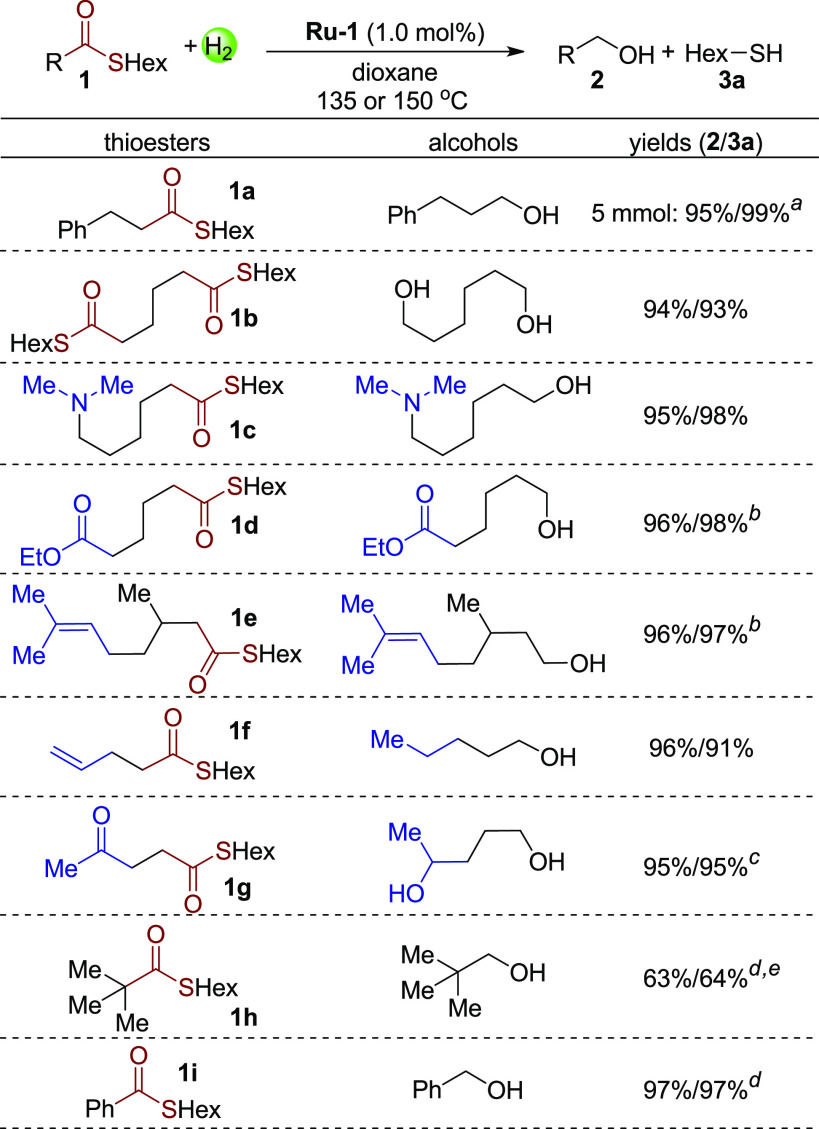
Hydrogenation of S-Hexyl Thioesters
Conditions: substrate (0.5 mmol), catalyst Ru-1 (1 mol%), dioxane (1 mL), 135 °C, 20 bar H2, 36 h. Yields of products were determined by NMR and GC using benzyl benzoate or 1,3,5-trimethoxybenzene as internal standards; isolated yields in parentheses. Unless otherwise noted, the conversion of the reaction is >99%.
1a (5 mmol), Ru-1 (0.2 mol%), dioxane (3 mL), 150 °C, 30 bar H2, 2 h.
150 °C, in the presence of 2% hexanethiol.
In the presence of 1 equiv of hexanethiol.
150 °C, 40 bar H2.
66% conversion of 1h.
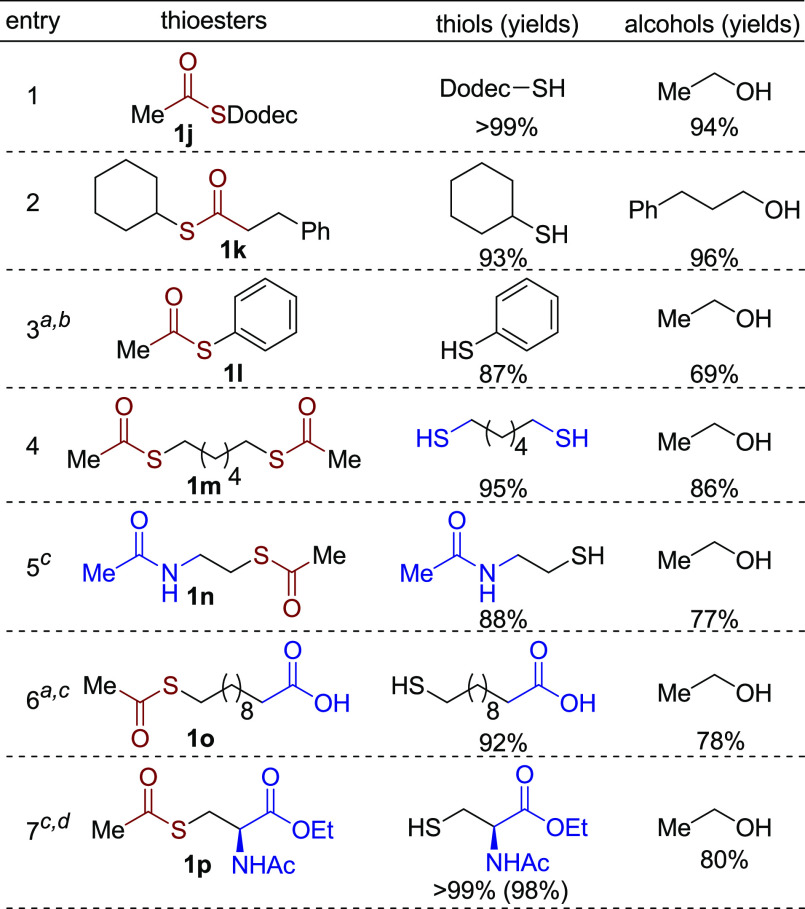
Next, thioesters based on different thiols were explored (Scheme 5). Thioesters derived from longer thiol chains or secondary thiols were hydrogenated to the corresponding thiols and alcohols in excellent yields under the optimal conditions (1j and 1k). However, for thiophenol-based thioesters, 40 bar H2 was needed to ensure high conversion (>90%), affording thiophenol in 87% yield (Scheme 5, entry 3). In addition, the dithiol-based thioester 1m was fully hydrogenated (95% yield of thiol). Similarly, functional groups on the thiol chain were examined. A tethered amide group was well tolerated under the reaction conditions, affording N-acetyl cysteamine in high yield (1n, 88%). Surprisingly, the reaction system is also compatible with a carboxylic acid group. Employing the thioester 1o, the corresponding thio-substituted carboxylic acid was produced in 92% yield at 150 °C under 40 bar H2 in spite of the possible deactivation effect of the catalyst by the carboxylic acid group.15 Finally, the cysteine-derived thioester 1p was also tested in the hydrogenation reaction (Scheme 5, entry 7). Satisfactorily, chiral N-acetyl cysteine ethyl ester16 was obtained with excellent chemoselectivity toward the thioester unit. Noteworthy, no racemization of the chiral center was observed (see Supporting Information, pp S33–S34 and Figure S48). In contrast, base-catalyzed hydrolysis of 1p does not only deprotect the ester group, but also causes elimination of the RS- group, forming olefinic side products.17 Significantly, this result indicates that the current system for selective hydrogenation of thioesters can serve as a deprotection strategy for the thiol group.
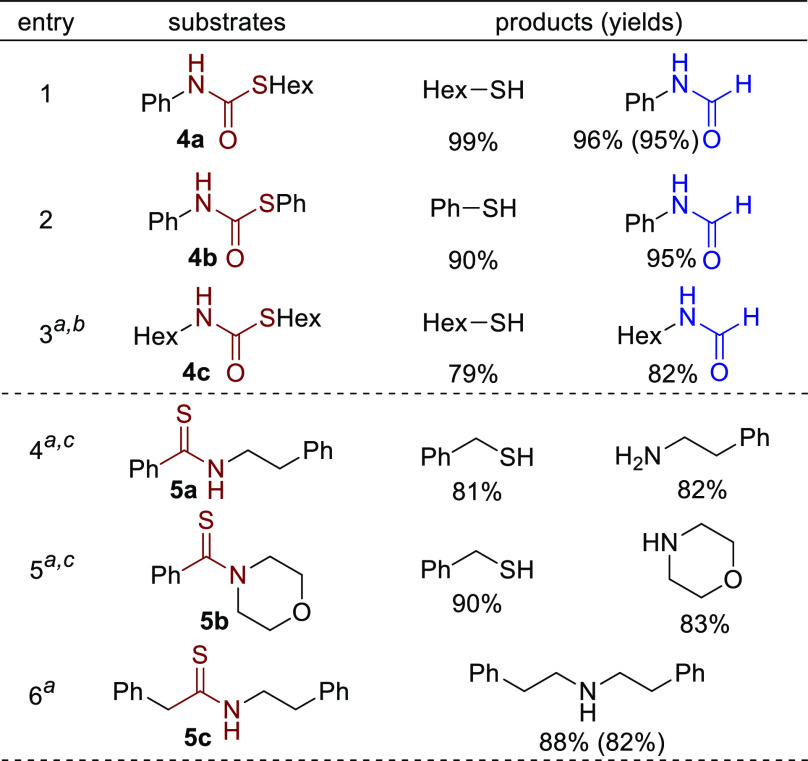
The success in efficient hydrogenation of thioesters and the tolerance toward the generated thiol motivated us to study other organosulfur compounds, which are also widely used and ubiquitous in natural products and pharmaceuticals.4,18 First, thiocarbamates were studied. To our surprise, all of the screened substrates were efficiently hydrogenated to the corresponding formamides and thiols, whereas in the case of the oxygen-based analogs, it is much harder to hydrogenate carbamates than amides and esters (Scheme 6, entries 1–3).5e,19 For thiocarbamate 4c with both alkyl chains attached to the S and O atoms, hydrogenation required higher temperature and pressure (entry 3, >80% conversion). We then turned our attention to organosulfur compounds containing a typical C=S bond.20 Three different types of thioamides were tested (entries 4–6). While benzyl thiol and amines were generated as the major products starting from thioamides 5a and 5b, most of thioamide 5c underwent hydrogenative desulfurization to afford the secondary amine21 in 88% yield. A possible reason for the observed chemoselectivity, i.e., cleavage of either C–N or C–S bond, is related to the stability of thioaldehyde intermediates. Specifically, thiobenzaldehyde is more stable than linear aliphatic thioaldehyde, which promotes its generation. Such chemoselectivity toward two types of products was also observed in the reduction of thioamides with amalgams22 (for proposed pathways and other unsuccessful examples, see the Supporting Information).
In conclusion, we have developed the unprecedented catalytic hydrogenation of thioesters to efficiently form thiols and alcohols, using a ruthenium acridine pincer complex as catalyst. Functional groups including amide, ester, carboxylic acid, and trisubstituted double bonds were tolerated, highlighting the excellent selectivity of the system. Since the catalyst is not deactivated by the generated thiol, the catalytic system was also utilized, for the first time, for the hydrogenation of thiocarbamates and thioamides, indicating a versatile range of applications. Other challenging reactions based on the developed system are being explored in our lab.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10884.
Experimental details, GC-MS/GC traces, characterization data, and NMR spectra (PDF)
The authors declare no competing financial interest.
This research was supported by the European Research Council (ERC AdG 692775). We thank Prof. Sergey Semenov and Dr. Yutao Sang for their help during the project. M.R. acknowledges the Zuckerman STEM Leadership Program for a research fellowship. D.M. is the Israel Matz Professorial Chair of Organic Chemistry.
Note that over time, the aldehyde further couples to form an ester in the absence of hydrogen gas.
With other classical catalysts developed in our group (in the presence of catalytic amount of base), the reaction could indeed generate some products; however, the conversion was inadequate (<35%), possibly due to poisoning of the catalyst by the generated thiol.
 Catalytic
Hydrogenation of Thioesters, Thiocarbamates,
and Thioamides
Catalytic
Hydrogenation of Thioesters, Thiocarbamates,
and Thioamides
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp