
A plethora of studies indicate that iron metabolism is dysregulated in Parkinson's disease (PD). The literature reveals well-documented alterations consistent with established dogma, but also intriguing paradoxical observations requiring mechanistic dissection. An important fact is the iron loading in dopaminergic neurons of the substantia nigra pars compacta (SNpc), which are the cells primarily affected in PD. Assessment of these changes reveal increased expression of proteins critical for iron uptake, namely transferrin receptor 1 and the divalent metal transporter 1 (DMT1), and decreased expression of the iron exporter, ferroportin-1 (FPN1). Consistent with this is the activation of iron regulator protein (IRP) RNA-binding activity, which is an important regulator of iron homeostasis, with its activation indicating cytosolic iron deficiency. In fact, IRPs bind to iron-responsive elements (IREs) in the 3ꞌ untranslated region (UTR) of certain mRNAs to stabilize their half-life, while binding to the 5ꞌ UTR prevents translation. Iron loading of dopaminergic neurons in PD may occur through these mechanisms, leading to increased neuronal iron and iron-mediated reactive oxygen species (ROS) generation. The “gold standard” histological marker of PD, Lewy bodies, are mainly composed of α-synuclein, the expression of which is markedly increased in PD. Of note, an atypical IRE exists in the α-synuclein 5ꞌ UTR that may explain its up-regulation by increased iron. This dysregulation could be impacted by the unique autonomous pacemaking of dopaminergic neurons of the SNpc that engages L-type Ca+2 channels, which imparts a bioenergetic energy deficit and mitochondrial redox stress. This dysfunction could then drive alterations in iron trafficking that attempt to rescue energy deficits such as the increased iron uptake to provide iron for key electron transport proteins. Considering the increased iron-loading in PD brains, therapies utilizing limited iron chelation have shown success. Greater therapeutic advancements should be possible once the exact molecular pathways of iron processing are dissected.
Parkinson's disease (PD) is the second most common severe neurodegenerative condition with a projected prevalence of 7.1 million people by 2025 [1]. PD is increasing in industrialized countries as the average lifespan rises, affecting 1% of those greater than 60 years old [[2], [3], [4]]. There is building evidence that the metabolism of iron is disturbed in PD, with the resulting oxidative stress playing a potential role in the death of critical dopaminergic neurons [5].
The precise roles of iron in the mechanisms involved in the pathobiology of PD are unclear and complex, requiring further elucidation. In fact, the exact molecular defect(s) within the substantia nigra pars compacta (SNpc) of a PD brain that leads to iron accumulation also remains unclear. However, progressive degeneration of dopamine neurons in the SNpc in PD is associated with the appearance of siderotic foci [6,7]. This has been suggested to be the result of dysregulation of iron metabolism, which could potentiate the observed oxidative stress that is at least partially due to dysfunctional mitochondria [8]. Disturbance in mitochondrial function, another key pathognomonic of PD, also has major effects on cellular iron metabolism, as the mitochondrion is a metabolic “hot spot” that is involved in the synthesis of heme and iron-sulfur cluster proteins [[9], [10], [11]]. Additionally, it is notable that the aggregation of α-synuclein that is a known factor in PD pathogenesis inhibits, iron release from the major iron storage protein, ferritin, by interfering with autophagy [12].
Studies have already attempted to treat PD by removing excess iron, with conservative regimens of iron chelating drugs resulting in some success in experimental models and clinical trials [6]. Considering this, the current review will first describe the mechanisms responsible for normal iron metabolism in cells. It will then assess the current state of knowledge regarding alterations of iron metabolism in PD and possible therapies using iron-binding pharmacotherapies that could be promising.
The serious consequences of PD on patients and their carers cannot be understated considering its striking effects on both motor and non-motor functions [13]. PD is a progressive, age-related, neurodegenerative disorder, characterized by three cardinal signs, namely: bradykinesia (slow movements), rigidity, and tremors at rest [14]. These symptoms occur when approximately 50–60% of dopaminergic neurons projecting from the SNpc to the striatum are lost and about 80–85% of dopamine levels in the striatum are depleted [4,[14], [15], [16], [17]]. PD also includes gastrointestinal symptoms such as constipation, nausea and vomiting. More recently it has been shown that sensory cells of the gut known as enteroendocrine cells contain α-synuclein [18] and synapse with enteric nerves, forming a neuroepithelial circuit [19]. Hence, this provides evidence for a tangible connection from the brain to the gut, with it being suggested that abnormal α-synuclein first develops in nerve-like enteroendocrine cells [18] and then spreads to the central nervous system (CNS) [20].
The neuropathology of PD is not only confined to the nigrostriatal pathway, as α-synuclein aggregates are found throughout the nervous system, including the cortex, amygdala, locus coeruleus, peripheral autonomic system, enteric nervous system, sympathetic ganglia, submandibular gland, cardiac and pelvic plexuses, the skin and adrenal medulla [[21], [22], [23], [24], [25]]. Unfortunately, the degeneration of the caudate nucleus and frontal cortex leads to deterioration of cognitive functions [26], while the non-motor symptoms of PD, including dysfunction of the autonomic pathway, sleep and olfactory function, can precede motor symptoms by greater than a decade [21,22,27]. PD also affects vision, with impairments of visual acuity, contrast sensitivity, color vision, and motion perception being suggested to be due to retinal degeneration [28]. Of interest, PD has also been associated with depression [29].
Lourenco Venda and colleagues [13] have aptly suggested that: “PD is an excellent example of a neurological disorder in which a complex mix of aging, genetic susceptibility and environmental insult converge to varying degrees along a spectrum to cause neurodegeneration and disease.” [13]. An important aspect of the pathobiology of PD is that it is likely more than one disease, with there being different clinical phenotypes, but also divergence at the etiological and histological levels [30,31].
Genetic background plays a substantial role in 5–10% of PD cases, while the remaining cases are of unknown etiology and designated as idiopathic [32], with a small proportion being linked to mutations in disease-causing genes designated PARK 1–18 [33]. The gene, PARK1, encodes α-synuclein, a ubiquitously expressed protein [13] that forms a large proportion of the Lewy body. The Lewy body is the “gold standard” neuro-histopathological indicator of PD and is discussed in greater detail below in Section 5.1. Mutations in the genes encoding: α-synuclein [34], leucine-rich repeat kinase 2 (LRRK2, encoded by the LRRK2/PARK8 gene) [35], vacuolar sorting protein 35 [36,37] and potentially ubiquitin carboxy-terminal hydrolase L1 (UCH-L1; a deubiquitinating enzyme encoded by the UCLH1 gene) [38], result in autosomal-dominant forms of familial PD. In contrast, mutations in Parkin (an E3 ubiquitin ligase encoded by the PARK2 gene) [39], DJ-1 (encoded by the PARK7 gene) [40] and phosphatase and tensin homolog deleted on chromosome 10 (PTEN)-induced putative kinase 1 (PINK1) (encoded by the PINK1 gene) [41], cause autosomal-recessive forms of PD. There are also a variety of other genes that have been indicated to be involved in the pathogenesis of PD, including those encoding proteins involved in intracellular trafficking and autophagosomal pathways [42,43].
In terms of environmental insults that induce PD, mercury, solvents, pesticides, iron, manganese and lead have been suggested to raise the risk of this condition [[44], [45], [46], [47], [48]]. However, investigations examining occupational exposure to metals have been inconclusive concerning PD development [49]. Other studies indicate the sensitivity of dopaminergic neurons to mitochondrial poisons, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [50], the pesticide rotenone and the redox-active herbicide, paraquat [[51], [52], [53]]. Clearly, MPTP-induced PD represents a useful model of this condition in cultured cells and animals, which is etiologically distinct from the PD observed in human patients. Considering that MPTP has been extensively used to generate experimental models of PD, its mechanism of activity is described in some detail below.
The conversion of MPTP to 1-methyl-4-phenylpyridinium (MPP+; Fig. 1A) is vital for its neurotoxic efficacy and this is catalyzed largely by brain monoamine oxidase type B in glial cells [54,55]. Then in dopaminergic neurons, MPP+ is effectively transported into these cells by high affinity dopamine transporters on the plasma membrane [56,57], but also into synaptic vesicles [58] (Fig. 1B). Once within the neuron, MPP+ is also transported into mitochondria by an energy-dependent mechanism [54,58] leading to inhibition of NADH-ubiquinone oxidoreductase I (Complex I), ATP depletion, mitochondrial membrane potential alterations and cell death via mitochondrial apoptosis [[59], [60], [61], [62]] (Fig. 1B). As part of mitochondrial apoptosis, there is cytochrome c release via the voltage-dependent anion channel (VDAC), that then initiates the caspase cascade and PARP activation [63]. Further, there is activation of JNK/c-Jun and also the tumor suppressor p53, leading to Bim and Bax expression, respectively [63,64]. Bim activates the translocation of BAX to the mitochondrion with BAX facilitating cytochrome c release via VDAC [65]. Considering this, depressed mitochondrial Complex-I activity has been identified in patient autopsy brain samples and also platelets in sporadic PD patients [[66], [67], [68], [69]].

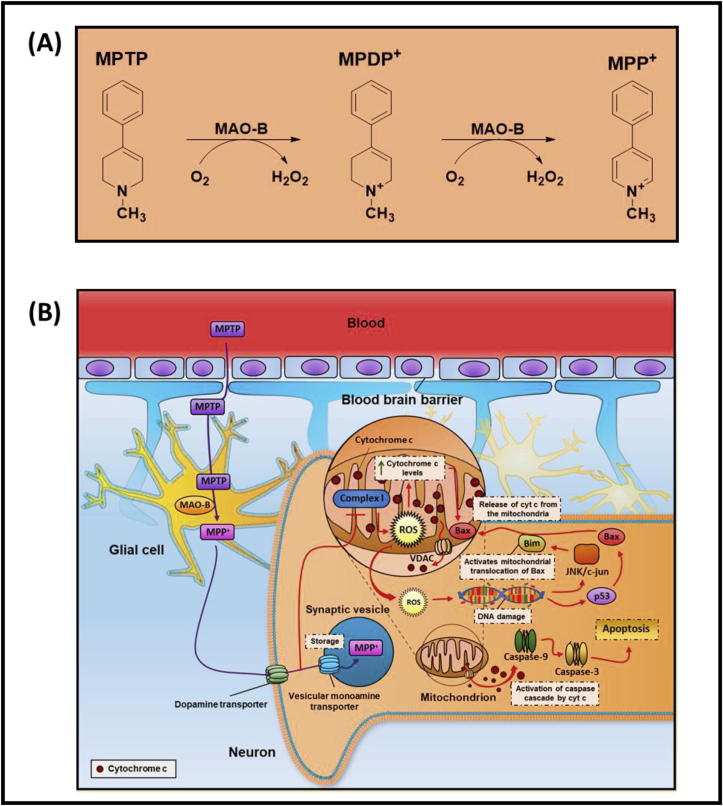
A. Schematic illustrating the mechanisms involved in the conversion of the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), into 1-methyl-4-phenyl-2,3-dihydropyridium (MPDP+) and then 1-methyl-4-phenylpyridinium (MPP+). This reaction is catalyzed by brain monoamine oxidase type B (MAO-B) with the generation of hydrogen peroxide.
1B. Schematic illustrating the processing of MPTP in the brain and also the multiple molecular targets of MPP+ in neurons. The conversion of MPTP to MPP+ is essential for its neurotoxicity and is catalyzed by brain monoamine oxidase type B (MAO-B) in glial cells. In dopaminergic neurons, MPP+ is transported into these cells by plasma membrane dopamine transporters, but also into synaptic vesicles by the vesicular monoamine transporter. An energy-dependent process leads to the transport of MPP+ into mitochondria, inhibiting Complex I and ATP synthesis resulting in mitochondrial apoptosis. As part of this latter process, there is cytochrome c release via the voltage-dependent anion channel (VDAC), that then initiates the caspase cascade and PARP activation. There is also activation of JNK/c-Jun and also the tumor suppressor p53, leading to Bim and Bax expression, respectively. Bim activates translocation of BAX to the mitochondrion with BAX facilitating cytochrome c release via VDAC.
Several in vitro studies using MPP+ and rotenone showed that these compounds can induce oxidative stress and apoptosis and other biochemical changes such as lipid peroxidation, protein oxidation and DNA damage that also occurs in idiopathic PD [70,71]. It has also been reported that the neurotoxicity of MPP+ is mediated by excessive production of nitric oxide by nitric oxide synthase [72,73]. Nitric oxide can react with superoxide to generate the cytotoxic agent, peroxynitrite (ONOO-) [74]. The latter then reacts irreversibly with tyrosine to generate nitrotyrosine that markedly affects protein function and has been suggested to play a role in the pathogenesis of several neurodegenerative diseases, including PD [75].
The potential role of nitric oxide is of interest to a key focus of the current review on PD and iron metabolism, as nitric oxide avidly binds cellular iron pools and alters the metabolism of this metal ion [[76], [77], [78]]. In fact, nitric oxide can result in the removal of this essential nutrient from iron-sulfur cluster proteins [79] and other critical iron-containing molecules such as ribonucleotide reductase, which is the rate-limiting step in DNA synthesis [80]. The overall effects of nitric oxide in cells are inhibition of mitochondrial respiration, ATP and DNA synthesis and finally cell death [[81], [82], [83], [84], [85]]. Upon binding iron, nitric oxide forms dinitrosyl dithiol iron complexes that form the largest proportion of nitric oxide in cells [86] and results in cellular iron depletion that is also cytotoxic [81,[83], [84], [85],[87], [88], [89]]. The molecular mechanism of iron release from cells mediated by nitric oxide is discussed in greater detail below in Section 3.3.
It has been reported that there are multiple mitochondrial defects in PD [90]. These defects include bioenergetic defects, mitochondrial DNA mutations, nuclear DNA gene mutations linked to mitochondria, alterations in mitochondrial dynamics, motility, size, morphology, trafficking or transport, and mutated mitochondrial genes [90]. This evidence of mitochondrial pathobiology is underlined by biochemical and genetic evidence demonstrating that the products of two genes mutated in autosomal recessive Parkinsonism, namely, Pink1 and Parkin, normally function together in the same pathway to control mitochondrial quality [91]. In this case, Pink1 accumulates on the outer membrane of damaged mitochondria and activates Parkin's E3 ubiquitin ligase activity and recruits Parkin to dysfunctional mitochondria. Then, Parkin ubiquitinates outer mitochondrial membrane proteins to trigger the selective autophagic process, mitophagy [92]. Additionally, considering PD models induced by rotenone or MPTP/MPP+, these agents lead to increased expression of α-synuclein and its pathological aggregation, which results in the death of dopaminergic neurons [[93], [94], [95]]. Regarding alterations in iron metabolism, MPP+ has been demonstrated to result in the up-regulation of transferrin receptor 1 [96], which is the major route of iron uptake by most cells [97] (as described in Section 3.2.1 below). The subsequent increased uptake of iron from transferrin was reported to then lead to oxidative stress and death via apoptosis [96].
The present treatments for PD result in only partial relief from disease symptoms without any influence on disease progression [98]. The mainstay pharmacological therapies involve dopamine replacement using the well-known drug, Levodopa (l-DOPA; L-3,4-dihydroxyphenylalanine), a dopamine precursor, which can permeate the blood-brain barrier and be converted by dopaminergic neurons to dopamine via dopamine decarboxylase [98]. Levodopa is usually administered with a dopamine decarboxylase inhibitor to prevent systemic metabolism and allow permeation of Levodopa across the blood brain barrier for conversion to dopamine by neurons. However, the long-term use of l-DOPA gives rise to motor alterations with dyskinesia and a decrease in efficacy [99]. Alternatively, dopamine agonist drugs can also be used for treatment. The loss of brain dopamine can be effectively suppressed by the use of catechol O-methyltransferase inhibitors and monoamine oxidase B inhibitors [7]. Of note, monoamine oxidase B degrades dopamine into dihydroxyphenyl acetic acid and hydrogen peroxide. More recently, a diverse variety of other agents have been developed for the potential treatment of PD and while these are beyond the scope of the review, interested readers are encouraged to examine the following articles for further information [100,101].
However, while the development of many therapeutics is still ongoing, further studies are required to understand the major pathological features of PD that include iron loading and oxidative stress. From these investigations, a better understanding could lead to novel targets and better therapeutics that may target defective iron metabolism.
Iron is a critical trace element for multiple biological mechanisms with its deficiency leading to cell death, but it is also cytotoxic when in excess [102]. Thus, organisms have developed multiple intricate mechanisms for the maintenance of iron levels within optimal levels by controlling cellular iron uptake, transport, storage and release [103]. Iron in the brain accounts for less than 2% of whole-body iron content, and interestingly, is distributed heterogeneously in different brain regions. Iron accumulates in the brain with normal aging [104], but larger increases by two-fold, occur in specific brain regions in PD [105,106] and Alzheimer's disease [107].
Unbound excess cellular iron, particularly that which is not bound to the iron storage protein, ferritin, can be cytotoxic, as it can be involved in the generation of reactive oxygen species (ROS) via Fenton and Haber-Weiss chemistry [108]. In fact, the accumulation of intracellular iron can lead to the relatively recently discovered process of ferroptosis [109], which is a unique non-apoptotic mechanism defined by iron-induced lipid ROS and depletion of plasma membrane unsaturated fatty acids [110]. For ferroptosis to occur there is a need for coincident glutathione (GSH) depletion with inactivation of the GSH-dependent enzyme, glutathione peroxidase 4 (GPx4), with the incorporation of oxidizable polyunsaturated fatty acids into phospholipids [111,112].
There is evidence that ferroptosis is a distinct mechanism that differs from the processes of apoptosis, necrosis, and autophagy when assessed via genetic, biochemical and morphological studies [109,111]. As an important differentiator to other cell death processes, using electron microscopy, ferroptotic cells demonstrate shrunken mitochondria, while this organelle is swollen in other forms of cell death [109]. A distinct set of genes regulate ferroptosis, including those encoding ribosomal protein L8 (RPL8), IRP2, and ATP synthase F0 complex subunit C3 (ATP5G3) [109]. More recently, other proteins have been suggested to participate in ferroptosis, including: phosphatidylethanolamine-binding protein 1 (PEBP1) [113], nuclear factor E2-related factor 2 (Nrf2) [114,115], and the tumor suppressor, p53 [116].
Iron is a double-edged sword and in contrast to the effects of excess iron as described above, iron deficiency results in the inhibition of DNA synthesis, G1/S arrest of the cell cycle with the induction of autophagy and apoptosis. The inhibition of DNA synthesis is mediated by the iron-containing enzyme, ribonucleotide reductase, which catalyzes the rate-limiting step of this process [117] and is one target of cellular iron chelation [118,119]. The inhibition of DNA synthesis induces cell cycle arrest [120], with iron-regulated changes in the expression of multiple proteins, including: down-regulation of cyclins D1-D3, cyclin-dependent kinase-2 and -4 [121] and up-regulation of p53 [122]. Additionally, iron depletion also leads to the dysregulation of the expression of the cyclin-dependent kinase inhibitor, p21 [123,124]. The deficiency of cellular iron also leads to the induction of autophagy via the activation of 5′-adenosine monophosphate-activated protein kinase (AMPK), which increases catabolism, while inhibiting anabolism [125]. This acts as an initial cell “rescue response” after iron-depletion that is an initial attempt to prevent cytotoxicity [126,127] (discussed in greater detail below in Section 3.4). Prolonged iron deficiency leads to endoplasmic reticulum stress, apoptosis and eventual cell death [128].
To inhibit toxic ROS generation due to superfluous unbound iron, organisms have developed distinct iron transport, storage and homeostasis mechanisms involving an interconnected web of proteins regulated by iron levels. The binding of iron to these proteins ensures that under physiological circumstances, very little iron is “free” and in a state not bound by proteins. Systemic iron levels in mammals are controlled, in part, by several hormones, including hepcidin and erythroferrone, that modulate iron absorption and recycling [129]. In terms of iron import and export across membranes, there are two transmembrane iron transporters, namely divalent metal transporter 1 (DMT1) that transports iron into cells [130,131], and ferroportin-1 (FPN1; SLC11A3) [132], which exports iron (Fig. 2). Once iron is imported into the cell via DMT1 to the time it is exported by FPN1, cytosolic iron is transported by chaperone proteins (see below) to ensure it reaches specific target proteins or organelles [133] (Fig. 2). However, cellular iron can be directly detected in the Fe+2 and Fe+3 states by Mössbauer spectroscopy [134], suggesting a dynamic equilibrium that has functional and toxicological consequences in terms of ROS generation.

Schematic illustration of normal iron metabolism in a generalized cell-type that constitutes all possible functions described in the text. Under physiological conditions, iron in the blood is transported by the iron transport protein, diferric transferrin (Fe-Tf). This latter interaction ensures that there is no free iron that could be redox active and induce cellular damage. The apo-transferrin which binds two atoms of iron to form holo-transferrin then binds to the transferrin receptor 1 on the plasma membrane of cells. The transferrin-transferrin receptor 1 complex is then internalized within cells by receptor-mediated endocytosis. In the endosomal compartment, iron is released from transferrin by a decrease in intra-vesicular pH. The iron released from transferrin is in the Fe+3 state and is thought to be reduced to Fe+2 by the ferrireductase STEAP3. The Fe+2 is then transported across the endosomal membrane by the divalent metal ion transporter (DMT1). It has been shown that the Fe+2 transported through the endosomal membrane can be bound by the iron chaperone, PCBP2, in its iron free form, namely apo-PCBP2. The apo-PCBP2 becomes bound to the N-terminal cytosolic region of iron-loaded DMT1 to receive ferrous iron forming holo-PCBP2. As such the iron is never in a free state and is delivered by PCBP1 or PCBP2 to apo-enzymes, or to the large, multimeric iron storage protein, ferritin. The release of ferritin-bound iron occurs via the process of autophagy (ferritinophagy) that leads to the breakdown of ferritin in the lysosome. Iron in the cytosol is released by ferroportin-1 (FPN1). The mechanism is thought to involve the interaction of cytosolic holo-PCBP2, with the C-terminal cytosolic region of iron-deplete FPN1. The catabolism of heme by the heme oxygenase 1 (HO1)/cytochrome P450 reductase (CPR) complex and its subsequent intracellular iron transport involves the formation of an HO1/CPR complex. PCBP2 binds this iron after association with HO1 and can then dissociate to then donate iron to other molecules, e.g., FPN1 for iron release from th + e cell.
Under physiological circumstances, iron in the blood is bound and transported to almost all cells by the protein, transferrin, which binds two atoms of iron [135]. Transferrin is highly expressed in adult and fetal liver, but also in a wide variety of sites throughout the brain, including the cerebellum, substantia nigra, frontal lobe, medulla oblongata, occipital lobe, etc. [136]. Under physiological conditions in humans, transferrin is only 30% saturated with iron to prevent the formation of toxic, non-transferrin-bound iron, which can be redox active [137,138]. In contrast, under conditions of iron overload, non-transferrin-bound iron can exist in the circulation [137]. In the duodenum or kidney, free ionic iron is absorbed from food by duodenal enterocytes of the small intestine [139], or re-absorbed from the urine, respectively, by DMT1 [130,140].
DMT1 has been reported to transport Fe+2, Zn+2, Mn+2, Co+2, Cd+2, Cu+2, Ni+2 and Pb+2 and is proton-coupled, with transport being dependent on membrane potential [130,141,142]. The iron in the gut lumen is predominantly in the Fe+3 state and is reduced to the Fe+2 state by the ferrireductase, duodenal cytochrome b reductase (Dcytb) enzymes [141,142]. For erythroblasts that actively synthesize hemoglobin and many other cell-types including cancer cells, the binding of transferrin to the transferrin receptor 1 on the plasma membrane is the predominant mechanism of iron uptake [[143], [144], [145]] (Fig. 2).
Transferrin bound to the transferrin receptor 1 is internalized into cells by receptor-mediated endocytosis, leading to the formation of an endosome [137,138,146] (Fig. 2). Upon acidification of the endosome, Fe+3 is released from transferrin by a decrease of endosomal pH [147] and is then reduced to Fe+2 by the ferrireductase, six-transmembrane epithelial antigen of the prostate 3 (STEAP3 [148]). The so-formed Fe+2 is then transported across the endosomal membrane by DMT1 into the cytoplasm [130,131] (Fig. 2). The transferrin receptor 1 is broadly expressed amongst different tissues, with the placenta, fetal liver and bone marrow having high levels, while relatively, the brain has far lower levels [149].
The transferrin receptor 1 is an interesting protein, as it appears to be capable of binding multiple other molecules including the soluble extracellular heavy chain of the iron storage protein ferritin [150]. However, it is unclear how ferritin acts to regulate iron uptake from transferrin via the transferrin receptor 1 and whether it plays a role in iron acquisition by erythroid cells remains a debated point [151]. Nonetheless, a recent cryo-electron microscopy study demonstrated the contact surfaces between heavy-chain ferritin and the transferrin receptor 1 ectodomain largely overlap with arenavirus- and Plasmodium vivax-binding regions [152]. This led to the speculation these pathogens have mimicked the ferritin-binding site. Another receptor for transferrin was also identified by Kawabata and colleagues and termed transferrin receptor 2 [153,154]. While the transferrin receptor 2 can result in iron uptake from transferrin [153], it is not regulated by cellular iron levels after iron depletion [154]. However, transferrin receptor 2 expression supports cellular growth and is regulated by the cell cycle, with its overexpression in Chinese hamster ovary cells leading to tumors in vivo [154]. Further work showed that transferrin receptor 2 also plays a role in the regulation of cellular iron metabolism, with the protein being part of the hepatocyte iron-sensing machinery (for review see Ref. [155]).
There are other less well characterized iron import proteins, including the transporters, Zrt-/Irt-like protein 8 (ZIP8; SLC39A8) and ZIP14 (SLC39A14), with some data demonstrating iron uptake via the L-type and T-type Ca+2 channels and the transient receptor potential cation channel, subfamily C, member 6 (TRPC6) [156]. The exact physiological roles of these mechanisms are unclear, as free iron in the general circulation is present at exceptionally low levels [137,138], as discussed above (Section 3.2.1).
Melanotransferrin (MTf) is a transferrin homolog, belonging to the same family as lactoferrin and transferrin, which are soluble proteins in the blood and interstitial fluid [157]. In contrast to the latter two proteins, MTf is membrane-bound [157] via a phosphatidylinositol anchor [158] with its expression being distributed broadly in human fetal and adult tissues, including the brain [136,149]. However, MTf levels are generally greater in human melanoma cells [159,160]. Despite the protein having one functional high-affinity iron-binding site [161], a series of studies assessing iron uptake by MTf in vitro in melanoma cells [136], as well as in vivo implementing both knockout mice [162] and transgenic MTf hyper-expression models [163], have not demonstrated a significant role for MTf in iron uptake. Even in melanoma cells, little iron uptake via MTf from small molecular weight iron complexes was observed [160,164], relative to the more prominent iron uptake from transferrin via the transferrin receptor 1 [144,165].
While MTf did not play an appreciable role in iron metabolism, it did have a significant role in enhancing melanoma cell proliferation [166,167]. It has been reported that MTf plays a role in Alzheimer's disease, with the protein being expressed on reactive microglia associated with amyloid plaques [168]. However, in contrast to reports that soluble serum MTf levels could be a marker for Alzheimer's disease [169,170], this could not be confirmed by later investigations [171]. While it is well known that transferrin cannot pass the blood brain barrier, it was suggested that a soluble form of MTf could achieve this [172]. Later work using the same soluble form of MTf could not repeat these studies, suggesting that like transferrin, MTf cannot efficiently cross the blood brain barrier and donate iron to the brain [173]. In fact, most soluble MTf was catabolized by the liver, with only a small amount of MTf entering the brain [173].
The role of non-transferrin-bound iron uptake mechanisms by cells could be pathophysiologically relevant during iron overload disease (e.g., β-thalassemia major). In these conditions, low molecular weight complexes of iron-citrate become significant due to the saturation of transferrin with iron [174]. Another source of iron for cells particularly for gut enterocytes is heme that can be absorbed from the gut via the heme carrier protein 1 (HCP1; encoded by the SLC46A1 gene [175]). However, this latter finding has been challenged [176] by a subsequent publication indicating the same protein (SLC46A1) acted more effectively as a folate transporter [177]. Free heme can also occur in the circulation after erythrocyte hemolysis that results in the release of hemoglobin and heme, which can be bound by circulating haptoglobin and hemopexin, respectively [178]. These complexes of hemoglobin with haptoglobin and heme with hemopexin are bound and internalized by the scavenger receptor, CD163, which are expressed on macrophages and peripheral blood monocytes that clear these potentially toxic molecules [[179], [180], [181], [182]].
The physiological iron export protein, FPN1 is involved in iron release from cells [132,183,184]. Ferroportin disease relates to a sub-type of hereditary hemochromatosis, where a loss of function mutation in this molecule results in tissue iron loading [[185], [186], [187]]. As part of the iron release process, iron transported through the membrane by FPN1 is thought to be Fe+2 [184]. Once it is transported to the extracellular milieu it is then thought to be oxidized by the action of the circulating ferroxidase ceruloplasmin, or the membrane-bound ferroxidase, hephaestin [188].
The role of ceruloplasmin in the process of cellular iron mobilization is strongly supported by many studies in vitro and in vivo [[189], [190], [191], [192]]. This includes work examining human aceruloplasminemia, where iron loading occurs in both neurons and glia of the striatum and dentate nucleus of the brain, the parenchymal cells of the liver, pancreas, heart, kidney, spleen and thyroid gland [193,194]. Ceruloplasmin was subsequently described to be involved in iron uptake by HepG2 cells under non-physiological conditions [195]. However, repetition of these studies using the same cell-type demonstrated this was not the case, with ceruloplasmin only playing a role in facilitating iron release [196]. The absence of ceruloplasmin leads to a marked increase in lipid peroxidation in the plasma [197]. Additionally, apart from ceruloplasmin and hephaestin, apo-transferrin has its own ferroxidase activity [198]. This later report suggests redundancy of ferroxidase activity due to its critical function, or potentially, its compartmentalization that requires specific ferroxidase activity relevant to the precise purpose needed.
Of note, FPN1 is not the only cellular iron export mechanism. Cellular iron release can also occur via the interaction of endogenous nitric oxide with iron pools leading to the formation of dinitrosyl dithiol iron complexes (DNICs) [89,199]. These complexes are then actively transported out of the cell via multi-drug resistance protein transporter 1 (MRP1) [200,201]. In fact, a coordinated system involving glutathione-S-transferase P1 and MRP1, which store and transport dinitrosyl dithiol iron complexes, respectively, is known to be involved in the intracellular storage and transport of nitric oxide [[200], [201], [202]]. The iron release via this system occurs in cells generating nitric oxide or when nitric oxide generating cells such as macrophages, interact with other cell-types e.g., tumor cells [203], or potentially in the central nervous system when activated microglia interact with neurons.
Irrespective of the source of iron or the mechanism of uptake of iron, the internalized iron is either utilized for metabolic needs such as for the synthesis of heme and non-heme iron-containing proteins, or is stored and detoxified in the 450 kDa protein, ferritin (Fig. 2) [204]. Ferritin is composed of 24 subunits of two types, heavy and light, that form a protein shell with an inner cavity, which can accommodate up to approximately 4,500 atoms of ferric iron [204]. The release of iron from ferritin is achieved by the catabolic process of autophagy that has been termed ferritinophagy, which is mediated by the ferritin chaperone, nuclear receptor coactivator 4 (NCOA4) [205,206]. Under conditions of iron overload, such as that found in hemochromatosis and β-thalassemia major, iron accumulates in a secondary form of iron storage known as hemosiderin. Histologically, hemosiderin reacts strongly with the Prussian blue stain (while the reaction with ferritin is weak) and is thought to be composed of partially degraded ferritin and iron generally bound within autophagic vacuoles (e.g., autolysosomes), and are known as siderosomes [207]. It has also been described that under heavy iron loading, hemosiderin accumulates in a process independent of ferritin breakdown [204]. Apart from cytosolic ferritin, an intronless gene encodes mitochondrial ferritin, which is required for iron storage in this organelle, with mitochondrial ferritin becoming loaded with iron in sideroblastic anemias [[208], [209], [210]].
Considering the cellular signaling involved in ferritin turnover and the release of iron from ferritin via autophagy (ferritinophagy), it is known that iron-deficiency mediated by iron chelators, such as desferrioxamine (DFO), results in the activation of the catabolic enzyme, 5ꞌ-adenosine monophosphate-activated protein kinase (AMPK; Fig. 3) [125]. The activation of the AMPK pathway by iron depletion has been demonstrated to be mediated by the upstream kinase, liver kinase B1 (LKB1). The activation of AMPK results in the inhibition of anabolic enzymes involved in fatty acid (e.g., acetyl CoA carboxylase 1) and protein synthesis (e.g., raptor), and activation of Unc-51 like kinase (ULK1) (Fig. 3) [125]. These effects inhibit fatty acid synthesis, suppress protein synthesis and induce autophagic activation, respectively. This AMPK-mediated response aims to rescue cellular iron deficiency, resulting in the liberation of iron via the catabolic breakdown of ferritin by autophagic processing (Fig. 3) [125].
![Schematic overview of the effect of iron depletion on the AMPK-dependent energy homeostasis pathway. The chelator, desferrioxamine (DFO), binds iron, which results in the inhibition of cellular energy generation due to the requirement for this metal for key enzymes required for electron transport (e.g., cytochromes and iron-sulfur proteins) and the activation of AMPK via LKB1. Activated AMPK leads to inhibition of raptor and acetyl CoA carboxylase 1 (ACC1) by their phosphorylation, which results in suppression of protein and fatty acid synthesis, respectively. The inhibition of these anabolic processes is important in the face of the cytotoxic insult that activates catabolism (e.g., via autophagy) to mobilize essential nutrients (such as iron) that are crucial for metabolic repair and function. As an integral part of this response, iron depletion also results in activation of ULK1 which leads to the activation of autophagy, which aims to recycle nutrients via catabolism of proteins such as the iron storage protein, ferritin [205]. This initial metabolic response of the cancer cell attempts to rescue the loss of metal ions and the repair of cytotoxic damage mediated through cellular metal ion chelation and the subsequent redox cycling of Dp44mT-metal ion complexes, respectively.](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr3.jpg)
Schematic overview of the effect of iron depletion on the AMPK-dependent energy homeostasis pathway. The chelator, desferrioxamine (DFO), binds iron, which results in the inhibition of cellular energy generation due to the requirement for this metal for key enzymes required for electron transport (e.g., cytochromes and iron-sulfur proteins) and the activation of AMPK via LKB1. Activated AMPK leads to inhibition of raptor and acetyl CoA carboxylase 1 (ACC1) by their phosphorylation, which results in suppression of protein and fatty acid synthesis, respectively. The inhibition of these anabolic processes is important in the face of the cytotoxic insult that activates catabolism (e.g., via autophagy) to mobilize essential nutrients (such as iron) that are crucial for metabolic repair and function. As an integral part of this response, iron depletion also results in activation of ULK1 which leads to the activation of autophagy, which aims to recycle nutrients via catabolism of proteins such as the iron storage protein, ferritin [205]. This initial metabolic response of the cancer cell attempts to rescue the loss of metal ions and the repair of cytotoxic damage mediated through cellular metal ion chelation and the subsequent redox cycling of Dp44mT-metal ion complexes, respectively.
Cells possess sophisticated regulatory processes to homeostatically maintain cellular iron levels via regulation of multiple proteins involved in iron uptake, release and storage [145]. At the cellular level, one of these regulatory processes is mediated post-transcriptionally by the iron regulatory proteins 1 and 2 (IRPs). This occurs via the binding of IRPs to iron-responsive elements (IREs; Fig. 4) in the untranslated regions (UTRs) of mRNAs encoding proteins involved in iron metabolism [211]. The IREs consist of a stem and a loop, with typical (Type I) IREs consisting of a consensus 5ꞌ-CAG(U/A)GN-3ꞌ sequence of 6 nucleotides forming the loop [[212], [213], [214]], such as those found in the 5ꞌ UTRs of ferritin H- and L-chain [208], FPN1 [132], mitochondrial aconitase (ACO2) mRNAs [215], hypoxia-inducible factor 2α (HIF2α [216] and erythroid δ-aminolevulinic acid synthase (eALAS) [217] (Fig. 4). Functional IREs that bind IRPs are also found in the 3ꞌ UTR of transferrin receptor 1 (that has 5 IREs [218]) and DMT1 mRNA (Fig. 4 [130,219,220]).
![Schematic illustration of the structures of iron-responsive elements (IREs). The IREs are mRNA motifs of two general types: (1) those containing the consensus 3ꞌ-CAG(U/A)GN-5ꞌ sequence (typical (Type I) IRE motif); and (2) those with a modified sequence (atypical (Type II) IRE motif) that also bind IRPs. Typical IREs in the 5′ UTR are found in the mRNAs of: human ferritin-H chain, human ferroportin1, human mitochondrial aconitase (mACO), human HIF2α and human erythroid δ-aminolevulinic acid synthase (eALAS). Typical IREs in the 3′ UTR are found in transferrin receptor 1 and Dmt1 mRNAs. Atypical IREs have been identified in the 5ꞌ UTR of α-synuclein and amyloid precursor protein (APP). Two predicted atypical IRE structures of α-synuclein have been reported (prediction #1 was from Refs. [223,394] and prediction #2 was from Ref. [224].](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr4.jpg)
Schematic illustration of the structures of iron-responsive elements (IREs). The IREs are mRNA motifs of two general types: (1) those containing the consensus 3ꞌ-CAG(U/A)GN-5ꞌ sequence (typical (Type I) IRE motif); and (2) those with a modified sequence (atypical (Type II) IRE motif) that also bind IRPs. Typical IREs in the 5′ UTR are found in the mRNAs of: human ferritin-H chain, human ferroportin1, human mitochondrial aconitase (mACO), human HIF2α and human erythroid δ-aminolevulinic acid synthase (eALAS). Typical IREs in the 3′ UTR are found in transferrin receptor 1 and Dmt1 mRNAs. Atypical IREs have been identified in the 5ꞌ UTR of α-synuclein and amyloid precursor protein (APP). Two predicted atypical IRE structures of α-synuclein have been reported (prediction #1 was from Refs. [223,394] and prediction #2 was from Ref. [224].
The structure of both the loop and the stem are vital for optimal IRP-binding [214]. A number of atypical (Type II) IREs have also been reported to be functional in terms of binding to IRPs and imparting the regulation by iron levels and are found in the 5ꞌ UTR of amyloid precursor protein mRNA [221,222] and α-synuclein mRNA [223,224]. The significance of the IRE in the regulation of α-synuclein expression and its potential role in the pathogenesis of PD will be discussed below in Section 5.1.
As a pertinent example of post-transcriptional regulation via the IRP-IRE mechanism and how this differs when the IRE is found in either the 3ꞌ- or 5ꞌ-UTR, the effect of iron on transferrin receptor 1 mRNA expression is first described [225] (Fig. 5). When iron levels are low, IRP1 and 2 bind to the 3ꞌ IREs in transferrin receptor 1 mRNA to stabilize it and increase its half-life [226]. This results in increased transferrin receptor 1 mRNA translation, which then increases iron uptake from transferrin to satisfy the cellular iron depletion (Fig. 5). On the other hand, ferritin mRNA contains an IRE within its 5ꞌ-UTR, and upon iron depletion, IRP1 and 2 binds to the 5ꞌ-IRE to sterically inhibit the translation of ferritin mRNA. In contrast, during iron repletion, IRP1 cannot bind to the 5ꞌ -IRE (due to the formation of an iron-sulfur cluster [4Fe–4S] in IRP1) leading to the translation of ferritin (Fig. 5). The resultant increase in ferritin protein levels then promotes the storage of increased iron levels [226]. Regarding IRP2, a different regulatory mechanism exists, where increased cellular iron levels induce proteasomal degradation of the protein [227]. It has been demonstrated that the SKP1-CUL1-FBXL5 ubiquitin ligase protein complex associates with and leads to iron-dependent ubiquitination and degradation of IRP2 with F-box substrate adaptor protein FBXL5 being degraded by iron and oxygen depletion [228].
![Schematic representation of the post-transcriptional function of iron regulatory protein 1 (IRP1) illustrating the conversion of this RNA-binding protein without an [Fe–S] cluster under iron-depletion to the inactive state with an [4Fe–4S] cluster after cellular iron repletion. Upon formation of the [4Fe–4S] cluster, the protein displays activity as the cytosolic aconitase. In this schematic, IRP1 is illustrated as being composed of two protein subunits linked by a hinge region that opens and closes to bind mRNA. This model was created as per the X-ray crystallography results of the closely homologous mitochondrial aconitase [554].](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr5.jpg)
Schematic representation of the post-transcriptional function of iron regulatory protein 1 (IRP1) illustrating the conversion of this RNA-binding protein without an [Fe–S] cluster under iron-depletion to the inactive state with an [4Fe–4S] cluster after cellular iron repletion. Upon formation of the [4Fe–4S] cluster, the protein displays activity as the cytosolic aconitase. In this schematic, IRP1 is illustrated as being composed of two protein subunits linked by a hinge region that opens and closes to bind mRNA. This model was created as per the X-ray crystallography results of the closely homologous mitochondrial aconitase [554].
As an additional layer of regulation, the DMT1 degradation system mediated by ubiquitin ligation occurs via an iron-dependent mechanism [229] and it is known that depending on the use of alternative promoters and alternative splicing of 3ꞌ exons, there are 4 DMT1 isoforms [230]. These include two DMT1 mRNAs that contain an IRE in the last exon and two that do not, with the C-terminal DMT1 protein isoforms being designated as I (or IRE +) and II (IRE -).
The expression of FPN1 is regulated post-transcriptionally by two major mechanisms, namely by the IRE-IRP interaction, and also by the peptide hormone, hepcidin [231,232]. Hepatocytes secrete low molecular weight hepcidin (2.8 kDa) into the blood, which is then bound by labile interactions to the high molecular weight carrier protein, α2-macroglobulin (725 kDa) [233,234]. The binding of hepcidin to α2-macroglobulin prevents the filtration of hepcidin through the kidney [233,234]. Hepcidin released from α2-macroglobulin can bind to ferroportin-1 to induce its internalization and degradation, resulting in inhibition of cellular iron export [231,232]. As could be expected, dysregulation of any of the transporters involved in iron uptake or iron efflux causes an iron deficiency or iron overload.
Both DMT1 and FPN1 have been suggested to transport ferrous iron, not ferric iron, and it has been speculated for many years that intracellular iron exists in equilibrium between these two redox states bound to ATP, citrate, amino acids, etc. [235]. Due to the redox properties of iron, such a pool would probably result in the generation of toxic ROS. However, studies in reticulocytes that actively synthesize heme for hemoglobin synthesis demonstrated that any low molecular weight iron present had the kinetics of an end product, rather than an intermediate [236]. More recently, it has been demonstrated that poly(rC)-binding proteins 1–4 (PCBP1-4) are involved in the intracellular transport of iron as chaperone molecules that donate iron to iron-containing proteins [[237], [238], [239], [240], [241], [242], [243], [244], [245], [246]]. The PCBPs are involved in transporting iron intracellularly and act as acceptors of iron from proteins and then mediate the transfer of this metal ion to other molecules [133]. For instance, the enzyme, heme oxygenase 1, degrades heme to liberate iron with the aid of cytochrome P450 reductase that acts as an electron donor. PCBP2 associates with heme oxygenase 1 and cytochrome P450 reductase as part of a metabolon [133]. When iron is released by the concerted action of the latter two enzymes, PCBP2 binds this iron and then dissociates from the metabolon [133] to transfer its bound iron to other acceptor proteins, e.g., FPN1, which then releases iron from the cell. Hence, under physiological conditions, iron is never free and is bound by proteins to ensure its efficient transport and storage.
Again, as part of the concept that there is little to no free iron in cells, it was first proposed by Ponka and colleagues in erythroid cells that a direct transfer of iron between transferrin bound to the transferrin receptor 1 in internalized endosomes and mitochondria may occur [[247], [248], [249]]. Such a process would account for the very rapid rate of iron uptake and heme synthesis in erythroid mitochondria [236]. This direct mechanism of iron transfer from transferrin to the mitochondria has been aptly termed the “kiss and run” hypothesis and has also been reported in non-erythroid cells [250]. More recent studies implementing erythroid cells suggest that the transferrin receptor 2 may also be trafficked via this route with interactions between lysosomes and mitochondria [251]. This process between transferrin receptor 2 and lysosomes has been suggested to involve the lysosomal ion channel, transient receptor potential mucolipin 1 (TRPML1), and mitofusin-2, a protein mediating organelle contacts [251]. Presumably, this latter process via the transferrin receptor 2 may act in a supplemental, but non-essential manner to supply iron relative to the process mediated by the transferrin receptor 1 [[242], [243], [244]]. This is suggested as it is well known that complete transferrin receptor 1 knockout mice are not viable, with the transferrin receptor 1 being essential for the development of erythrocytes and the nervous system [252].
Considering models of PD, there is evidence that transferrin accumulates in dopaminergic neurons with the transferrin receptor 2 possessing an uncharacterized mitochondrial targeting sequence that was suggested to result in transferrin-bound iron delivery to the mitochondrion [253]. Furthermore, in the human substantia nigra, transferrin receptor 2 expression was found associated with the mitochondria of dopaminergic neurons, there being a dramatic increase of oxidized transferrin in the substantia nigra of PD patients [253]. This latter report in neurons supports results observed examining the kiss and run hypothesis of iron uptake by the transferrin receptor 1 and transferrin receptor 2 described above in erythroid and non-erythroid cells. To account for the effective transfer of cellular iron without ROS generation, both the interaction of organelles (e.g., mitochondrion and endosomes) and chaperones could be involved in efficient intracellular trafficking of iron [133].
The brain has highly specialized iron metabolism that is distinctly different to general body iron metabolism and these differences are summarized in Table 1. For example, under physiological circumstances, the protein transferrin cannot enter the brain from the blood due to the blood-brain barrier and the blood-cerebrospinal barrier [254,255]. Due to this, iron transport in the brain is mediated by transferrin synthesized by oligodendrocytes and choroid plexus epithelial cells [256,257], with transferrin expression being marked throughout the brain [136]. The transferrin in the blood donates its iron to the brain via the transferrin receptor 1 of endothelial cells at these brain barriers [254,255] and the rate of transferrin transport through these brain barriers is very low in marked contrast to iron [258]. Iron uptake from transferrin in the brain of the rat is regulated at the whole animal level, with iron deficiency or iron loading in the animal leading to increased or decreased iron uptake by the brain [259]. This effect is probably mediated by the regulation of transferrin receptor 1 expression on the luminal endothelial cells of capillaries that form the blood brain barrier [259]. In iron overload conditions where non-transferrin-bound iron is present in the plasma, iron fails to accumulate in the brain, as demonstrated in a mouse hemochromatosis model [260], but also in hemochromatosis patients [261].

| Systemic iron metabolism | Brain iron metabolism | |
|---|---|---|
| 1 | Systemic iron levels exist mostly as transferrin-bound iron in the blood [137]. | Iron levels exceed the iron-binding capacity of transferrin in the brain, hence, significant non-transferrin-bound iron exists [258]. |
| 2 | The protein, transferrin, binds two iron atoms and transports iron in the blood to all areas in the body, except to sanctuary sites such as the brain [135]. | Transferrin cannot enter the brain from the blood due to the blood-brain barrier and the blood-cerebrospinal barrier [254,255]. The brain has its own locally synthesized transferrin [137]. |
| 3 | Most iron uptake by cells occurs through transferrin-bound iron uptake mediated by the interaction of diferric transferrin with the transferrin receptor 1 [146]. | While transferrin cannot cross the blood-brain barrier, non-transferrin-bound iron uptake by the astrocytes in the brain is mediated, in part, by DMT1 [280]. It is also postulated that other modes of non-transferrin-bound uptake may play a role, such as via the Zn+2 transporter, ZIP14 [280]. Neurons in the brain take up iron from locally synthesized transferrin [256,257]. |
| 4 | Transferrin is predominantly synthesized by the liver for maintaining systemic iron homeostasis [136]. | The only source of transferrin in the brain is via its synthesis by oligodendrocytes and choroid plexus epithelial cells [256,257]. |
| 5 | Generally low levels (~50 μM) of ascorbic acid are present in the blood [270]. | It is estimated that mammalian brain can contain up to 8 times the concentration of ascorbic acid than plasma iron levels (200–400 μM) [268]. This may prevent the toxicity associated with high non-transferrin-bound iron by maintaining ferrous iron, which prevents redox cycling [268]. |
| 6 | The ferroxidase, ceruloplasmin, in the blood plays a role in systemic iron mobilization [[189], [190], [191], [192]]. | In the brain, a unique ceruloplasmin is linked to the plasma membrane by a glycosylphosphatidyl inositol anchor [281,282] and is involved in iron efflux from astrocytes [283]. |
Within the extracellular compartment of the brain, non-transferrin bound iron could bind to transferrin secreted from oligodendrocytes and choroid plexus epithelial cells [256,257]. This binding is likely to be of considerable importance since neurons internalize diferric transferrin by receptor-mediated endocytosis [[262], [263], [264]]. In contrast to neurons, cell-types of non-neuronal origin, such as astrocytes and oligodendrocytes, show very little transferrin receptor 1 expression in vivo [264,265] and may take up non-transferrin-bound iron by a non-transferrin receptor 1-mediated process. However, when compared in vitro, enriched cultures of astrocytes, neurons, or glia could all take up iron from non-transferrin-bound iron [266].
It has been demonstrated that the concentrations of iron in the cerebrospinal fluid exceed the iron-binding capacity of transferrin [267], suggesting the presence of potentially dangerous non-transferrin-bound iron. These studies were confirmed in rats, where while iron in the plasma was almost completely precipitated with anti-transferrin antibodies, in the cerebrospinal fluid, a significant proportion of iron was not precipitated [258]. In fact, depending on the age of the animal, 80–93% of iron in cerebrospinal fluid was absorbed with anti-transferrin antibody and 1 and 5% with anti-ferritin antibodies. While no iron in the blood plasma passed through a 30,000 molecular weight cut-off filter, the fraction of iron from the cerebrospinal fluid passing through the filter was ~5%, 10% and 15%, when the rats were 15-, 20- and 56-days old, respectively [258].
These studies indicate surprising differences in the iron metabolism of the mammalian brain relative to other tissues in the body where non-transferrin-bound iron in the blood and interstitial fluid is very low [137,138]. Another important difference is implicated by the demonstration that the mammalian (rat) brain contains 8 times the concentration of the antioxidant ascorbic acid than the plasma (200–400 μM) [268], which may act to prevent the toxicity of non-transferrin-bound ferrous iron [267]. These high levels of ascorbate are also significant from the point of view that ascorbate is required for effective iron uptake from transferrin by acting as an intracellular reductant [269,270].
As glia (astrocytes, microglia and oligodendrocytes) have been indicated to be involved in brain iron metabolism and the pathogenesis of PD, a brief overview of their roles is described below.
Astrocytes are the most abundant cell-type in the CNS with multiple physiological functions that include cellular support during CNS development, ion homeostasis, uptake of neurotransmitters, neuromodulation and neuroprotection being crucial modulators of synaptic, neuronal and cognitive function [271]. It has been reported that astrocytes cover 95% of the capillary surface of the blood-brain barrier and may act as an iron transport conduit into the brain and also to regulate this process [15,[272], [273], [274], [275], [276]]. In fact, DMT1 expression has been detected in the end foot processes of astrocytes that contact endothelial cells [277]. The ability of astrocytes to obtain iron through their direct interaction with endothelial cells has been speculated to be the reason for their apparent lack (or very low levels) of transferrin receptor 1 expression [276,278]. While DMT1 plays some role in iron uptake from non-transferrin iron sources in activated astrocytes [279], other modes of non-transferrin-mediated iron uptake may potentially also occur in astrocytes via the Zn+2 transporter, ZIP14 [280], and resident transient receptor potential channels (TRPCs) in quiescent astrocytes [279].
Another little known, but highly interesting observation is that astrocytes express a unique form of ceruloplasmin, which is linked to the plasma membrane by a glycosylphosphatidylinositol anchor [281,282]. The ferroxidase activity of this protein converting Fe+2 to Fe+3 may be key for iron transport and delivery of iron to the brain and could also aid in the prevention of ROS as found for its counterpart protein in the serum [197]. In accordance with its ferroxidase function, this membrane-anchored form of ceruloplasmin was demonstrated to be involved in iron efflux from astrocytes [283], again supporting the role of astrocytes as iron transport conduits from endothelial cells. This role is important, as later studies by the same group demonstrated that decreased iron release from astrocytes induced by a conditional knockout of FPN1, depressed re-myelination by oligodendrocyte precursor cells [284]. Furthermore, this depressed iron release from astrocytes decreased the expression of cytokines in microglial cells, which are involved in re-myelination [284].
Astrocytes can also become activated to protect or act in a detrimental way depending on the stimulus, with the molecules being released from these cells having neurotrophic or inflammatory effects [15,[272], [273], [274], [275], [276]]. In fact, astrocytes have been reported to possess immune and inflammatory activities similar to microglia. The activation of astrocytes leads to reactive astrogliosis that can be abundant in atypical PD [285], which acts to limit pathology and aid repair and can be mediated by the binding of extracellular α-synuclein [286]. Astrocytes have also been suggested to clear and internalize extracellular α-synuclein that could be beneficial for SNpc neurons, but also may lead to the up-regulation of the expression of inflammatory cytokines, including interleukin-1β and tumor necrosis factor-1α [287]. Iron in excess has been reported to activate microglia and astrocytes that could then, in turn, act on dopaminergic neurons to induce neurodegeneration [288].
Considering oligodendrocytes, it is well known they are involved in myelin synthesis and immunohistochemical studies have demonstrated that they express transferrin and ferritin, but are also the principal cells staining for iron in the brain [289,290]. There is also evidence that oligodendrocytes may take up iron from ferritin in interstitial fluid or cerebrospinal fluid potentially via T cell immunoglobulin and mucin domain-containing protein 2 (Tim 2) [291], although the relative importance of this pathway is not clear relative to that mediated by transferrin. In fact, while transferrin has been identified in oligodendrocytes, it has been reported that there is no transferrin receptor 1 expression in these cells [292]. However, this does not agree with reports of transferrin being found within oligodendrocytes in humans, rats and chickens [[293], [294], [295], [296]]. This latter observation could suggest transferrin within endosomes, or alternatively, transferrin being synthesized by these cells. It has been suggested that while transferrin can be synthesized by oligodendrocytes, it is not secreted, with only the choroid plexus fulfilling this function [297]. However, transferrin expression, like that of transferrin receptor 1, is widely expressed throughout the brain [136], with transferrin receptor distribution detected by both 125I-labeled transferrin and anti-transferrin receptor monoclonal antibody binding being almost indistinguishable [292].
Irrespective of the mechanism of iron uptake by oligodendrocytes, deficiency of iron leads to hypo-myelination, suggesting the important role of iron in this process [246], with myelination and motor coordination being increased in transferrin over-expressing transgenic mice [298]. Furthermore, it is well known that iron deficiency during human development leads to motor and behavioral issues persisting into adulthood, demonstrating the important function of iron in the CNS [299]. This is because of the critical role of iron for neuron function, as reflected by transferrin receptor expression [300].
Microglia are macrophage-like cells and contribute to dopaminergic neuron degeneration in PD via inflammatory activation [301] that may occur after exposure to mutated and over-expressed α-synuclein [302] or other inflammatory mediators e.g., lipocalin 2 [303]. In contrast, activated microglia can also participate in neuroprotection, with these two contrasting effects being dependent on differential activation states namely the classical M1 phenotype that results in pro-inflammatory cytokines and the M2 anti-inflammatory phenotype [304,305]. Iron has been demonstrated to induce selective and progressive dopaminergic neurotoxicity in rat neuron-microglia-astroglia cultures [288]. The activation of microglia by elevated iron levels results in increased mRNA and protein levels of the p47 and gp91 subunits of the superoxide generating enzyme, NADPH oxidase 2 (NOX2), leading to increased ROS [288]. This response is intriguing, as in other cell-types iron-depletion rather than iron-loading has been demonstrated to up-regulate NOX2 expression through a hypoxia-inducible factor-1α (HIF-1α)-mediated mechanism [306]. From these results, it can be speculated that NOX2 expression is probably under different regulatory mechanisms depending on the cell-type.
The role of iron in the pathogenesis of PD appears complex and multi-factorial. Iron is an essential cofactor for the enzyme, tyrosine hydroxylase, which catalyzes the rate-limiting step in the biosynthesis of dopamine [307], which is the neurotransmitter that declines in this disease. As such, it is known that tyrosine hydroxylase activity is increased dose-dependently by iron levels in vitro [308]. Additionally, it has been demonstrated in PD patients using inductively coupled plasma spectroscopy that iron levels increase by 31–35% in the SNpc, while no significant alterations are observed in the substantia nigra pars reticularis [309]. These latter studies have been strongly supported by broad variety of other techniques demonstrating selective-increased levels of iron in the SNpc. These methods include histochemistry and biochemical methods demonstrating an increased ratio of Fe+3 to Fe+2 [310], magnetic resonance imaging using the R2* [311], R2ꞌ [312], quantitative susceptibility mapping [313], and advanced magnetic resonance imaging using 3D-enhanced T2 star weighted angiography (ESWAN [314]), or transcranial sonography [315].
Dopaminergic neurons in the SNpc of PD patients have been reported to have high ROS levels due to the enzymatic and non-enzymatic metabolism of dopamine, which generates superoxide anions, hydrogen peroxide and hydroxyl radicals [14]. The increased levels of iron present could then potentiate cytotoxic ROS generation by the Fenton and Haber-Weiss reactions. It has been reported in PD animal and cell models that the increased iron levels are correlated with increased and decreased expression of the iron importer (DMT1) and iron exporter FPN1, respectively [[316], [317], [318], [319]]. Additionally, a DMT1 mutation (G185R) that impairs iron transport in Belgrade rats and mk/mk mice also protects them against the Parkinsonism-inducing neurotoxins, MPTP and 6-hydroxydopamine [318]. The increased iron and DMT1 levels were also confirmed in post-mortem PD patient brains [318]. To understand the mechanism involved, studies assessing IRPs demonstrated their activation, suggesting cytosolic iron deprivation [318].
Why does increased dopamine biosynthesis, which is likely potentiated by the accumulated iron in the SNpc, lead to neurodegeneration? This might be explained by the cytotoxic potential of dopamine, as it can readily auto-oxidize in the presence of iron and can also be metabolically deaminated to generate toxic dopamine metabolites (quinones) and ROS. The generation of hydrogen peroxide has been suggested to result in mitochondrial damage with this oxidant leading to the oxidation of GSH to glutathione disulfide (GSSG), which may participate in thiol-disulfide interchange to result in protein mixed disulfides. These latter species could reversibly inhibit thiol-dependent enzymes that affect their function [14,320]. Therefore, excessively synthesized dopamine and a failure to properly store dopamine into synaptic vesicles may lead to oxidative stress, terminal degeneration, and eventually cell death [14].
Oxidative stress is a crucial mediator in the pathogenesis of PD, with increased levels of lipids, proteins, and nucleic acid oxidation products having been demonstrated in the PD SNpc [14,321]. In fact, increased levels of oxidants such as copper and iron together with decreased antioxidants such as GSH and GPx4 have been documented in the SNpc of PD patients [322,323].
There is evidence that environmental toxins may play a role in the pathogenesis of PD. Related to this, is the high incidence of a complex of neurodegenerative diseases (now known as amyotrophic lateral sclerosis and Parkinsonism-dementia complex; ALS-PDC) with similarities to ALS, AD, and PD that were observed amongst the Chamorro people of Guam [324]. This was thought to be due to the non-proteogenic amino acid, β-methylamino-l-alanine, a neurotoxin, which was ingested through the consumption of flying foxes that ate cycad seeds [324]. Considering this in relation to the iron-loading observed, β-methylamino-l-alanine has been suggested to transport iron into the brain and disrupt the correct biosynthesis of proteins [325]. However, amino acids are poor chelators of iron, and therefore, its neurotoxic role is likely more aligned to its ability to disturb protein synthesis or folding.
There are a variety of molecular alterations in PD that appear to play a role in the pathogenesis of the condition and have potential roles in iron metabolism or oxidative stress. Of all the proteins indicated to be involved in PD, evidence for a role in the pathogenesis of the disease and linkage to iron metabolism is most comprehensive for the 140 amino acid presynaptic protein, α-synuclein [326]. Point mutations (A53T, A30P, E46K) or over-expression of α-synuclein due to gene multiplication can result in an early onset autosomal dominant form of PD, supporting that increased gene dosage and altered protein structure may accelerate disease onset and progression significance of this protein in disease pathogenesis [[327], [328], [329], [330], [331]].
The expression and co-aggregation of α-synuclein are at least partly responsible for the decrease of dopaminergic neurons in the SNpc [332]. Research over the past two decades has indicated that α-synuclein is a significant regulator of PD pathology in genetic and sporadic cases with oligomeric and aggregated α-synuclein playing a pathogenic role [333]. The misfolding and subsequent self-association of fibrillar α-synuclein results in Lewy bodies and Lewy neurites [334] that are the classic neuropathological hallmark of PD. Lewy bodies are eosinophilic inclusions in neurons composed of a dense core and surrounding halo from which fibrils radiate. Lewy neurites are composed of α-synuclein that becomes misfolded into amyloid structures in axons and neurites [335].
However, Lewy bodies not only consist of α-synuclein, with these inclusions being composed of up to 90 molecules, including proteins linked to this condition, such as DJ-1, LRRK2, Parkin, and Pink-1, but also mitochondria-related proteins, proteins involved in the ubiquitin-proteasome system, autophagy, and aggresome formation [336]. It has been suggested that α-synuclein monomers condense via oligomeric intermediates, into amyloid fibrils to cause the cytotoxicity observed towards neurons in the SNpc. Recently, it has been shown that prefibrillar α-synuclein oligomers (but not fibrils) are toxic in vivo and that α-synuclein oligomers may interact with, and potentially disrupt, membranes [335,337,338].
Triggers that initiate α-synuclein co-aggregation include environmental toxins [339], metal ions [340], mutations and amplifications in the α-synuclein gene [328,341] and other gene products that exacerbate α-synuclein co-aggregation [342]. Considering the importance of α-synuclein aggregation and Lewy body formation in PD development, recently through the utilization of mass spectrometry, many promising compounds have been discovered as a potential therapeutics by acting as α-synuclein aggregation inhibitors [343]. In PD patients, there is evidence of nitration of α-synuclein [[344], [345], [346]]. Nitration via tyrosine oxidation induces its aggregation [347], which could potentially prevent its ability to associate and regulate synaptic vesicle pools and mobilization at nerve terminals [348,349].
The aggregation of α-synuclein to form Lewy bodies is associated with a broad spectrum of brain pathologies known as the synucleinopathies, which include PD, but also dementia with Lewy bodies, Lewy body variant of Alzheimer's disease, multiple system atrophy, and neurodegeneration with brain iron accumulation type I. Thus, factors that regulate α-synuclein expression are relevant to the pathogenesis of PD and other synucleinopathies.
A single intra-striatal injection of synthetic, misfolded α-synuclein into multiple strains of wild-type mice initiates a neurodegenerative cascade characterized by the accumulation of intracellular Lewy body and Lewy neurite pathology, the selective loss of SNpc dopaminergic neurons, and impaired motor coordination [350]. In fact, misfolded α-synuclein has been reported to spread from cell-to-cell in a prion-like manner acting as a template for other endogenous α-synuclein molecules [[351], [352], [353], [354]]. Thus, it appears that certain α-synuclein conformations are directly toxic to neurons and may potentially propagate Lewy pathology in the nervous system [355]. It has also been described that the transfer of α-synuclein from neurons to astrocytes results in an inflammatory response [287].
Analogous to the aggregation of α-synuclein in PD to form Lewy bodies, it is of interest that in Creutzfeldt–Jakob disease there is a toxic aggregation of prion protein to prion protein scrapie [356]. Furthermore, like PD, there is an imbalance of iron homeostasis in diseased brains despite the distinctly different pathologies and sites affected in the brain (the parenchyma relative to the SNpc) [357]. Both prion protein [358] and α-synuclein play functional roles in iron metabolism (as described below).
Lewy bodies trigger multiple mechanisms in the brain including mitochondrial dysfunction, generation of ROS, JNK pathway activated apoptosis, microglia-triggered inflammation and disruption of protein degradation pathways [359]. Metals such as iron, copper and manganese can also play a role in the co-aggregation of α-synuclein and iron and copper can also lead to ROS generation due to the Haber-Weiss and Fenton reactions. In fact, metal ions especially iron, cause fibrilization of α-synuclein through disruption of their N- and C-terminal regions or via metal-catalyzed oxidation [360].
The exact physiological and pathophysiological role of α-synuclein remains unclear with many functions being suggested. For example, α-synuclein is localized in presynaptic terminals and in synaptosomal preparations with the protein playing a role in membrane association upon the formation of amphipathic helical structures [361]. There is also compelling lines of evidence indicating that mitochondrial dysfunction and α-synuclein synaptic deposition may play a primary role in PD, although the temporal order of these changes remains unclear [362]. It could also be significant that α-synuclein has homology to the chaperone, 14-3-3 [363]. In fact, α-synuclein possesses chaperone-like functions with this protein capable of inhibiting thermally-induced protein precipitation [364]. In addition, α-synuclein can bind to 14-3-3 with both α-synuclein and 14-3-3 binding to many of the same proteins [365]. The homologous domains of α-synuclein and 14-3-3 contain residues that for 14-3-3 mediate protein-binding interactions [365].
Considering the many possible functional roles of α-synuclein, substantial evidence has accumulated from many investigators strongly implicating the role of α-synuclein in neuronal iron metabolism and/or its direct binding of iron and copper [366]. As such, due to the focus of this review, the potential role of α-synuclein in iron metabolism will be reviewed below.
Studies have suggested that α-synuclein can regulate dopamine production in cells via its interaction with tyrosine hydroxylase, the rate-limiting enzyme that converts tyrosine to L-3,4-dihydroxyphenylalanine (l-DOPA) [367,368]. Additionally, α-synuclein over-expression in cells reduces the activity of the tyrosine hydroxylase promoter [369], leading to decreased expression of tyrosine hydroxylase mRNA and protein [370,371]. It has been reported that α-synuclein also directly binds tyrosine hydroxylase, depressing its phosphorylation and inhibiting its activation by promoting protein phosphatase 2A activity [367,372]. Dysfunction of α-synuclein is therefore likely to alter cellular iron homeostasis and also dopamine metabolism. This is important, as tyrosine hydroxylase is the rate-limiting step in dopamine generation and requires iron in its active site, with iron regulating its activity. In fact, human tyrosine hydroxylase activity in vitro can be stimulated >13-fold in the presence of Fe+2 [308]. While no significant differences could be shown for the Km values of tyrosine hydroxylase in the SNpc of PD patients, the activity of tyrosine hydroxylase was half of that of controls [308] probably due to the loss of dopaminergic neurons. Brain iron deficiency in neonatal mammals is associated with reduced expression of dopamine receptors D1 and/or D2 [373,374] and dopamine transporter in the brain, reducing overall dopamine functioning [375,376].
Interestingly, α-synuclein over-expression results in increased iron levels and its redistribution in neurons [377]. Supporting these observations above and the relationship to iron metabolism is the finding that α-synuclein can bind iron and copper and acts as an NADH-requiring ferrireductase that associates with membranes as a tetramer [378,379]. However, while α-synuclein binds to iron and copper, the exact physiological role mediated by this binding and the precise stoichiometry of binding to copper and/or iron and its relative affinity varies amongst different reports [[380], [381], [382], [383]]. This may be due to a dependence on α-synuclein conformation, self-association, interaction with lipid and mutational status. Considering this, Davies and colleagues [378] have speculated the ferrireductase activity of α-synuclein could be required for reducing Fe+3 to Fe+2 that is essential for the Fe+2-dependent enzyme, tyrosine hydroxylase [384]. As α-synuclein becomes aggregated to form Lewy bodies and Lewy neurites, it could lose ferrireductase activity preventing l-DOPA formation. In accordance with this suggestion, dopamine synthesis in α-synuclein knockout mice is decreased [385].
On the other hand, increased α-synuclein v activity of α-synuclein that generates excess Fe+2 results in ROS generation and damage to tyrosine hydroxylase and also the affected cells [378]. Considering that α-synuclein binds to tyrosine hydroxylase to modulate its function [367] further studies are required to fully understand its biological role. This is particularly apparent considering the effects of α-synuclein expression on tyrosine hydroxylase expression and activity [367,372,386]. However, as discussed by Perez and Hastings [387], since dopamine and its metabolites can generate highly reactive quinones and ROS, a failure to regulate their levels through tyrosine hydroxylase could lead to disturbed packaging of dopamine into vesicles leading to neurotoxicity.
It has also been reported that α-synuclein interacts with clathrin and promotes clathrin-mediated endocytosis [388]. In fact, α-synuclein reversibly associates with membranes and helps regulate membrane polyunsaturated fatty acid composition that modulates clathrin-mediated endocytosis [388]. Since transferrin is endocytosed after binding to the transferrin receptor 1 via a clathrin-mediated process [389], it is possible that α-synuclein could alter iron uptake through this mechanism.
Considering this latter hypothesis, Singh and colleagues examining the role of α-synuclein in retinal pigment epithelial cells have demonstrated that α-synuclein silencing results in an accumulation of transferrin-transferrin receptor 1 complexes in recycling endosomes, suggested alterations in vesicular trafficking [390]. Additionally, α-synuclein over-expression can increase transferrin receptor 1 expression and also ferritin levels, which is consistent with the elevated transferrin-mediated iron uptake observed [390]. As part of this study, α-synuclein knockout mice demonstrated marked down-regulation of ferritin in the retina, as well as other tissues that utilize transferrin-bound iron for their needs. These authors suggest retinal iron dyshomeostasis occurs in the retina of PD due to impaired or altered function of α-synuclein and its effects on transferrin-transferrin receptor 1 recycling. These observations link the known function of α-synuclein in vesicular transport [391] to dysfunctional retinal iron metabolism.
Later studies by the Singh laboratory indicated that α-synuclein over-expression also resulted in iron-rich ferritin due to decreased autophagic catabolism in the outer retina in vivo and retinal-pigment-epithelial cells in vitro [12]. Of note, α-synuclein expression did not alter the expression of the ferritin chaperone, NCOA4, which mediates ferritinophagy [205], but disrupted lysosomal hydrolase transport to the autophagic apparatus, resulting in suppression of ferritin catabolism [12]. The failure of cells over-expressing α-synuclein to efficiently degrade and recycle iron in ferritin to functional cell pools could explain the up-regulation of the transferrin receptor 1 previously demonstrated by the same group of investigators [390]. Additionally, exogenous iron in vitro stimulated exosome-mediated release of ferritin and α-synuclein to the extracellular milieu, further aggravating retinal iron dyshomeostasis, prompting the authors to suggest this could contribute to the PD-associated retinal degeneration observed. Hence, multiple lines of evidence indicate a relationship between α-synuclein expression and iron metabolism [12].
The role of α-synuclein in iron metabolism is further supported by studies demonstrating that its expression is down-regulated by decreasing cellular iron levels [392,393]. Significantly, it has been discovered that an atypical IRE is present in the 5ꞌ-UTR of α-synuclein mRNA (Fig. 4) that could be important for regulating its expression (prediction #1 of the IRE stem-loop structure was from Refs. [223,394,395], while prediction #2 was from Refs. [224,394]). Rogers and colleagues [394] have indicated that their sequence alignment of the α-synuclein mRNA 5ꞌ-CAGUG-3ꞌ stem-loop motif reveals homology against the 5ꞌ -CAG(U/A)UG-3ꞌ sequence in mRNAs encoding ferritin-H and -L, ferroportin-1, and erythroid δ-aminolevulinc acid synthetase (eALAS). However, the loop formed in the α-synuclein IRE is composed of 5 nucleotides (Prediction #1; Fig. 4), which is clearly distinct to the consensus 5ꞌ-CAG(U/A)GN-3ꞌ sequence of 6 nucleotides found in the loop of most IREs that bind IRPs (Fig. 4).
There are also distinct differences in the stem of the α-synuclein IRE (Prediction 1 and 2), relative to canonical IREs, where consistently there is 5 nucleotides between the loop and the single bulge (Fig. 4). Indeed, for the α-synuclein IRE (Prediction 1), there is 4 nucleotides between the bulge and the loop, while for α-synuclein IRE (Prediction 2), there is 2 nucleotides between the loop and the first bulge and then another 2 nucleotides to the next bulge (Fig. 4) Since IRP-binding is markedly affected by both the structure of the stem and the loop of the IRE [214], it can be concluded that both predictions of the α-synuclein IRE are atypical (Fig. 4). Prediction #2 of the α-synuclein IRE [224] is the most atypical, with a loop of only 3 nucleotides, making it far less likely to efficiently bind IRPs (Fig. 4).
Consistent with the post-transcriptional role of iron in α-synuclein expression, cytoplasmic iron deficiency that is known to be induced through mitochondrial ferritin over-expression [396], resulted in down-regulation of α-synuclein protein levels [392]. This mode of regulation of α-synuclein is also supported by studies demonstrating that treatment of cells with the permeable iron chelator, N,N-bis(2-hydroxybenzyl)ethylenediamine-N,N-diacetic acid (HBED), or a transferrin receptor 1 antibody (that inhibits transferrin receptor 1-mediated iron uptake) also resulted in inhibition of α-synuclein expression/aggregation [393]. Both these latter investigations using over-expression of mitochondrial ferritin and cell permeable chelators are consistent with the regulation of translation by the IRP-IRE mechanism, although additional investigation is required to prove this.
IREs are found in the 5ꞌ UTRs of mRNAs of both α-synuclein [223,394,395] and amyloid precursor protein (APP) [221] (Fig. 4). Both of these proteins primarily form the amyloidogenic proteins in PD and Alzheimer's disease that precipitate to form β-pleated oligomers of amyloid and insoluble α-synuclein fibrils [330,397,398]. There is also some sequence similarities comparing the 5ꞌ UTRs of the α-synuclein and APP transcripts, another comparison that provides a further link between these proteinopathies [394,399,400]. It has been speculated for both PD and Alzheimer's disease that the atypical IRE sequences in α-synuclein and APP may be responsive to excess brain iron in a potential feedback loop to accelerate neuronal ferroptosis (a form of iron-mediated cell death) leading to cognitive decline and proteinopathy [401]. However, β-amyloid and α-synuclein rarely co-localize in brain amyloid deposits [402]. Notably, α-synuclein is widely expressed throughout the brain, while its oligomerization and fibrillization occur in the SNpc. As such, Finkelstein and colleagues [403] suggest its pathological effects could be due to its combination with other local factors such as the abundance of iron and/or the reducing or iron chelating properties of dopamine [346,404,405].
α-Synuclein is a member of a protein family that also includes β- and γ-synuclein, all of which are expressed within the brain [406]. While β- and γ-synuclein show marked protein sequence homology, they do not contain typical or atypical IREs in their UTRs and are not found in Lewy bodies or neurites [394,406]. Additionally, considering alternatively spliced α-synuclein mRNAs that do not possess IRE sequences, these also do not form fibrilized α-synuclein, while encoding similar sized proteins of 112 and 126 amino acids versus the 140 amino acid species [394,406,407].
Considering this, the level of α-synuclein expression is a significant determinant of the rate of its fibrillization and neurotoxicity [408]. This is underlined by the fact that multiplication of the α-synuclein gene locus results in dominantly inherited PD with dementia in correlation with a gene-dosage effect [328]. Polymorphisms in the α-synuclein promoter region and in a distal enhancer impact α-synuclein protein levels and increase the risk of developing PD [[409], [410], [411]]. Hence, decreasing the expression of α-synuclein has been touted as a useful disease-modifying strategy [[412], [413], [414]]. As pharmacological iron chelation has demonstrated to be successful in decreasing α-synuclein expression in vitro [393], this further substantiates the therapeutic potential of suitable regimens of limited iron chelation (see Section 7 below).
DJ-1 has been linked to several diseases including hereditary PD [415] and cancer [416,417]. The protein, DJ-1, encoded by the gene DJ-1 (also known as PARK7) is the cause of a rare, familial form of PD [418,419]. In some patients, the DJ-1/PARK7 gene is deleted [418], while the DJ-1 point mutations, M26I and L166P, alter the proteins’ stability leading to enhanced DJ-1 degradation and are loss of function mutations [420]. The reasons for PD development in other DJ-1 point mutation carriers remains unclear. In terms of the cellular distribution of DJ-1, it is predominantly cytosolic with some mitochondrial localization and exists in the form of a homodimeric protein that is ubiquitously expressed in the brain and peripheral tissues [421,422].
DJ-1 has been suggested to be involved in multiple functions, including: neuroprotection from oxidative stress [423,424], signal transduction [425], mitochondrial quality control [417], 20 S proteasome function [426], mitochondrial function [427], Ras-dependent cell transformation [416], fertility [428], control of protein-RNA interactions [429], regulation of androgen receptor signaling [430,431], proteolysis [432,433], a protein chaperone role [434] and it may also interact with the PD-linked proteins, Pink1 and Parkin [435,436]. However, its antioxidant defense roles appear compelling, with wild-type DJ-1 functioning through multiple specific mechanisms to sense oxidative stress that protects dopaminergic neurons against neurodegeneration in PD in vitro and in vivo [[437], [438], [439], [440]].
DJ-1 regulates a master transcription factor that is involved in antioxidant defense, Nrf2, through the PI3-kinase-AKT pathway by DJ-1-dependent inactivation of PTEN [441], and the tumor suppressor, p53. DJ-1 regulates signal mediators of oxidative stress, such as PTEN and ASK1, via direct interactions with these proteins. In fact, DJ-1 drives AKT-mediated cell survival as part of the p53-AKT axis. It has been reported that DJ-1 and p53 function are linked, with p53 preventing the cells accumulation of DJ-1 protein, whereas loss of p53 leads to stabilization and enhancement of DJ-1 expression [442].
The DJ-1 protein appears also to play a role as a redox sensor, with its levels increasing upon oxidative stress [443]. It is known that DJ-1 can alter the metabolism of GSH as well as the expression of heat-shock proteins and uncoupling proteins [444]. Other studies on postmortem tissue from PD patients demonstrated decreased levels of reduced GSH, increased oxidized DJ-1 that play a role in antioxidant defense, increased mitochondrial superoxide dismutase (SOD1) activity and selective impairment of complex I of the mitochondrial respiratory chain in the SNpc, indicating redox stress [445]. These alterations in PD show similarities with the activity of the selective nigral toxin, MPTP, in animals [446]. Examining the brain and peripheral tissues in young and aged mice and also a mouse model of PD induced by MPTP demonstrated that physiological metabolism, aging, and the neurotoxin MPTP, altered oxidized DJ-1 levels in PD-related brain sites (SNpc, olfactory bulb, and striatum), the heart, and skeletal muscle where the mitochondrial load is high [446]. These data suggest a role for DJ-1 in antioxidant defense and/or mitochondrial function in these tissues.
DJ-1 participates in a number of signaling pathways, including reaction to oxidative stress and control of mitochondrial quality. In fact, Surmeier and colleagues [447,448] have demonstrated that the knockout of DJ-1 decreases expression of two mitochondrial uncoupling proteins (namely, UCP4 and UCP5) that are mitochondrial ion channels (Fig. 6). Ca+2 from the endoplasmic reticulum influxes into the mitochondrion as a result of a complex formed between the endoplasmic reticulum Ca+2 channel, inositol 1,4,5-triphosphate receptor (IP3R), and VDAC1-mediated by the mitochondrial chaperone, glucose-regulated protein 75 (GRP75) [449]. The reduced expression of UCP4 and 5 impairs Ca+2-induced uncoupling of electron transport and increases the oxidation of matrix proteins in SNpc dopaminergic neurons [447,448].
![Schematic demonstrating Ca+2 and potentially Fe+2 uptake by L-type Ca+2 channel in dopaminergic neurons [156] and the regulatory function of DJ-1 in attenuating mitochondria oxidant stress via the mitochondrial uncoupling proteins (UCP4 and UCP5). The release of Ca+2 from the endoplasmic reticulum (ER) into the mitochondrion is facilitated by the complex formed between the endoplasmic reticulum Ca+2 channel, inositol 1,4,5-triphosphate receptor (IP3R), and VDAC1-mediated by the mitochondrial chaperone, glucose-regulated protein 75 (GRP75) [449]. The influx of Ca+2 during pacemaking can lead to a high metabolic load resulting in the accumulation of ROS via oxidative phosphorylation [447]. In non-parkinsonian neurons, DJ-1 increases UCP4 and UCP5 expression, these uncoupled proteins play an important role in protecting the mitochondrion against stress and damage by suppressing ROS production [447]. Both UPC4 and UPC5 may also inhibit ROS-mediated cytochrome c release and its neuronal apoptotic pathways [63].](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr6.jpg)
Schematic demonstrating Ca+2 and potentially Fe+2 uptake by L-type Ca+2 channel in dopaminergic neurons [156] and the regulatory function of DJ-1 in attenuating mitochondria oxidant stress via the mitochondrial uncoupling proteins (UCP4 and UCP5). The release of Ca+2 from the endoplasmic reticulum (ER) into the mitochondrion is facilitated by the complex formed between the endoplasmic reticulum Ca+2 channel, inositol 1,4,5-triphosphate receptor (IP3R), and VDAC1-mediated by the mitochondrial chaperone, glucose-regulated protein 75 (GRP75) [449]. The influx of Ca+2 during pacemaking can lead to a high metabolic load resulting in the accumulation of ROS via oxidative phosphorylation [447]. In non-parkinsonian neurons, DJ-1 increases UCP4 and UCP5 expression, these uncoupled proteins play an important role in protecting the mitochondrion against stress and damage by suppressing ROS production [447]. Both UPC4 and UPC5 may also inhibit ROS-mediated cytochrome c release and its neuronal apoptotic pathways [63].
In these latter studies, it was demonstrated that physiological autonomous pacemaking (that is, neurons capable of periodic spiking in the absence of synaptic input) by dopaminergic neurons resulted in unusual engagement of plasma membrane, voltage-gated, L-type Ca+2 channels [447,448]. This resulted in a transient, mild mitochondrial depolarization or uncoupling mediated by DJ-1, which acts as an antioxidant defense mechanism. These studies led to the suggestion that drugs antagonizing Ca+2 entry through L-type channels may be a novel neuroprotective strategy for idiopathic and familial forms of PD [447,448].
Regarding this latter mechanism, pacemaker neurons in the SNpc require considerable mitochondrial oxidative phosphorylation to generate enough ATP to enable Ca+2 influx through these L-type channels and also its subsequent extrusion [447,448]. This induces mitochondrial electron fluxes that result in considerable redox stress and it is known that Ca+2 influx into mitochondria contributes to oxidative phosphorylation [450]. These studies support a role for mitochondria in the pathogenesis of PD [8] and particularly mitochondrial-driven oxidative stress in dopaminergic neurons and the ability of DJ-1 to limit this. It has been underlined that this pathophysiological mechanism is highly specific for neurons of the SNpc, which constitute an extremely small fraction (0.0001%) of total neurons and are central to the genesis of PD [447,448]. Surmeier and colleagues [447,448] have indicated that these neurons are unique, especially in terms of their tremendous axon length (470,000 μm) and axonal field [451], with each axon supporting approximately 370,000 synapses [452]. This has been suggested to potentiate cellular stress due to the requirement for marked axonal trafficking and mitochondrial activity required to maintain this massive axonal field [447,448].
The above studies of Surmeier and colleagues [447,448] are of significant interest and could also point towards a potential mechanism of neuronal iron-loading in the SNpc in PD. As described above, since the cerebrospinal fluid contains non-transferrin-bound iron, it is possible to suggest other mechanisms of iron transport apart from the classical receptor-mediated uptake mechanism of transferrin. Considering this, it has been demonstrated in neuronal cells that iron can compete with Ca+2 for entry into cells [453]. Furthermore, analogous to Ca+2 uptake, the uptake of iron was inhibited by the specific L-type Ca+2 channel blocker, nimodipine, and enhanced by the L-type voltage Ca+2 channel activator, FPL 64176 [453]. Hence, since the engagement of plasma membrane L-type Ca+2 channels occurs upon autonomic pacemaking in dopaminergic neurons [447,448], it can be hypothesized that the uptake of non-transferrin-bound iron, may lead to the cellular iron loading observed in PD. Further, this redox active iron could then aggravate the oxidative stress caused, leading to ferroptosis. As such, the use of high affinity, L-type Ca+2 channel inhibitors could be an interesting new therapeutic modality for PD treatment.
DJ-1 is a homodimeric 23 kDa protein found in the cytosol and mitochondrion [421,454] and is a member of the DJ-1/Hsp31/PfpI superfamily [455]. The expression of DJ-1 appears ubiquitous, with it being identified in the brain and peripheral tissues [454]. The ability of DJ-1 to induce activation of different transcriptional factors and change redox balance could protect neurons against aggregation of α-synuclein-induced neurodegeneration [456]. It has been shown that cells with a high level of DJ-1 are resistant to both oxidative stress and neurotoxins such as 6-hydroxydopamine, while lower expression of DJ-1 makes cells vulnerable to oxidative stress [419,457]. In fact, the loss of DJ-1 function results in oxidative stress-induced cell death [419], with the protein being suggested to have atypical peroxiredoxin-like peroxidase activity [458].
DJ-1 possesses three redox-sensitive cysteine residues Cys-46, Cys53 and Cys-106 that can be oxidized after exposure to hydrogen peroxide and are involved in its dimerization and functional properties [459]. The Cys-106 residue is the most sensitive to oxidation and forms a cysteine-sulfinic acid upon exposure to ROS [459]. Oxidation of Cys-106 has also been indicated to be important for DJ-1's relocalization to mitochondria in response to oxidative stress [460,461].
Bjorkblom and colleagues have indicated that in vitro DJ-1 binds metal ions (copper and mercury) and prevents cellular damage from metal-induced cytotoxicity [437]. This protective mechanism of DJ-1 was lost when assessing DJ1 with the PD mutations at A104T and D149A. It was subsequently described that a redox response pathway may exist between DJ-1 and superoxide dismutase 1, which was suggested based upon the interaction of these two proteins with copper and their association with familial PD and amyolateral sclerosis [462]. These studies demonstrated a novel copper-binding site in DJ-1 involving Cys-106 that could transfer Cu+2 to SOD1, suggesting a role for DJ-1 as a copper chaperone. The binding of copper by DJ-1 was also reported by different investigators using mass spectrometric analysis and X-ray crystal analysis. These studies demonstrated that one Cu(I) ion was bound per DJ-1 homodimer via juxtaposed cysteine residues at the homodimer interface leading to a subfemtomolar dissociation constant (Kd = 6.41 × 10-16 M).
Further investigation by others indicated that amongst a range of metal ions (i.e., Zn+2, Mn+2, Fe+2, Co+2, Ni+2 and Cu+2) it was demonstrated that Zn+2 binds to DJ-1 with the greatest selectivity [463]. X-ray crystallography demonstrated that Zn+2 was coordinated to DJ-1 via Cys106 and Glu18 suggesting a potential role for this metal ion in the stabilization or regulation of DJ-1 [463]. However, later studies by others using in-cell NMR demonstrated that DJ-1 did not bind Cu+2 or Zn+2, nor did DJ-1 act as a potential chaperone to provide Cu+2 to SOD1, or obtain Cu+2 from the copper chaperone of SOD1 [464].
Clearly, these latter studies were performed directly in cells while previous investigations used isolated proteins, suggesting the possibility that the protein is not metallated under physiological conditions. Nonetheless, further experiments are required to independently repeat these data, as the consequences for understanding DJ-1 function are important. While DJ-1 did not bind metal ions, the same investigation directly examining cells [464] demonstrated that oxidative treatment caused the complete and selective oxidation of C106 to sulfinic acid, consistent with the reported role of DJ-1 as a redox sensor. Collectively, there is evidence that DJ-1 can bind metal ions, although their role in the function of the protein remains less clear.
Neuromelanin is an insoluble, black pigment, which accumulates in neurons of the SNpc and locus coeruleus of healthy human brains [465,466]. Histologically, examining normal SNpc from aged human patients, blue iron deposits (stained by Prussian blue) can be found throughout the brain parenchyma, including glial cells, with neuromelanin in dopaminergic neurons being stained brown by Fontana Masson silver staining, but with apparently no iron staining (Fig. 7A and B). However, neuromelanin-free neurons can also be found in the SNpc from aged normal patients and can contain iron deposits, probably suggesting the presence of hemosiderin that can be derived from the breakdown of ferritin (Fig. 7C).
![The distribution of iron in the normal SNpc and the iron-binding properties of neuromelanin. (A, B, C) Histochemistry using modified Perls' Prussian blue staining for detection of ferric iron deposits in normal human SNpc tissue sections from an 88 year-old patient [555]. (A) Low power light microscopy demonstrating blue iron deposits are abundant throughout the parenchyma including glial cells of the SNpc, but are seemingly absent in brown coloured neuromelanin-containing neurons (scale bar = 100 μm). (B) Absence of iron staining in neurons containing neuromelanin at higher magnification show. (C) Neuromelanin-free neurons in the SNpc also contain large amounts of iron deposits (see arrowhead). Panels A, B, C are modified from Ref. [556]. (D, E) Neuromelanin containing organelles from normal human SNpc from an 89 year-old patient observed using transmission electron microscopy. (D) Classical structure of neuromelanin-containing organelles in SNpc neurons that contain dark neuromelanin (arrow) that is associated with lipid bodies (arrowhead) [467,469]. (E) Elemental iron distribution map obtained by energy-dispersive X-ray spectroscopy indicates substantial iron (red area) within the neuromelanin (scale bar = 500 nm). This observation demonstrates the ability of neuromelanin to sequester iron [469]. Taken from Refs. [467,468] with permission. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr7.jpg)
The distribution of iron in the normal SNpc and the iron-binding properties of neuromelanin. (A, B, C) Histochemistry using modified Perls' Prussian blue staining for detection of ferric iron deposits in normal human SNpc tissue sections from an 88 year-old patient [555]. (A) Low power light microscopy demonstrating blue iron deposits are abundant throughout the parenchyma including glial cells of the SNpc, but are seemingly absent in brown coloured neuromelanin-containing neurons (scale bar = 100 μm). (B) Absence of iron staining in neurons containing neuromelanin at higher magnification show. (C) Neuromelanin-free neurons in the SNpc also contain large amounts of iron deposits (see arrowhead). Panels A, B, C are modified from Ref. [556]. (D, E) Neuromelanin containing organelles from normal human SNpc from an 89 year-old patient observed using transmission electron microscopy. (D) Classical structure of neuromelanin-containing organelles in SNpc neurons that contain dark neuromelanin (arrow) that is associated with lipid bodies (arrowhead) [467,469]. (E) Elemental iron distribution map obtained by energy-dispersive X-ray spectroscopy indicates substantial iron (red area) within the neuromelanin (scale bar = 500 nm). This observation demonstrates the ability of neuromelanin to sequester iron [469]. Taken from Refs. [467,468] with permission. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The neuromelanin pigment can be observed to be within organelles of varying size (0.5–3 μm) surrounded by a double membrane and associated within this vesicle with lipid bodies, protein accumulations and iron [467,468]. Transmission electron microscopy of aged human SNpc demonstrates the classical structure of the neuromelanin-containing organelles in neurons containing dark neuromelanin (arrow) that is associated with lipid bodies [467,469] (see arrowhead; Fig. 7D). Assessing these deposits further using energy dispersive X-ray spectroscopic assessment indicates that there is substantial iron in neuromelanin (see arrow denoting red deposits), which demonstrates its ability to bind iron under physiological circumstances (Fig. 7E) [469]. In humans, neuromelanin pigmentation increases steadily, appearing approximately at three years of age with its levels peaking between 60-70 years of age [470,471].
However, in PD there is a marked reduction of neuromelanin to 50% of that found in age-matched controls [471], while multiple studies have demonstrated that total iron levels increase up to 35% [71,309,310,472,473]. The death of dopaminergic neurons containing large quantities of iron probably leads to the release of neuromelanin extracellularly that is then processed and broken down by microglia. This may account for the loss of the neuromelanin stained neurons in PD. In fact, the SNpc has demonstrated increased iron staining of the neuropil with iron-positive microglial cells associated with extracellular neuromelanin [474]. In the SNpc region of PD patients, iron accumulation induces oxidative stress and reduction in the levels of neuromelanin that are visible by transcranial sonography well before the clinical manifestation of the disease develop [315]. It has been known for over 30 years that the neuromelanin-pigmented sub-population of dopamine-containing mesencephalic neurons in PD appears to be selectively vulnerable in this disease [475]. This conclusion is supported by evidence that MPTP and its metabolite, MPP+, the latter of which causes nigral neuron death, binds to neuromelanin [315].
Neuromelanin appears to be a mixture between eumelanin, but also pheomelanin (a red pigment), which can be generated when there is excess cysteine, leading to 5-S-cysteinyl DOPA that then reacts with benzothiazine [476]. Currently, it remains unclear if the reactions for generating neuromelanin are enzymatically driven. The enzyme, tyrosinase, catalyzes the oxidation of l-tyrosine to L-3,4-dihydroxyphenylalanine (DOPA or Levodopa), which is a precursor for neuromelanin [465]. However, tyrosinase is not detectable in the SNpc and this is consistent with the fact that albinos maintain pigmentation in this region of the brain [465]. In fact, unlike the skin that possesses specialized melanocytes that synthesize melanin, the SNpc does not possess this cell-type, and as such, a different process is involved in its formation. Considering this it has been demonstrated in vitro that neuromelanin could be generated non-enzymatically via iron-mediated oxidation and polymerization of l-DOPA [477].
Since neuromelanin is derived, at least in part, from l-DOPA, it has been proposed that it could act as a reversible store of this essential neurotransmitter [478]. Neuromelanin has also been reported to become crosslinked to α-synuclein [479], although the exact pathophysiological significance remains unclear. While the precise function of neuromelanin remains unclear, it may exert protection from the redox activity of the increased iron levels observed in PD by directly binding and sequestering it, along with other metal ions including manganese, but also potential toxins and xenobiotics [468,480]. However, such binding of metal ions would need to be bound by neuromelanin in a way that would prevent ROS generation. The fact the neuromelanin is often found in double membrane-bound organelles next to lipid bodies [467,468] could suggest it is a “final destination” for an unwanted item.
The iron in neuromelanin is thought to be bound by the dihydroxy catechol groups of 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid, resulting in the formation of Fe+3-oxo/hydroxy clusters [[481], [482], [483]]. A schematic of a speculated, potential structure of neuromelanin is shown in Fig. 8 [467,468]. It is important to note that information regarding the exact coordination sites and form of iron bound to the neuromelanin polymer still remains uncertain. Like other melanins, the exact 3-dimensional structure of neuromelanin and its iron complexes have not been solved, as it forms a black insoluble precipitate that is difficult to crystallize. However, chemical degradation studies have indicated it is a polymer that is biosynthesized from 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid, which are derived from dopamine and DOPA, respectively [476,484]. Unfortunately, to add to the uncertainty regarding its structure, neuromelanin polymer also contains benzothiazine units, together with lipid and protein components [[485], [486], [487]] and is similar to the brown/black eumelanin pigment found in skin and hair. This observation led to the suggestion that neuromelanin may provide a nidus for iron mineralization, somewhat like that observed in ferritin.
![Schematic line drawing suggesting the possible structure of the iron-binding sites in neuromelanin as predicted from spectroscopic and other studies. Modified from Refs. [467,468] with permission. Of note, the exact structure and coordination geometries of neuromelanin with iron remain unknown, as it is a black pigment that is not readily crystallizable to enable characterization.](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr8.jpg)
Schematic line drawing suggesting the possible structure of the iron-binding sites in neuromelanin as predicted from spectroscopic and other studies. Modified from Refs. [467,468] with permission. Of note, the exact structure and coordination geometries of neuromelanin with iron remain unknown, as it is a black pigment that is not readily crystallizable to enable characterization.
It remains unclear why the dopaminergic neurons in the SNpc would require an additional iron storage mechanism when there is already ferritin? As such, the physiological role that neuromelanin provides as an iron-binding entity in SNpc neurons suggests a different role in iron storage or metabolism that is different to that found for ferritin.
It has been hypothesized that normally, neuromelanin is only 50% saturated with iron, which maintains its chelation capacity to prevent neurons from iron-catalyzed ROS generation [485,488,489]. However, the decrease of neuromelanin levels in the neurons of PD patients, suggests that it may become saturated with iron preventing its ability to buffer free iron that could lead to toxic, iron-catalyzed ROS and neuronal death [465,485,488,489]. Again, while this hypothesis is feasible, it does not take into account the iron storage role of ferritin, which should be up-regulated via the IRP-IRE mechanism (Fig. 5, Fig. 6) when intracellular iron levels increase to protect neurons against cytotoxic ROS generation.
Considering this, it has been demonstrated that ferritin metabolism and especially ferritinophagy are disturbed by α-synuclein expression, resulting in the accumulation of iron-rich ferritin in the outer retina in vivo and retinal-pigment-epithelial cells in vitro [12]. This disturbance in metabolism via α-synuclein appears to be related to a failure in autophagy-mediated by the lysosome, as the expression of Rab1a that mediates dynamic intracellular vesicular transport [490], restored ferritinophagy [12]. Whether the marked disturbance in ferritin metabolism due to α-synuclein expression leads to neuromelanin acting as an alternative iron sink remains a possible speculation to explain its iron-binding role in PD. This line of reasoning indicates that further investigation is necessary to understand the mechanisms involved in PD pathobiology.
In addition to PD, it is interesting from a perspective viewpoint to note that a number of severe neurodegenerative conditions such as Huntington's disease [491], Alzheimer's disease [492,493], multiple sclerosis [494,495], amyotrophic lateral sclerosis [495,496], neurodegeneration with brain iron accumulation type 1 [497], as well as the neuro-degenerative and cardio-degenerative conditions such as Friedreich's ataxia, share a common feature, disturbance of iron metabolism and oxidative stress [493]. For many of these diseases, the precise mechanistic role of iron in their pathogenesis remains uncertain. Additionally, as an integral part of this, it is not clear if the increased iron present is the result or the cause of the pathobiology.
In contrast, for the large majority of Friedreich's ataxia patients, the mechanism of the iron-loading involves the decreased expression of the FRDA gene, which encodes the mitochondrial protein, frataxin [9,10]. The decreased expression of frataxin leads to multiple defects in iron-sulfur cluster and heme metabolism that results in mitochondrial iron-loading in some tissues, especially the heart [498,499]. Moreover, for one specific form of neurodegeneration with brain iron accumulation (NBIA), that is, neuroferritinopathy, dysregulation of ferritin homeostasis is certainly pathogenic. This disease results from autosomal dominant mutations in the gene encoding ferritin-L chain, with the patients presenting with some features of Parkinsonism [500].
Iron chelators represent a class of therapeutics that were originally developed for the treatment of iron overload diseases that have been acquired after multiple blood transfusions. These conditions include β-thalassemia major [501], sickle cell disease [502], diamond-blackfan anemia [503] and myelodysplastic syndromes [504]. For iron overload disease, the hexadentate hydroxamate ligand, desferrioxamine (DFO; Fig. 9A), has been the “gold standard” for clinical treatment for over 50 years [505,506]. However, this agent suffers from being orally ineffective and requires long and intensive subcutaneous infusions of 12–24 h/day, 5–6 days per week [505,506]. Moreover, the large size of DFO (Mr: 657 Da) and its hydrophilicity limits membrane permeability and access to potentially toxic intracellular iron pools [507].
![Line drawings of the molecular structures of: (A) Desferrioxamine (DFO); (B) Deferiprone; (C) Deferasirox; (D) pyridoxal isonicotinoyl hydrazone (PIH); (E) (5-[4-(2-hydroxyethyl) piperazine-1-ylmethyl]-quinoline-8-ol) (VK28); (F) N-(8-hydroxy-5-quinolylmethyl)-N-methyl-2-propynylamine M30; (G) 5-((methylamino)methyl)-8-hydroxyquinoline) (Q1); (H) 5-chloro-7-iodo-8-hydroxyquinoline (clioquinol); (I)tris-dopamine ligand 1,3,5-tris[(3,4-dihydroxyphenethyl)carbamido]benzene; (J) (S)–N6-(2-(4-([2,2′-bipyridin]-5-yl)piperazin-1-yl)ethyl)-N6-ethyl-4,5,6,7-tetrahydrobenzo [d]thiazole-2,6-diamine (D607); and (K) 5,7-dichloro-2-((ethylamino)methyl)-8-hydroxy-3-methylquinazolin-4(3H)-one (PBT434).](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr9.jpg)
Line drawings of the molecular structures of: (A) Desferrioxamine (DFO); (B) Deferiprone; (C) Deferasirox; (D) pyridoxal isonicotinoyl hydrazone (PIH); (E) (5-[4-(2-hydroxyethyl) piperazine-1-ylmethyl]-quinoline-8-ol) (VK28); (F) N-(8-hydroxy-5-quinolylmethyl)-N-methyl-2-propynylamine M30; (G) 5-((methylamino)methyl)-8-hydroxyquinoline) (Q1); (H) 5-chloro-7-iodo-8-hydroxyquinoline (clioquinol); (I)tris-dopamine ligand 1,3,5-tris[(3,4-dihydroxyphenethyl)carbamido]benzene; (J) (S)–N6-(2-(4-([2,2′-bipyridin]-5-yl)piperazin-1-yl)ethyl)-N6-ethyl-4,5,6,7-tetrahydrobenzo [d]thiazole-2,6-diamine (D607); and (K) 5,7-dichloro-2-((ethylamino)methyl)-8-hydroxy-3-methylquinazolin-4(3H)-one (PBT434).
The latter pharmacological issues have led to the development of orally active iron chelators, with better physiochemical properties. Table 2 provides a summary of such properties of chelators that have been tested in PD models or patients. These agents include the smaller, more permeable, bidentate and tridentate ligands, namely 1,2-dimethyl-3-hydroxy-4(1H)-pyridinone (Deferiprone Mr: 139 Da; Fig. 9B) and Deferasirox (Mr: 373 Da; Fig. 9C), respectively [508]. Having three clinically available chelators has enabled the implementation of combination schedules that have been utilized for some patients with β-thalassemia and sickle cell disease [509]. Deferasirox [510] and especially Deferiprone have been the subject of considerable controversy over the years [511,512]. However, both compounds have demonstrated efficacy and tolerability in patients, although their pharmacological activity needs to be carefully monitored for optimal results and to minimize side effects.
| Name of chelator | Molecular weight | denticity | Log P (calc) | Tested in Parkinson's Models | Tested in Parkinson's Patients |
|---|---|---|---|---|---|
| Desferrioxamine (DFO) | 657 | hexadentate | –0.41 | – | – |
| Deferiprone | 139 | bidentate | –1.44 | [543,544]. | [496,542,544,545]. |
| Deferasirox | 373 | tridentate | 4.98 | [543]. | – |
| Pyridoxal isonicotinoyl hydrazone: (PIH) | 286 | tridentate | 0.43 | – | – |
| 5-[4-(2-hydroxyethyl) piperazine-1-ylmethyl]-quinoline-8-ol): (VK28) | 285 | bidentate | 3.68 | [522,523]. | – |
| N-(8-hydroxy-5-quinolylmethyl)-N-methyl-2-propynylamine: (M30) | 226 | bidentate | 1.89 | [523]. | – |
| 5-((Methylamino)methyl)-8-hydroxyquinoline): (Q1) | 188 | bidentate | 1.29 | [524]. | – |
| 5-Chloro-7-iodo-8-hydroxyquinoline: (clioquinol) | 305 | bidentate | 3.64 | [525,526]. | – |
| 1,3,5-tris[(3,4-dihydroxyphenethyl)carbamido]benzene: (Tris-dopamine ligand) | 616 | tris(bidentate) | 3.16 | – | – |
| (S)–N6-(2-(4-([2,2′-bipyridin]-5-yl)piperazin-1-yl)ethyl)-N6-Ethyl-4,5,6,7-tetrahydrobenzo [d]thiazole-2,6-diamine: (D607) | 478 | bidentate | 4.22 | [532,533]. | – |
| 5,7-Dichloro-2-((ethylamino)methyl)-8-hydroxy-3-methylquinazolin-4(3H)-one: (PBT434) | 302 | bidentate | 1.72 | [403]. | – |
Other chelators that showed potential include the tridentate ligand, pyridoxal isonicotinoyl hydrazone (PIH; Fig. 9D) and its analogues, which demonstrated marked activity in vitro at chelating and mobilizing cellular iron [[513], [514], [515], [516]], in vivo in animal models [517,518] and also in humans [519]. Pyridoxal isonicotinoyl hydrazone is mentioned here because of its ability to permeate the mitochondrion and remove excess iron in vitro in a cellular model [520], or in vivo in the muscle creatine kinase frataxin knockout mouse model of Friedreich's ataxia that suffers mitochondrial iron-loading of the heart [521].
Other experimental agents of the hydroxyquinoline group have also been developed, including 5-[4-(2-hydroxyethyl) piperazine-1-ylmethyl]-quinoline-8-ol (known as VK-28; Fig. 9E), which is a chelator with a hydroxyquinoline backbone that is permeable to the blood brain barrier and demonstrates antioxidant and iron chelation efficacy [522]. The chelator, N-(8-hydroxy-5-quinolylmethyl)-N-methyl-2-propynylamine (known as M30; Fig. 9F), when administered via the oral route prevents the loss of tyrosine hydroxylase-positive neurons and attenuated iron accumulation and microglial activation in the PD mouse model induced with lactacystin [523]. Oral administration of the hydroxyquinolone analogue, 5-((methylamino)methyl)-8-hydroxy quinoline (known as Q1; Fig. 9G) to mice protected SNpc neurons against oxidative damage and MPTP-induced death [524].
The anti-protozoal and anti-fungal drug, 5-chloro-7-iodo-8-hydroxyquinoline (known as Clioquinol; Fig. 9H) and is also an effective iron chelator that was more recently demonstrated to inhibit MPTP-induced neurotoxicity in mice and reverse PD-like motor pathology in tau KO mice [525,526]. However, clioquinol in humans and animal models is associated with subacute myelo-optico-neuropathy [527], and as such, its toxicological profile is unclear. Dopamine and l-DOPA are both bidentate chelators (catechols) owing to their vicinal hydroxyl groups on the benzyl ring. Considering these properties, recent studies have developed a tris(dopamine) derivative, 1,3,5-tris[(3,4-dihydroxyphenethyl)carbamido]benzene, containing three dopamine moieties attached to a trimesic acid molecular scaffold (Fig. 9I) [528]. This new ligand has an affinity similar to that of diethylenetriaminepentaacetic acid for Fe+3 and also exhibits high antioxidant efficacy and is tolerable in vitro in cell culture [528].
As PD and also Alzheimer's disease appear to be complex and multi-factorial conditions, ligands have been designed to demonstrate polyfunctional activity to target pathological hallmarks of these diseases [529,530]. For instance, multi-functional chelators that are neuroprotectants by having both monoamine oxidase inhibitor activity with antioxidant iron chelator capacity [531]. A bi-functional ligand designed for PD treatment, namely (S)–N6-(2-(4-([2,2′-bipyridin]-5-yl)piperazin-1-yl)ethyl)-N6-ethyl-4,5,6,7-tetrahydrobenzo [d]thiazole-2,6-diamine (D-607; Fig. 9J), is based on the addition of 2 fragments, namely the Fe+2 chelator bipyridyl and also a pramipexole moiety that acts as a agonist of dopamine D2/D3 receptor [532,533]. The specific synthesis of an Fe+2 chelator was indicated as important since the ratio of Fe+3:Fe+2 from 2:1 in normal subjects is shifted to 1:2 in PD patients [534].
Additionally, a variety of mechanistic studies with D-607 [532,533] demonstrated this agent was able to: (1) act as a D2/D3 receptor agonist; (2) possess antioxidant activity that inhibits Fe+2-mediated redox cycling that would prevent GSH depletion and lipid peroxidation; (3) restore PC12 cell viability to control levels upon treatment of the redox active neurotoxin, 6-hydroxydopamine; (4) rescue dopaminergic neurons from MPTP toxicity in mice; (5) impart mitochondrial protection; (6) prevent α-synuclein aggregation potentially through the ability to bind iron [532], as iron-depletion decreases α-synuclein expression [392,393] and may prevent fibrilization of this protein [535]; and (7) via its ability to bind iron, D607 prevented proteasomal-mediated degradation of prolyl hydroxylases, which led to the up-regulation of the neuroprotective transcription factor, hypoxia-inducible factor-1α, that prevents MPTP toxicity [536] (Fig. 10).
![Schematic illustration of the effects of the dual-acting D2/D3 agonist and iron chelating ligand, D-607, and its multiple molecular targets in neurons affected by PD [532,533]. These include its ability to: (1) act as a D2/D3 receptor agonist; (2) bind Fe+2 and inhibit redox cycling that would prevent glutathione (GSH) depletion and lipid peroxidation; (3) bind iron and prevent the prolyl hydroxylase mediated degradation of hypoxia-inducible factor-1α (HIF1α); and (4) prevent α-synuclein aggregation as iron-depletion decreases α-synuclein expression [392,393] and may prevent fibrilization of this protein [535].](/dataresources/secured/content-1766030145342-4b23ff53-1c5e-4801-9fc1-7d7c0a96e6d6/assets/gr10.jpg)
Schematic illustration of the effects of the dual-acting D2/D3 agonist and iron chelating ligand, D-607, and its multiple molecular targets in neurons affected by PD [532,533]. These include its ability to: (1) act as a D2/D3 receptor agonist; (2) bind Fe+2 and inhibit redox cycling that would prevent glutathione (GSH) depletion and lipid peroxidation; (3) bind iron and prevent the prolyl hydroxylase mediated degradation of hypoxia-inducible factor-1α (HIF1α); and (4) prevent α-synuclein aggregation as iron-depletion decreases α-synuclein expression [392,393] and may prevent fibrilization of this protein [535].
Finkelstein and colleagues have developed a novel quinazolinone compound, 5,7-dichloro-2-((ethylamino)methyl)-8-hydroxy-3-methylquinazolin-4(3H)-one (known as PBT434; Fig. 9K). In vitro, PBT434 was far less potent than Deferiprone or Desferrioxamine at lowering cellular iron levels, yet was found to inhibit iron-mediated redox activity and iron-mediated aggregation of α-synuclein, a protein that aggregates in the neuropathology. In vivo, PBT434 did not deplete tissue iron stores in normal rodents, yet prevented loss of SNpc neurons, lowered nigral α-synuclein accumulation, and rescued motor performance in mice exposed to the Parkinsonian toxins, 6-OHDA and MPTP, and in a transgenic animal model (hA53T α-synuclein) of PD. These positive results were also associated with reduced markers of oxidative damage and increased levels of the iron exporter, ferroportin-1, and DJ-1. These authors have suggested that ligating a pool of pathological iron that is not held in high-affinity complexes SNpc neurons can maintain their viability [403].
Apart from iron overload, other diseases including infectious diseases, cancer, and neurodegenerative conditions, have been suggested to also possibly benefit from treatment with iron chelators [501]. For example, in terms of neuro-degenerative and cardio-degenerative conditions, chelators such as Deferiprone and PIH have been implemented in experimental models in vitro and in vivo for the treatment of Friedreich's ataxia [521,537]. While the chelators effectively decreased the mitochondrial iron-loading observed in a mouse heart model of Friedreich's ataxia, this treatment only partially ameliorated the pathology observed, namely weight loss and the hypertrophic cardiomyopathy [521]. This suggested that the iron loading contributed to the cardiac pathology, but was not the key molecular event responsible for the disease process.
Initial studies examining Friedreich's ataxia patients demonstrated that after a 6-month treatment with 20–30 mg/kg/d of Deferiprone in 9 adolescent patients, there was decreased iron loading in the dentate nuclei as demonstrated by magnetic resonance imaging (MRI) [537]. Deferiprone caused no apparent hematological or neurological toxicity, while reducing neuropathy and ataxic gait in the youngest patient [537]. Brain iron loading also occurs in familial and idiopathic ALS, prompting studies with Deferiprone in a murine preclinical model and a pilot clinical trial, which demonstrated favorable results [496]. In fact, using an ALS mouse model (i.e., Sod1G86R mice), orally administered Deferiprone was demonstrated to increase mean life span, while 23 ALS patients enrolled in the clinical trial receiving the agent orally (30 mg/kg bid), did not suffer anemia after 12 months of treatment [496]. While this safety trial was conducted with a small number of patients, Deferiprone treatment was associated with slower disability progression (as judged by the ALS Functional Rating Scale) and weight loss. MRI of the cervical spinal cord, medulla oblongata, and motor cortex, as well as cerebrospinal fluid, demonstrated lower iron levels and decreased oxidative stress after Deferiprone treatment [496]. As observed for Friedreich's ataxia, Deferiprone demonstrated safety at low doses and considerable safety without systemic iron depletion.
Considering the earlier studies above examining Friedreich's ataxia patients, there was a solid foundation for treating PD patients with chelation therapy. As described in detail in Section 7.1, a number of studies, in vitro in cell culture, in vivo in animal models and also patients indicate the beneficial effects of iron chelation therapy. The major aim is to remove the excess iron without inducing whole body iron depletion. This is achievable using the correct dose, as demonstrated in models of other diseases [538,539] and clinical trials in other conditions [537]. As described above, the SNpc of PD patients has markedly higher iron levels than normal age-matched controls that could lead to cytotoxicity and loss of dopaminergic neurons. In contrast, iron deficiency can also lead to dopaminergic neurodegeneration in mice [540], demonstrating that the action of iron is comparable to a “double-edged sword”, with a fine balance of its levels being essential.
Pioneering studies demonstrated that intracerebroventricular injection of DFO protects against the dopaminergic neurodegeneration induced by 6-hydroxydopamine, suggesting that iron chelators with appropriate permeability across the blood brain barrier could be useful neuroprotective drugs for PD [541]. Deferiprone has been considered as a potential treatment for PD due to: (1) the low molecular weight of the agent and relative high lipophilicity, which enable facile membrane permeability; (2) high affinity for iron and ability to remove iron from the iron-binding sites of transferrin; (3) permeation of the blood-brain barrier; (4) acceptable toxicological profile; and (5) high iron chelation efficacy [542]. Considering the advantages of Deferiprone and its widespread clinical use, a number of studies have assessed its therapeutic value for treating PD. Using the 6-OHDA rat model of PD, it has been demonstrated that systemic administration of Deferasirox (20 mg/kg), Deferiprone (10 mg/kg), or DFO (30 mg/kg), significantly prevented the decrease in dopaminergic neurons and striatal dopamine levels. The local administration to the rat brain striatum of DFO or Deferasirox before treatment with 6-hydroxy dopamine (6-OHDA), inhibited hydroxyl radical generation which is consistent with the ability of the ligands to bind redox active iron [543].
Similarly, studies with other iron chelators have also demonstrated efficacy in terms of their effects on PD. The ligand, VK28, when administered via the intra-cerebroventricular or intra-peritoneal route to rats was able to demonstrate protection against intracerebroventricular 6-hydroxydopamine-induced striatal dopaminergic lesions [522]. The novel mitochondrial iron chelator, 5-((methylamino)methyl)-8-hydroxyquinoline, protects against mitochondrial-induced oxidative damage and neuronal death [524].
Devos and colleagues in 2014 examined the effects of Deferiprone in cells, animals and a preliminary clinical trial [544]. Assessing cells, Deferiprone significantly reduced labile iron in oxidation-stressed cells, while in animals it improved motor function and increased striatal dopamine. A pilot, double-blind, placebo-controlled randomized clinical trial was performed using early-stage Parkinson's patients on stabilized dopamine regimens using oral Deferiprone (30 mg/kg/day) [544]. Based on a 6-month delayed-start design, early-start patients (n = 19) compared to delayed start patients (n = 18) responded significantly quicker and sustainably to treatment in terms of SNpc iron deposits (R2* MRI) and the Unified PD Rating Scale motor indicator of disease progression. Safety was maintained throughout the trial with three rapidly resolved neutropenia cases being evident [544].
In a more recent 2017 study, a randomized, double-blind, placebo-controlled trial with Deferiprone was performed using 22 early-onset PD patients [545]. The administration scheme consisted of Deferiprone at 10 or 15 mg/kg bid or placebo for 6 months. Patients were assessed for PD severity, cognitive function, depression rating and quality of life. Iron levels were examined in the SNpc, dentate and caudate nucleus, red nucleus, putamen and globus pallidus by T2* MRI at baseline and after 3 and 6 months of therapy. Deferiprone decreased dentate and caudate nucleus iron levels and was well tolerated [545]. However, SNpc iron levels decreased in only 3 patients and there was a trend for improvement in motor-Unified PD Rating Scale (UPDRS) scores and quality of life.
Collectively, the studies above provide strong preliminary evidence for larger, rigorously controlled preliminary trials especially with Deferiprone to definitively answer the question if conservative iron chelation could be a useful therapeutic option for PD.
PD is a complex, multifactorial disease that can result from insults on various molecular targets that leads to the common symptoms of the condition. Nonetheless, there are a range of distinct interconnections with the metabolism of iron. As dopaminergic neuron death in the SNpc appear to be highly specific to PD, distinct physiological differences that separate these neurons from other parts of the brain are important to identify. In fact, landmark physiological studies have demonstrated that SNpc neurons uniquely show autonomous pacemaker activity that could result in their distinct vulnerability [447,448]. This is mediated by the opening of L-type Ca+2 channels that leads to the sustained entry of Ca+2 into the cytoplasm of these neurons and mitochondrial stress and ROS generation due to the high energy demand required for this activity, but also the subsequent extrusion of Ca+2 [447,448]. Over short period of time, this may be tolerated especially through mechanisms mediated by DJ-1 to uncouple electron transport to overtly prevent mitochondrial ROS production [447,448].
However, over the longer-term, and through the reported ability of L-type Ca+2 channels to take up Fe+2, as well as Ca+2 [453], this could result in iron loading to facilitate cytotoxic ROS generation. Moreover, the activity of L-type Ca+2 channels may lead to toxic iron loading by these neurons, as it remains unknown if these channels are modulated by cellular iron levels. This mechanism of non-transferrin-bound iron uptake is possible, in contrast to the general circulation, the brain interstitial fluid contains a significant quantity of non-transferrin-bound iron, as discussed previously (see Section 3.7.1).
As part of the mitochondrial stress and bioenergetic deficit induced by the unique autonomous pacemaker activity of SNpc neurons [447,448], mechanisms may be induced as part of a rescue attempt to facilitate mitochondrial function by increasing iron uptake via the transferrin - transferrin receptor 1 pathway and reducing iron release from cells via ferroportin-1. Such a response is observed in diseases such as Friedreich's ataxia, where impaired mitochondrial iron metabolism due to the loss of frataxin, results in decreased expression of heme- and iron sulfur cluster-containing proteins, including succinate dehydrogenase, that leads to a mitochondrial bioenergetic deficit [10,498,521]. In this case, there is a marked alteration of iron trafficking away from the cytosol, with it being vectorized towards the mitochondrion. This is facilitated by increased IRP RNA-binding activity resulting in the up-regulation of the transferrin receptor 1 and the increased expression of other proteins that facilitate intracellular iron trafficking [447,448]. At the same time, there is down-regulation of FPN1, which is involved in iron release [10,498,521]. Hence, due to the mitochondrial metabolic and bioenergetic insufficiency induced by frataxin loss, iron uptake is increased as a rescue response to deliver iron to the mitochondrion that is critical for the synthesis of heme and iron-sulfur cluster proteins involved in electron transport and ATP generation.
Similar metabolic compensation has been reported in PD neurons and models, including increased IRP-RNA-binding activity [546], up-regulation of the transferrin receptor 1 [390] and a decrease in ferroportin1 expression to prevent iron release [319]. Hence, in addition to the role of L-type Ca+2 channels facilitating iron uptake from non-transferrin bound iron, there could also be increased transferrin iron uptake via the transferrin receptor 1 and a decrease iron release via FPN1. This again is suggested to act as a rescue response to ameliorate mitochondrial metabolic stress due to the unique autonomous pacemaker function of dopaminergic SNpc neurons [447,448].
Another fascinating development is the role of nitric oxide in the pathogenesis of PD and the tight interrelationship between nitric oxide and iron metabolism that could lead to mitochondrial stress and the yet to be deciphered roles of dinitrosyl dithiol iron complexes (composed of nitric oxide, iron and GSH), which are the major form of intracellular nitric oxide within cells [86]. Considering this, it is known that PD is characterized by decreased total GSH levels that specifically occurs in SNpc dopaminergic neurons [[547], [548], [549], [550]], with the magnitude of the depletion being in parallel with disease severity [540, 543. This decrease in GSH levels proceeds measurable loss in mitochondrial complex I activity or striatal dopamine content [547,550]. It has also been demonstrated that the GSH depletion in dopaminergic cells is via a NO-mediated pathway, which is independent of peroxynitrite generation [551]. Such an effect could be mediated through the ability of NO to form dinitrosyl dithiol iron complexes in dopaminergic neurons after the binding of iron from proteins involved in electron transport such as complex I that are then transported out of the cell via MRP1 leading to GSH efflux. As such, the use of nitric oxide inhibitors against inducible and neuronal nitric oxide synthase has been suggested as potential PD therapeutics [552,553].
The protective role of neuromelanin in the pathogenesis of PD has been suggested for many years, although it remains unknown how exactly this pigment captures iron and why, as all cells have the specialized iron storage protein, ferritin. This suggests that neuromelanin has a specialized function in SNpc neurons that is distinct from ferritin. Considering this, neuromelanin is thought to be generated through a process of dopamine polymerization when this neurotransmitter is in excess to prevent the generation of redox active quinones. In terms of how it captures iron, it is known that neuromelanin enters the process of autophagy and may bind iron after it is released from ferritin during its proteolysis in the autolysosome. Alternatively, neuromelanin may bind iron through interactions with iron-binding chaperone proteins, such as the PCBPs.
The complex molecular mechanisms involving iron and redox stress in the pathogenesis of PD remain intriguing and their dissection will lead to the development of novel chemotherapeutics that will enable more advanced treatment regimens. The key involvement of iron in the generation of cellular ROS certainly provides clues not only for the generation of advanced therapeutics, but also the development of detection modalities that may be used early to detect susceptibility in certain patients. For example, the continued assessment of advanced magnetic resonance imaging techniques based on brain iron levels has been indicated to be useful in providing information on PD progression and treatment.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
1
3
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
21
22
23
24
25
26
27
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
112
113
114
115
116
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
236
237
238
239
240
241
242
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
470
471
472
473
474
475
476
477
478
479
480
481
482
483
484
485
486
487
488
489
490
491
492
493
494
495
496
497
498
499
500
501
502
503
504
505
506
507
508
509
510
511
512
513
514
515
516
517
518
519
520
521
522
523
524
525
526
527
528
529
530
531
532
533
534
535
536
537
538
539
540
541
542
543
544
545
546
547
548
549
550
551
552
553
554
555
556
D.R.R. sincerely appreciates career support from the
 Parkinson's disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies
Parkinson's disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
