
Boon Cher Goh and Shazib Pervaiz contributed equally as senior authors.
Apart from its physiological role in inflammation and immunity, the nuclear factor-kappa B (NF-κB) protein complex has been implicated in tumorigenesis and its progression. Here, we provide evidence that a pro-oxidant milieu is an upstream effector of oncogenic NF-κB signaling. Through pharmacological or genetic inhibition of SOD1, we show that elevated intracellular superoxide (O2•-) mediates sustained IKK phosphorylation, and induces downstream degradation of IκBα, leading to the nuclear localization and transcriptional activation of NF-κB. Mechanistically, we show that such sustained NF-κB signaling is a function of protein phosphatase 2A (PP2A) inactivation brought about by the nitrative modification of its substrate-binding sub-unit B56γ. Importantly, the pro-oxidant driven NF-κB activation enhances the migratory and invasive potential of cancer cells. In summary, our work highlights the critical involvement of O2•--dependent peroxynitrite production in inhibiting PP2A-mediated dephosphorylation of IKK, thereby facilitating cancers to acquire an invasive phenotype. Given that NF-κB is a key player of chronic inflammation and carcinogenesis, our work unravels a novel synergistic node involving O2•--driven redox milieu and deregulated PP2A as a potential therapeutic target.
Reactive oxygen species (ROS) are important secondary messengers involved in cellular signaling and regulation of various biological processes. As such, perturbation of cellular redox states has been implicated in the etiology of many disease states including cancer. While aberrantly high ROS levels could indiscriminately induce cellular damage, recent studies demonstrated that altered redox signaling could also induce neoplastic transformation of normal cells as well as fuel the malignancy of transformed cells through activation of various pro-survival or anti-apoptotic signaling networks [[1], [2], [3], [4], [5], [6], [7], [8]]. To that end, our previous reports provided evidence to link an increase in intracellular O2•- as well as mitochondrial redox metabolism to the death inhibitory activity of Bcl-2 [[9], [10], [11]]. The crosstalk between O2•--mediated pro-survival signaling and Bcl-2 was shown to involve serine 70 phosphorylation of Bcl-2 (Bcl-2pSer70) as well as its interaction with active GTPase Rac1 [[12], [13], [14]]. Furthermore, manipulating intracellular O2•- inhibited death receptor apoptosis via the upregulation of cFLIP (cellular FLICE-like Inhibitory Protein) [15]. Moreover, we demonstrated that O2•- could regulate the protein stability and cellular half-life of the oncoprotein c-Myc [16]. Notably, unlike most physiological ROS signaling that involves the dismutation of O2•- to H2O2, the pro-survival and/or death inhibitory effect of an increase in intracellular O2•- was associated with its reaction with nitric oxide (NO) to generate peroxynitrite (ONOO−) [[15], [16], [17]]. These findings suggest that cell fate decisions appear to be a function of a specific redox milieu jointly maintained by the activities of SOD(s) and iNOS. Interestingly, the master transcription factor NF-κB was implicated in ONOO−-induced upregulation of cFLIP [15], which prompted us to question whether the pro-survival redox environment effected by ONOO− also influenced the activation and/or transcriptional activity of NF-κB and if so explore the underlying mechanism. To that end, a bi-directional crosstalk between NF-κB and cellular redox environment has been demonstrated whereby oxidative stress can activate or inhibit NF-κB and similarly NF-κB activation can serve as pro- or anti-oxidant in a cell type or context dependent manner (reviewed in Ref. [18]).
In its inactive state, NF-κB is sequestered in the cytoplasm by its physical interaction with IκBα, a member of the IκB family of inhibitory proteins. NF-κB activation is triggered by a plethora of stimuli including pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα), and interleukin-1 (IL-1), viruses, toll-like receptors and ROS [19,20]. This involves a series of upstream phosphorylation events targeting the various protein sub-units involved in NF-κB activation and regulation such as phosphorylation-mediated activation of IKKβ at serine 177 and 181 (S177/181) residues [21,22]. Phospho-activated IKKβ subsequently phosphorylates IκBα at serine 32 and serine 36 (S32/36), resulting in its ubiquitination and proteasomal degradation. This in turn releases NF-κB (p65/p50 sub-units) for translocation to the nucleus to activate its transcriptional targets, which include genes involved in cellular redox metabolism and its regulation as well as those that promote cancer cell survival, proliferation, angiogenesis, epithelial to mesenchymal transition (EMT) and metastasis [[18], [19], [20],23,24].
Whilst most studies pertaining to the interplay between cellular redox status and NF-κB activation and/or its regulation seem to highlight the direct or indirect effect of ROS such as H2O2 or NO on the upstream kinases, there is a relative lack of clarity whether the effect is exclusively on the kinases or also involves phosphatases that regulate these phosphorylation sites. To that end, the protein phosphatase 2A (PP2A) regulates serine phosphorylation sites of both IKK and IκBα [25]. Notably, our recent findings unraveled a novel mechanism of nitration-dependent inactivation of PP2A in the sustained phosphorylation of Bcl-2 (Bcl-2pSer70) and c-Myc (c-MycpSer62), which involved O2•--mediated ONOO− signaling [16,17]. On the backdrop of these findings, we set out to delineate the molecular mechanism(s) underlying the crosstalk between cellular redox metabolism and NF-κB activation. Here, we provide evidence that a pro-oxidant state triggers NF-κB activation via ONOO−-mediated sustained phosphorylation of IKK and IκBα, which stimulates cell migration and invasion potentially via upregulation of EMT inducers, Snail and Slug. Notably, the sustained phosphorylation is a function of nitration-mediated inactivation of PP2A, thus highlighting a novel mechanism of redox-induced upregulation of NF-κB signaling.
The effect of an increase in intracellular O2•- upon pharmacological inhibition of SOD1 activity with diethyldithiocarbamate (DDC) on the nuclear localization and transcriptional activity of NF-κB was investigated in human osteosarcoma U2OS cells. Results showed that DDC-induced increase in intracellular O2•- (Fig. 1A) was associated with enhanced nuclear localization of the NF-κB sub-units p65 and p50 (Fig. 1B). Similarly, the increase in nuclear localization of the non-canonical NF-κB sub-unit, RelB, was also observed upon treatment with DDC (Supplementary Fig. 1). The promoter binding and transcriptional activity of NF-κB p65 were subsequently assessed using an ELISA-based transcription factor binding assay and luciferase reporter assay, respectively. An increase in NF-κB binding and transcriptional activity was observed with increase in intracellular O2•- (Fig. 1C–D). To ascertain the role of DDC-induced O2•- in the transcriptional activity of NF-κB, cells were treated with JSH-23, a specific inhibitor of nuclear localization of NF-κB p65 [26]. Indeed, pre-treatment with JSH-23 abrogated the effect of DDC-induced O2•- on NF-κB translocation, binding and transcriptional activity (Fig. 1B–D). NF-κB has been shown to promote cancer progression through its ability to activate downstream genes that are essential for promoting cancer cell survival, invasion and metastasis (Fig. 1E). Validating the increase in the transcriptional activity, increase in intracellular O2•- induced the transactivation of NF-κB target gene products such as the EMT inducers Snail and Slug, and anti-apoptotic proteins Bcl-xL and XIAP, which could be reversed upon the addition of JSH-23 (Fig. 1F). Taken together, these data show that an increase in intracellular O2•- is a signal for the activation of NF-κB to upregulate its downstream transcriptional targets.

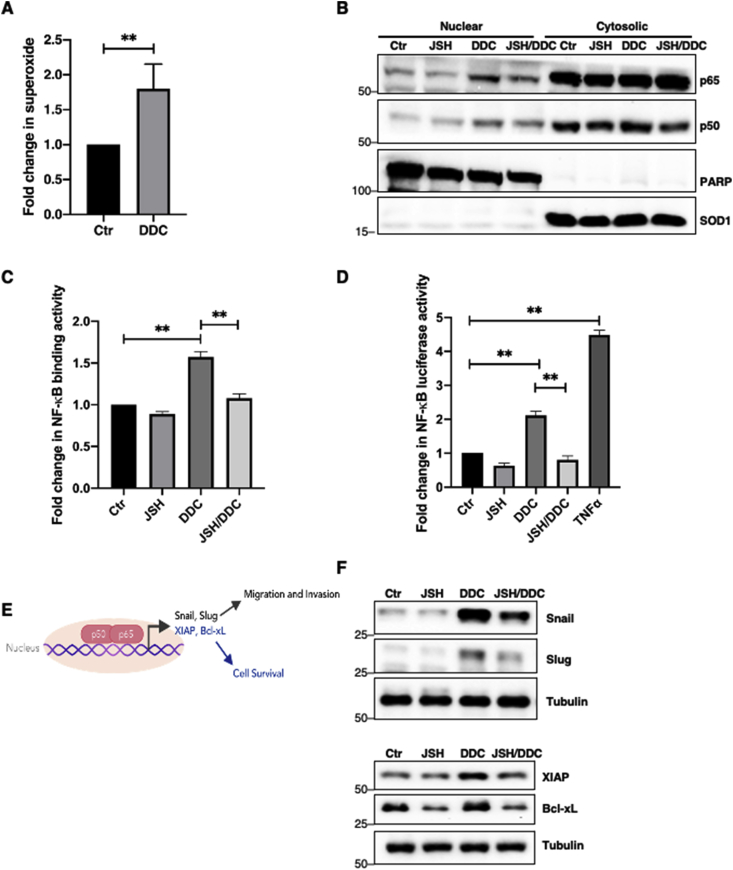
O2•-induces NF-κB localization, transcriptional activity and upregulates its transcriptional target protein levels.
A. Lucigenin chemiluminescence assay detecting intracellular O2•- level following 4-h treatment with 100 μM DDC in U2OS cells (** = p < 0.01)
B. Western blot analysis showing p65, p50, PARP (nuclear fraction control marker) and SOD1 (cytosolic fraction control marker) following nuclear-cytosolic fractionation after 12-h pre-treatment with 25 μM JSH-23 and subsequent 4-h treatment with 100 μM DDC in U2OS cells
C. Measurement of NF-κB transcription factor binding activity following nuclear-cytosolic fractionation after 12-h pre-treatment with 25 μM JSH-23 and subsequent 4-h treatment with 100 μM DDC in U2OS cells (** = p < 0.01)
D. Measurement of NF-κB luciferase reporter activity following 12-h pre-treatment with 25 μM JSH-23 and subsequent 4-h treatment with 100 μM DDC or treatment with 5 μg/ml TNFα (positive control) in U2OS cells (** = p < 0.01)
E. Schematic diagram illustrating some NF-κB transcriptional targets involved in invasion, migration and cell survival
F. Western blot analysis showing Snail, Slug, XIAP, Bcl-xL and tubulin following 12-h pre-treatment with 25 μM JSH-23 and subsequent 4-h treatment with 100 μM DDC in U2OS cells.
Since NF-κB activation is dependent on the upstream phosphorylation of IKK (p-IKKα/β) and serine 32 and 36 of IκBα (pSer32/36IκBα) that leads to the degradation of the NF-κB suppressor IκBα, we subsequently evaluated the effect of O2•- on p-IKKα/β, pSer32/36IκBα and total IκBα levels. Augmentation of intracellular O2•- by increasing doses of DDC (50–200 μM) was accompanied by increase in p-IKKα/β, and pSer32/36IκBα as well as a reciprocal decrease in IκBα levels (Fig. 2A–B). Correspondingly, an increase in phosphorylation of IKK and IκBα, and degradation of IκBα could be corroborated using small interfering RNA (siRNA) mediated silencing of SOD1 to increase intracellular O2•- (Fig. 2C–D). To further validate the role of O2•- in promoting p-IKKα/β and pSer32/36IκBα, cells were pre-treated with Tiron, a scavenger of O2•-. Indeed, scavenging O2•- reduced p-IKKα/β and pSer32/36IκBα levels and restored the expression of total IκBα to levels comparable with lysates from untreated control cells (Fig. 2E–F). Collectively, these data demonstrate that elevated intracellular O2•- level could regulate NF-κB through the increase in p-IKKα/β and pSer32/36IκBα coupled with the degradation of total IκBα.
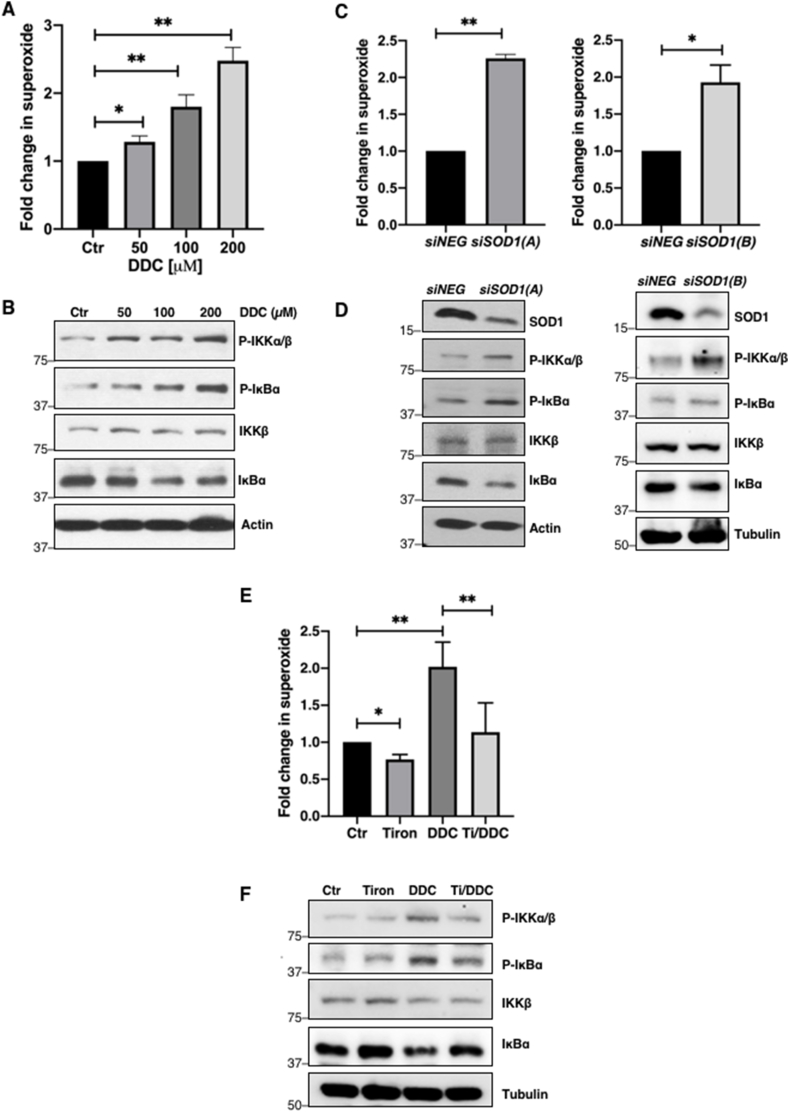
O2•-upregulates phosphorylation of IKK and pSer32/36IκBα
A. Lucigenin chemiluminescence assay detecting intracellular O2•- level following 4-h treatment with increasing concentrations of DDC (50–200 μM) in U2OS cells (* = p < 0.05, ** = p < 0.01)
B. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα and actin following 4-h with DDC (50–200 μM) in U2OS cells
C. Lucigenin chemiluminescence assay detecting intracellular O2•- level (RLU/μg protein) 48-h after transfection with control siRNA or siSOD1 sequences (100 nM) in U2OS cells (* = p < 0.05, ** = p < 0.01)
D. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα, actin and tubulin 48 h after transfection with control siRNA or siSOD1 sequences (100 nM) in U2OS cells
E. Lucigenin chemiluminescence assay detecting intracellular O2•- level following 1-h pre-treatment with 5 mM Tiron (Ti) and subsequent 4-h treatment with 100 μM DDC in U2OS cells (* = p < 0.05, ** = p < 0.01)
F. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα and tubulin following 1-h pre-treatment with 5 mM Tiron (Ti) and subsequent 4-h treatment with 100 μM DDC in U2OS cells.
Peroxynitrite (ONOO−) formation stemming from the reaction between O2•- and NO occurs rapidly at diffusion controlled rates [27]. Apart from increased O2•- levels, a readily available pool of intracellular NO is required for the generation for ONOO−. Majority of intracellular NO production relies on a tightly controlled enzymatic reaction catalyzed by nitric oxide synthases (NOS); to date, three isoforms have been identified – endothelial NOS (eNOS), inducible NOS (iNOS), and neuronal NOS (nNOS) [28]. On the other hand, intracellular O2•- is either dismutated into H2O2 by the activities of SODs or reacts with nitric oxide (NO) to produce the relatively more reactive ONOO−. Since intracellular O2•- levels are increased via pharmacological inhibition of SOD1 activity or its knockdown, we surmised that the reaction with NO is likely favoured to generate ONOO−. To ascertain if ONOO− was involved in the sustained p-IKKα/β and pSer32/36IκBα levels, we first investigated the effect of FeTPPS, a ONOO− decomposition catalyst. Notably, pre-treatment of cells with FeTPPS not only blocked the effect of DDC-induced increase in O2•- on p-IKKα/β and pSer32/36IκBα levels, but also restored total IκBα levels (Fig. 3A). In addition, treatment of cells with increasing concentrations of exogenous ONOO− increased p-IKKα/β and pSer32/36IκBα levels and led to a reciprocal decrease in IκBα (Fig. 3B). Lastly, cells treated with pharmacological NO donor/inducer, sodium nitroprusside (SNP), also affected p-IKKα/β and pSer32/36IκBα and total IκBα in a manner similar to that observed with DDC-induced increase in O2•- or upon treatment with pure ONOO− (Fig. 3C). Together, these data indicate a role for ONOO− as the downstream effector of O2•- in inducing NF-κB signaling.
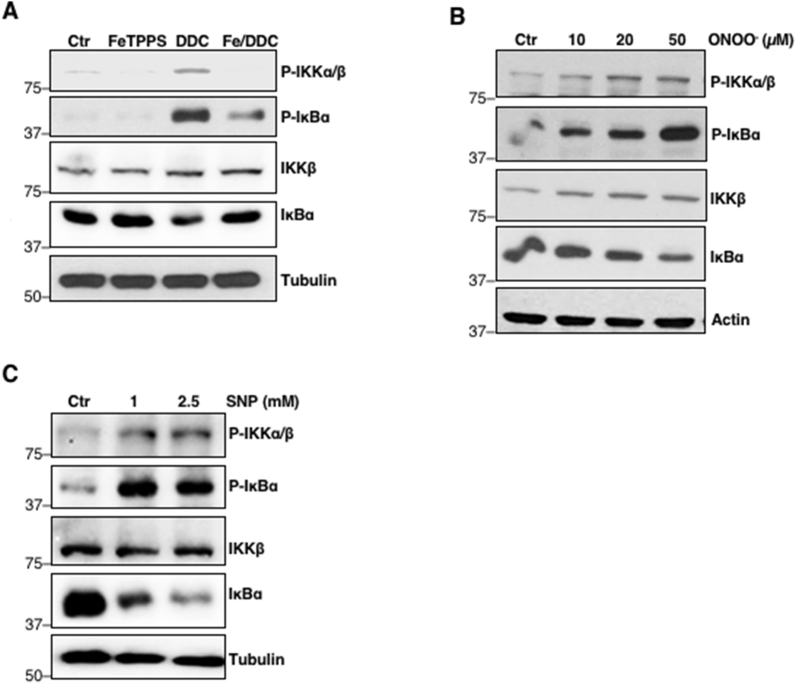
Upregulation of p-IKKα/β and pSer32/36IκBα is mediated by ONOO−
A. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα and tubulin following 2-h pre-treatment with 50 μM FeTPPS (Fe) and subsequent 4-h treatment with 100 μM DDC in U2OS cells
B. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα and actin following 2-h treatment with increasing concentrations of ONOO− (10–50 μM) in U2OS cells
C. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα and tubulin following 6-h treatment with increasing concentrations of SNP (1–2.5 mM) in U2OS cells.
Having demonstrated that O2•-/ONOO− activates NF-κB signaling, we next examined the possible mechanism(s) underlying the increase in p-IKKα/β in a pro-oxidant milieu. Canonical activation of NF-κB requires the initial phospho-activation of upstream kinase, IKK, implying that either redox activation of kinases and/or inactivation of protein phosphatases could result in ROS-mediated constitutive NF-κB signaling. Interestingly, we recently elucidated a novel mechanism underlying the upregulation of Bcl-2pSer70 and c-MycpSer62, which involved peroxynitrite (ONOO−)-mediated inactivation of PP2A [16,17]. The ONOO−-induced nitrative modification on B56δ or B56α regulatory subunit prevented the assembly of functional PP2A holoenzyme, thereby resulting in accumulation of Bcl-2pSer70 or c-MycpSer62, respectively [16,17]. Accordingly, we asked if a similar PP2A-dependent mechanism was responsible for increased p-IKKα/β levels and NF-κB activity in our model. Firstly, we investigated the involvement of PP2A using a pharmacological inhibitor of PP2A, okadaic acid (OA). Cells were treated with varying doses of OA and p-IKKα/β and pSer32/36IκBα levels were monitored. Indeed, increasing doses of OA (10–40 nM) upregulated p-IKKα/β and pSer32/36IκBα levels in a dose-dependent manner, and correspondingly downregulated total IκBα as well as upregulated NF-κB target proteins (Fig. 4A).
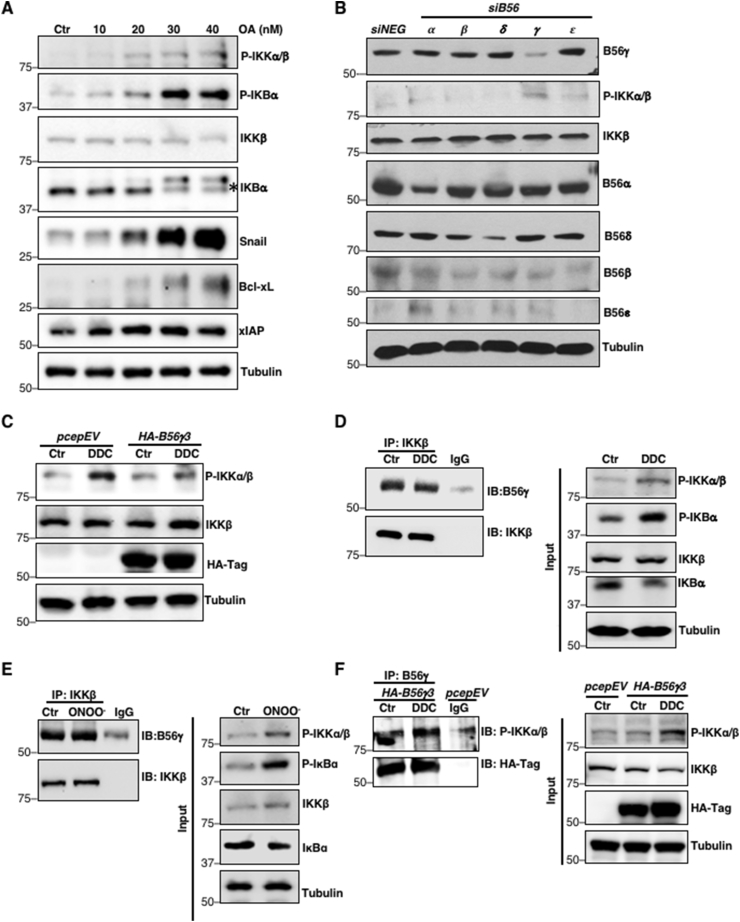
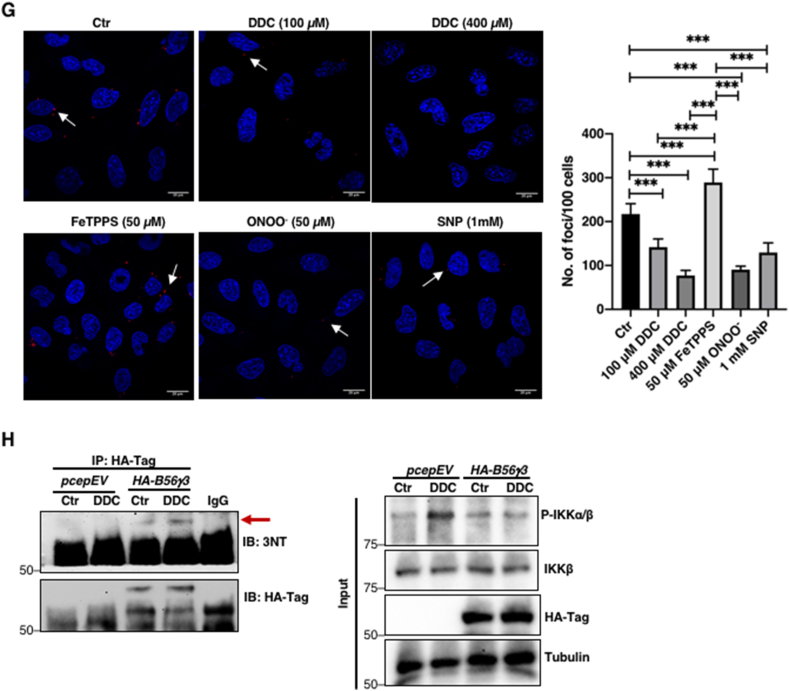
Accumulation of IKK phosphorylation is a function of redox-inactivation of B56γ3-containing PP2A holoenzyme (PP2AB56γ3)
A. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα, Snail, Bcl-xL, XIAP and tubulin following 6-h treatment with increasing concentrations of okadaic acid (10–40 nM) in U2OS cells
B. Western blot analysis showing p-IKKα/β, IKKβ, siB56α, siB56β, siB56δ, siB56γ, and siB56ε and tubulin 48-h after transfection with control siRNA or siB56α, siB56β, siB56δ, siB56γ, and siB56ε (100 nM) in U2OS cells (n = 2)
C. Western blot analysis showing p-IKKα/β, IKKβ, HA-Tag and tubulin following 4-h treatment with 100 μM DDC in U2OS cells transiently transfected with empty vector pCEP4 or HA-B56γ3 (2 μg) (n = 2)
D. Western blot analysis showing the immunoprecipitation of IKKβ and immunoblotting of B56γ following 4-h treatment with 100 μM DDC in U2OS cells (n = 2)
E. Western blot analysis showing the immunoprecipitation of IKKβ and immunoblotting of B56γ following 2-h treatment with 50 μM ONOO− in U2OS cells (n = 2)
F. Western blot analysis showing the immunoprecipitation of B56γ and immunoblotting of p-IKKα/β following 4-h treatment with 100 μM DDC in U2OS cells transiently transfected with HA-B56γ3 (8 μg) (n = 2)
G. Proximity ligation assay (PLA) showing red foci (dots) indicating endogenous interaction between PP2A catalytic subunit (PP2Ac) and IKKβ following 4-h treatment with 100 or 400 μM DDC, 6-h treatment with 50 μM FeTPPS, 2-h treatment with 50 μM ONOO− or 6-h treatment with 1 mM SNP, respectively, in U2OS cells. Random representative fields were imaged and the total number of foci in 100 random cells were counted (*** = p < 0.001)
H. Western blot analysis showing the immunoprecipitation of HA-B56γ3 and immunoblotting of 3-nitrotyrosine (3-NT) following 4-h treatment with 100 μM DDC in U2OS cells transiently transfected with HA-B56γ3 (n = 2).
PP2A is a ubiquitous heterotrimeric phosphatase comprised of a core enzyme, formed by the scaffolding subunit (PP2A-A) and the catalytic subunit (PP2A-C), which is recruited to its target proteins via a wide array of B regulatory subunits that confer substrate specificity. Hence, we first set out to identify the specific B56 regulatory subunit that interacts with and regulates the phosphorylation of IKKβ. Genetic knockdown of PP2A B56γ increased p-IKKα/β more drastically (Fig. 4B) compared to the knockdown of other B56 isoforms (α, β, δ, ε). Overexpression of HA-B56γ3 also reverted DDC-induced increase in p-IKKα/β level compared to the same treatment of cells transfected with the empty pCEP4 vector (Fig. 4C). To further ascertain if B56γ regulatory subunit is the interacting partner of IKKβ, we immunoprecipitated IKKβ. Co-immunoprecipitation (co-IP) analysis revealed interaction between B56γ and IKKβ which was not disrupted by augmentation of intracellular O2•- or ONOO− (Fig. 4D–E). Similarly, the interaction between B56γ and p-IKKα/β was not affected upon augmentation of intracellular O2•- level (Fig. 4F). Notably, proximity ligation assay (PLA) of IKKβ and PP2A catalytic subunit (PP2Ac) showed significantly fewer foci in DDC, ONOO− and SNP-treated cells compared to untreated control cells, thus suggesting that the recruitment of B56γ-IKKβ complex to PP2Ac was disrupted in the presence of elevated intracellular ONOO− (Fig. 4G). On the other hand, cells incubated with FeTPPS had significantly more foci as compared to cells treated with DDC, ONOO−, SNP or control (Fig. 4G) lending further support to this argument. Importantly, the concurrent decline in B56γ-PP2Ac interaction and upregulation of p-IKKα/β and pSer32/36IκBα levels were accompanied by the presence of nitrated B56γ3 subunit (Fig. 4H). These data collectively implicate O2•-/ONOO−-mediated inhibition of PP2AB56γ3 in the activation of NF-κB via increased p-IKKα/β and pSer32/36IκBα.
Aside from the regulation by PP2A, the phosphorylation status of IKKβ could also be influenced by the activities of its reported kinase(s) such as protein kinase C (PKC). Past studies demonstrated that PMA, a PKC activator shown to produce O2•- as a by-product of PKC-mediated activation of NADPH oxidase (NOX), induces NF-κB DNA binding and transcriptional activity [[29], [30], [31]]. Since PMA can augment O2•- production through PKC-induced activation of NOX, we also questioned whether PKC-mediated increase in kinase activation involved a redox-dependent mechanism. Results show that, while significant upregulation of p-IKKα/β and pSer32/36IκBα and a reciprocal downregulation of total IκBα was observed upon PMA treatment, there was only a moderate increase in intracellular O2•- production unlike in cells treated with DDC (Supplementary Figs. 2A–B). Notably, scavenging O2•- (Tiron) neither had a significant effect on PMA-induced p-IKKα/β and pSer32/36IκBα levels nor restored total IκBα (Supplementary Figs. 2C–D). Similarly, pre-treatment of cells with FeTPPS had no effect on PMA-induced p-IKKα/β and pSer32/36IκBα levels and was unable to restore total IκBα level (Supplementary Fig. 2E). These data appear to rule out the intermediary involvement of intracellular ROS/RNS in PKC-mediated induction of p-IKKα/β and pSer32/36IκBα as well as degradation of IκBα.
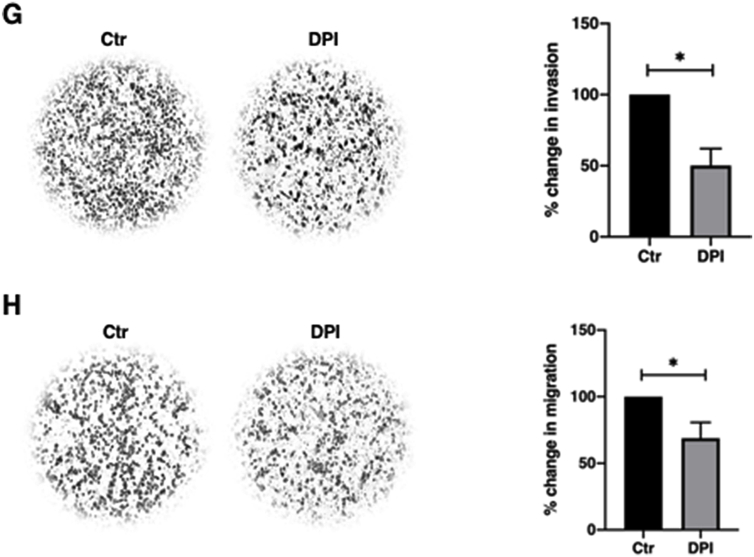
Since ROS have been shown to promote tumor progression and aberrant NF-κB activity drives tumor metastasis, we next examined the effect of an increase in intracellular O2•- on cancer cells’ migratory and invasive potentials. Interestingly, MDA-MB-231 triple negative breast cancer (TNBC) cell line possessed higher basal intracellular levels of O2•- and exhibited significantly stronger migratory and invasive capacities, compared to the ER+ luminal MCF-7 breast cancer cells (Supplementary Figs. 3A–C). In addition, an increase in intracellular O2•- by pharmacological inhibition of SOD1 activity or gene knockdown of SOD1 significantly enhanced invasiveness and migration of MCF-7 cells (Fig. 5A–D). Notably, the enhanced cell migration and invasion potentials also correlated with increased p-IKKα/β and downregulation of total IκBα levels following DDC treatment (Supplementary Fig. 4A). Conversely, MDA-MB-231 cells treated with the O2•- scavenger (Tiron) displayed a significant reduction in invasiveness, a mild reduction in migratory capacity (Fig. 5E–F) and lower p-IKKα/β and pSer32/36IκBα levels as compared to untreated control cells (Supplementary Fig. 4B), thereby reinforcing the critical involvement of NF-κB signaling in driving O2•--induced increase in cell migration and invasion. Similarly, treatment with NOX inhibitor, diphenyleneiodonium (DPI), impeded the invasive and migratory capabilities of MDA-MB-231 cells (Fig. 5G–H). Furthermore, corroborating the results of the invasion assay, decrease in intracellular O2•- significantly reduced matrix metalloproteinases (MMP) activity, one of the important drivers of cell invasion (Supplementary Figs. 5A–B).
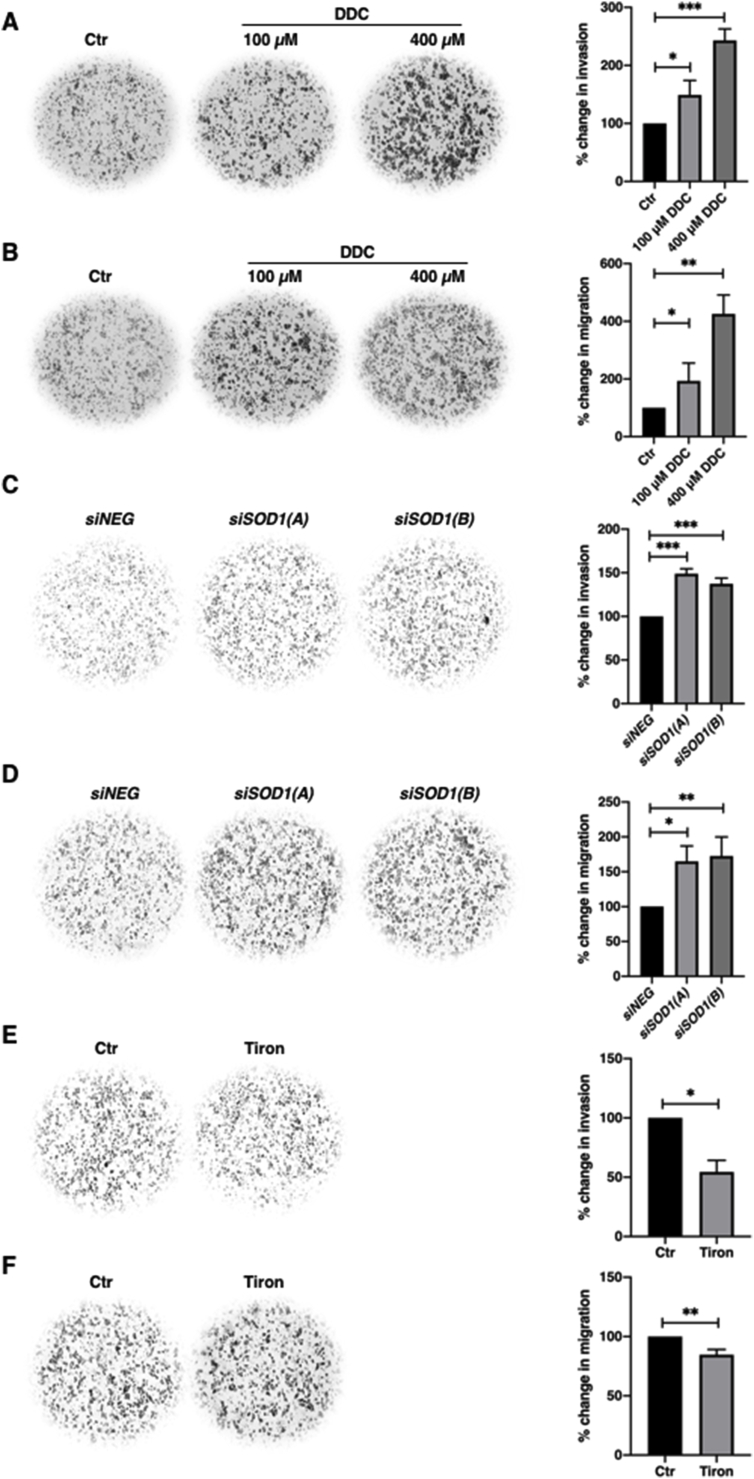
Intracellular O2•-stimulates cancer cell invasion and migration
A. 24-hour Matrigel invasion assay following 6-h treatment with 100 or 400 μM DDC in MCF-7 cells. Invaded cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in invasion of the average number of cells of 4 random fields. (* = p < 0.05, *** = p < 0.001)
B. 24-hour Boyden chamber migration assay following 6-h treatment with 100 or 400 μM DDC in MCF-7 cells. Migrated cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents the percentage change in migration of the average number of cells of 4 random fields (* = p < 0.05, ** = p < 0.01)
C. 24-hour Matrigel invasion assay following 48-h transfection with control siRNA or siSOD1 (100 nM) in MCF-7 cells. Invaded cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents the percentage change in invasion of the average number of cells of 4 random fields (*** = p < 0.001)
D. 24-hour Boyden chamber migration assay following 48-h transfection with control siRNA or siSOD1 (100 nM) in MCF-7 cells. Migrated cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents the percentage change in migration of the average number of cells of 4 random fields (* = p < 0.05, ** = p < 0.01)
E. 24-hour Matrigel invasion assay following 6-h treatment with 5 mM Tiron in MDA-MB-231 cells. Invaded cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in invasion of the average number of cells of 4 random fields (* = p < 0.05)
F. 24-hour Boyden chamber migration assay following 6-h treatment with 5 mM Tiron in MDA-MB-231 cells. Migrated cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in migration of the average number of cells of 4 random fields (** = p < 0.01)
G. 24-hour Matrigel invasion assay following 12-h treatment with 5 μM DPI in MDA-MB-231 cells. Invaded cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in invasion of the average number of cells of 4 random fields (* = p < 0.05)
H. 24-hour Boyden chamber migration assay following 12-h treatment with 5 μM DPI in MDA-MB-231 cells. Migrated cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in migration of the average number of cells of 4 random fields (* = p < 0.05).
Having shown that intracellular increase in O2•- concurrently enhanced invasion and migration potentials and increased p-IKKα/β and pSer32/36IκBα levels, we further questioned whether this was an effect of NF-κB activation. Indeed, pre-incubation of MCF-7 cells with NF-κB inhibitor JSH-23 abrogated O2•- -induced increase in cell invasion and migration (Fig. 6A–B). In addition, gene knockdown of IKKβ reverted DDC-induced upregulation of pSer32/36IκBα, rescued IκBα levels and functionally abolished the stimulatory effect of O2•- on cell migration and invasion (Fig. 6C–E). It should also be pointed out that pre-treatment with JSH-23 blocked O2•--induced upregulation of Snail and Slug (Fig. 1F), transcription factors involved in priming EMT, which is critical for migration and metastasis.
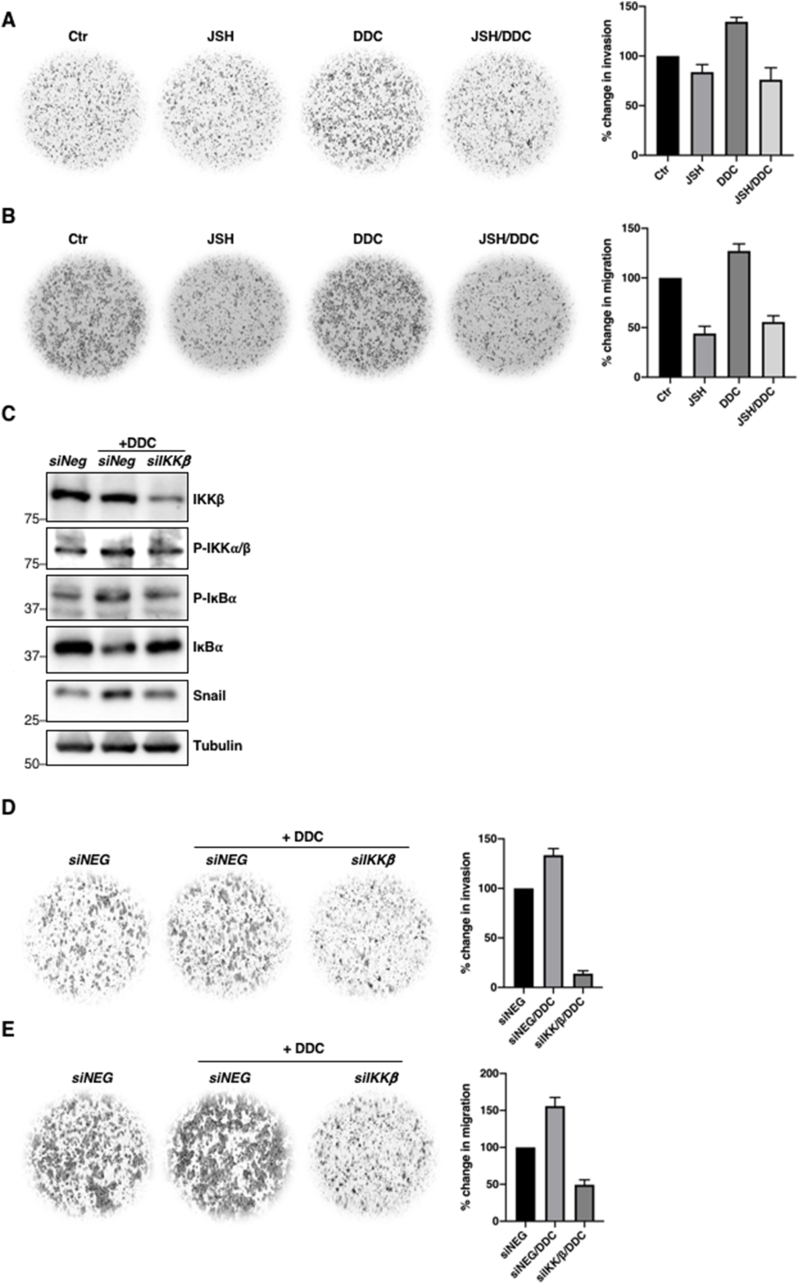
O2•--induced migration and invasion potential is a function of NF-κB activation
A. 24-hour Matrigel invasion assay following 12-h pre-treatment with 25 μM JSH-23 and subsequent 4-h treatment with 100 μM DDC in MCF-7 cells. Invaded cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in invasion of the average number of cells of 4 random fields (n = 2)
B. 24-hour Boyden chamber migration assay following 12-h pre-treatment with 25 μM JSH-23 and subsequent 4-h treatment with 100 μM DDC in MCF-7 cells. Migrated cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in migration of the average number of cells of 4 random fields (n = 2)
C. Western blot analysis showing p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα, Snail and tubulin following 48-h transfection with control siRNA or siIKKβ in MCF-7 cells (n = 2)
D. 24-hour Matrigel invasion assay following 48-h transfection with control siRNA or siIKKβ (100 nM) in MCF-7 cells. Invaded cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in invasion of the average number of cells of 4 random fields. (n = 2)
E. 24-hour Boyden chamber migration assay following 48-h transfection with control siRNA or siIKKβ (100 nM) in MCF-7 cells. Migrated cells were stained and cells from 4 random fields were counted using Fiji Image J software. The graph represents percentage change in migration of the average number of cells of 4 random fields (n = 2).
Finally, corroborating past report [16], increased O2•- levels via SOD1 inhibition significantly blunted the death-inducing effects of DNA-damaging agents, doxorubicin and etoposide, which correlated with increased NF-κB signaling and upregulation of its downstream targets Bcl-xL and XIAP (Supplementary Figs. 6A–C), thus validating our previous work linking elevated intracellular O2•- to apoptosis resistance in cancer cells [12,[15], [16], [17]]. Collectively, these findings testify to the intermediary role of NF-κB activation in the positive regulation of cell invasion, migration and survival upon an increase in intracellular O2•-.
So far we showed that a pro-oxidant milieu resulting from SOD1 inhibition or downregulation correlated with increased NF-κB activation and acquisition of an invasive/migratory phenotype. Intriguingly, we found that invasive MDA-MB-231 cells exhibited significantly lower SOD1 level as compared to non-invasive MCF-7 cells. Importantly, the lower SOD1 level observed in MDA-MB-231 cells correlated with higher basal intracellular O2•- level (Supplementary Fig. 3A), an increase in p-IKKα/β and a reciprocal decrease in total IκBα levels (Fig. 7A). This is in line with the stimulatory effect of DDC-induced O2•- on NF-κB activation (shown in the earlier figures), thereby suggesting that the migratory and invasive phenotype observed in MDA-MB-231 cells could be attributed to increased NF-κB signaling due to the constitutively higher intracellular O2•- level.
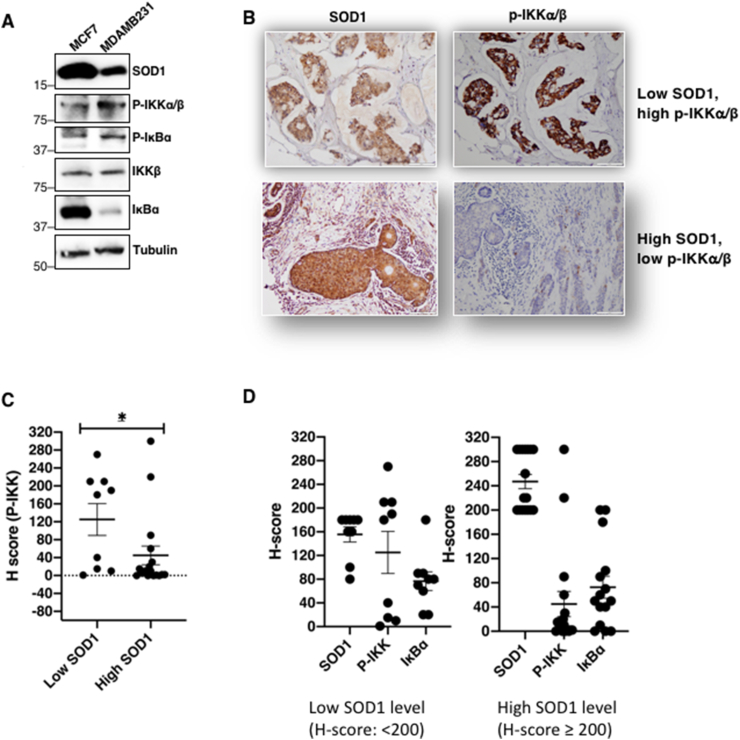
SOD1 expression negatively correlates with p-IKKα/β levels and tumor invasiveness
A. Protein expression levels of SOD1 in MCF-7 and MDA-MB-231 cells. Whole cell lysates were collected and immunoblotted for SOD1, p-IKKα/β, pSer32/36IκBα, IKKβ, IκBα and tubulin (n = 2)
B. Immunohistochemistry (IHC) staining of SOD1 and p-IKKα/β levels in patient-derived breast carcinoma tissue microarray. Representative sample of tissues with low SOD1/high p-IKKα/β levels (patient #1; top panel) and high SOD1/low p-IKKα/β levels (patient #39; bottom panel)
C. H-scores of p-IKK in low SOD1 [H < 200] (n = 9) and high SOD1 [H-score ≥ 200] (n = 16) tissue samples (* = p < 0.05)
D. H-scores of SOD1, p-IKKα/β and IκBα in low SOD1 [H < 200] and high SOD1 [H-score ≥ 200] tissue samples.
Importantly, immunohistochemistry (IHC) staining of patient-derived breast carcinoma tissues revealed that tissues with low SOD1 level (H-score: <200) had high p-IKKα/β whereas tissues with high SOD1 level (H-score: ≥200) expressed low p-IKKα/β (Fig. 7B–C). However, due to the small sample size, there was no significant correlation between low SOD1 level to low IκBα, or vice versa (Fig. 7D). These data with clinical samples, albeit in a small sample size, provide clinical proof-of-concept to our in vitro findings indicating that low SOD1 level (increase in O2•-) correlates with increased NF-κB activation, and is worthy for further exploration to provide clinical relevance.
Reports over the years have highlighted the dichotomy of redox signaling in cancer cell fate and state determination. While an overwhelming increase-also observed upon exposure of cancer cells to some chemotherapeutic agents-activates cell execution and tumor regression, there is also evidence to link a mild increase in ROS to processes that promote the initiation and/or progression of cancer [7,9,[15], [16], [17],32]. Furthermore, a more dynamic and temporal effect is beginning to emerge, whereby the effect of an increase or decrease in intracellular ROS could affect cell survival, transformation and metastasis in distinctly different manners [33]. Coupled to that is the distinctly different readouts of cancer cell fate and state, depending upon the specific ROS in question, such as O2•- or its dismutation product H2O2 [34]. It is also noteworthy that, under states of low SOD expression, an increase in intracellular O2•- was shown to endow cancer cells with a survival advantage [[15], [16], [17]], an effect linked to its reaction with NO to generate ONOO− [28,35]. As a matter of fact, the latter reaction is an order of magnitude faster than the dismutation of O2•- conversion to H2O2, and the involvement of ONOO− via its ability to target protein thiols and tyrosine residues to induce reversible or irreversible modifications with pathological sequelae has been demonstrated (elegantly reviewed in Ref. [28]).
The observed differences in the effect(s) that an altered redox milieu exert on cancer cell survival and metastasis are also influenced by the state of the intracellular anti-oxidant circuitry, often deregulated during transformation and again upon the acquisition of metastatic potential. To that end, the master transcription factor NF-κB plays a critical role in the crosstalk between cellular redox state and pro-carcinogenic processes. NF-κB is a redox-sensitive protein complex that regulates an extensive list of genes involved in anti-oxidant responses, cell fate and state transition and carcinogenesis [20,36]. Although several studies, with the majority examining the effects of increased intracellular H2O2, showed that ROS could modulate NF-κB, it has been especially challenging to characterize the effects of ROS due to conflicting observations reported by various groups [[36], [37], [38], [39], [40]]. This is, in part, due to the fact that ROS could influence NF-κB signaling at multiple points–through direct regulation of NF-κB heterodimers or by indirect regulation of upstream components including IKK and IκBα–and often in opposing and context-dependent manners i.e. increased ROS levels could activate or repress NF-κB activity [18,41]. Results presented here highlight a novel mechanism of redox-dependent activation of NF-κB as it relates to cancer cell invasion, migration and survival. An intracellular redox milieu driven by an increase in O2•- induces post-translation modification of IKKβ, specifically upregulation of p-IKKα/β, leading to downstream IκBα phosphorylation and degradation, which could be inhibited by the decomposition of ONOO−. While ROS-mediated effects on NF-κB have been extensively reported, the effects of RNS (reactive nitrogen species) still remain less well-understood with distinctly opposing activities attributed to NO (suppressing) and ONOO− (activating) [42]. Moreover, the effect of ONOO− has been mostly associated with activation of p38 MAPK-dependent phosphorylation and/or nitration of IκBα [43]. In comparison, our work provides a novel kinase-independent effect of ONOO− in mediating the sustained phosphorylation of IKK and IκBα to promote NF-κB activation, thus unraveling another potential node in the oncogenic signaling promoted by O2•- driven redox milieu.
The sustained phosphorylation of IKK and IκBα described in this report is not a function of a direct activation of a kinase but via mechanisms that disrupt the regulation of the phosphorylation sites. In this regard, PKC-mediated p-IKKα/β [44] does not seem to involve the intermediary role of cellular ROS; PMA-induced a dose dependent increase in p-IKKα/β and pSer32/36IκBα and inhibitors of O2•- or ONOO− neither rescued phosphorylation nor restored total IκBα levels. Based on these results, it seems plausible that PMA-induced increase in O2•- in the presence of normal SOD activity favors H2O2, rather than ONOO− generation, and as such the difference in the mechanism of NF-κB activation. In contrast, work reported here highlights an alternative pathway for the sustained phosphorylation involving inactivation of the phosphatase regulating serine phosphorylation(s) on IKKβ and IκBα [45]. Our results demonstrate sustained p-IKKα/β and degradation of IκBα to trigger NF-κB signaling as a consequence of redox-mediated inactivation of the putative tumor suppressor PP2A [46]. PP2A activity maintains homeostasis by regulating Ser/Thr phosphorylation(s) on proteins such as Akt/PKB, c-Myc, ERK, IKK and Bcl-2 to prevent aberrant cell proliferation, cell cycle progression, immune signaling, migration and invasion [16,17,29,[47], [48], [49], [50]]. In this regard, nitrative modification of the B56α and δ substrate-binding sub-units, which inhibited their complexing with the A and C sub-units, resulted in sustained Bcl-2pSer70 and c-MycpSer62 and promoted cell survival [16,17]. Of note, the susceptible tyrosine residue within the B56 sub-units is highly conserved, which is exemplified by the nitration of the B56γ3 sub-unit, reported in this study, to inhibit PP2A-mediated regulation of phosphorylation of IKKβ and subsequent NF-κB nuclear localization and activation of its transcriptional targets. This is, indeed, distinctly different from the reported effect of ONOO− on NF-κB activation via nitrative modification of the IκBα sub-unit [43].
Considering the role that unregulated NF-κB activation plays in the setting of chronic inflammation and several key processes associated with carcinogenesis and/or its progression [20,51], the stimulatory effect of an increase in intracellular O2•- provides yet another evidence that specific redox milieu facilitates oncogenic signaling. Under conditions of SOD1 inhibition, the propensity to generate ONOO− and its downstream effect(s) on various cell survival and/or oncogenic pathways have been reported by our group [[15], [16], [17]]. Notably, a recent study demonstrated that cellular ROS levels in pancreatic ductal adenocarcinoma (PDAC) cells are dynamically and delicately controlled to promote early carcinogenesis as well as metastasis [33]. Deregulation of the redox control upon deletion of the p53-target antioxidant protein, TIGAR, enhanced overall survival and metastatic potential of PDAC cells [33]. Similarly, loss of SOD3 (similar to our model of SOD1 inhibition) was previously shown to associate with increased aggressiveness in a model of pancreatic cancer [52]. Corroborating these findings, we provide evidence to link constitutively higher intracellular ROS (specifically O2•-) and lower SOD1 expression to the invasive nature of TNBC MDA-MB-231 cells, compared to the luminal MCF-7 cells. Moreover, a switch to a more aggressive (invasive and migratory) phenotype was obtained upon affecting an increase in intracellular O2•- in the luminal MCF-7 cells, thus corroborating the involvement of a O2•- driven redox milieu in cancer cells’ invasive and migratory capacity [34,52,53]. A recent study has also linked increased cell proliferation with elevated O2•- levels, albeit accompanied by higher SOD1 expression in ErbB2-driven primary mammary tumors [54]. Notably, inhibiting NF-κB activation rescued the effect on cell invasion and migration, as well as cell survival, which provides testimony to an intricate crosstalk between a redox state that favors the reaction of O2•- with NO (compromised SOD activity) and NF-κB activation in promoting drug resistance and fuelling cancer progression. These effects most likely could be explained by the induction of NF-κB target genes such as Bcl-xL and XIAP that promote apoptosis resistance as well as Snail and Slug that are involved in cell state regulation (EMT).
These findings have translational and therapeutic relevance. Data from a small number of clinical samples (breast CA TMA) seem to support the association between low SOD1 and high p-IKK score, compared to the high SOD1 expression. A similar correlation between low SOD1 expression and high Bcl-2pSer70 was reported in human lymphomas [17], which was also a function of nitrative modification of the substrate-binding sub-unit of PP2A, leading to its inactivation. Interestingly, a highly recurrent PP2A mutation has been reported to promote endometrial carcinogenesis [55]. The intertwined networking of the three nodes, namely a permissive redox environment, PP2A deregulation and NF-κB activation, lends itself to be exploited as a biomarker of aggressive/advanced disease as well as a therapeutic target. From the standpoint of targeted therapeutics, NF-κB inhibition would not be possible because of the profound role it plays in a host of physiological processes. PP2A reactivation has emerged as an attractive strategy due to the realization that its deregulation promotes oncogenic signaling [46,56,57]. Supporting this, FTY720, a PP2A activating drug, has demonstrated anti-leukemic activity [58], and a combination approach with standard anti-cancer agents significantly enhanced apoptosis sensitivity of leukemia cells ex vivo [59]. As for modulating the cellular redox environment, clinical studies with the use of anti-oxidants have not yielded promising results, which could very well be due to the nature of the ROS species being targeted. Based on our findings, it is plausible that inhibiting or scavenging intracellular ONOO− could be a novel strategy to specifically tailor the intracellular milieu. To that end, SOD mimetics such as the manganese porphyrin-based (MnP) compounds have shown great potential and are being clinically evaluated [60,61]. Notably, the direct anti-oxidant effect of the MnP compounds such as MnTBAP is believed to be due to the ability to decompose ONOO− [62]. As such, a combination approach involving PP2A activating drug(s) and compounds specifically targeting ONOO− such as SOD mimetics could bode well as a promising therapeutic approach to combat processes associated with redox-driven aberrant NF-κB activation.
In summary, our work highlights a novel signaling node involving redox-mediated inactivation of PP2A to relieve the breaks on NF-κB activation, which endows cancer cells with heightened invasive and migratory capacity and the ability to evade execution (Fig. 8). These results unravel a redox-vulnerability which could potentially be targeted with therapeutic strategies including judicious inclusion of PP2A activator and/or anti-oxidants in treatment regimes.
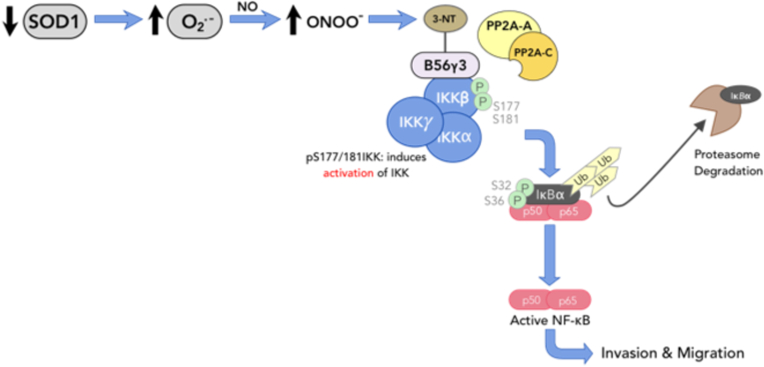
Schematic summary of O2•-/ONOO−-mediated activation of NF-κB via redox inactivation of PP2A promotes invasiveness.
Downregulation or inhibition of SOD1 induces a pro-oxidant milieu favoring elevated intracellular O2•- upregulates ONOO− level through its reaction with NO. Resulting downstream ONOO− nitrates and inactivates B56γ3-containing PP2A holoenzyme assembly by preventing the recruitment of PP2A AC catalytic core to IKKβ. This consequentially results in accumulation of p-IKKα/β leading to activation of NF-κB signaling which promotes tumor cell invasion and migration potential.
Human U2-OS, MCF-7 and MDA-MB-231 were purchased from ATCC. Human U2-OS human osteosarcoma cells were cultured in McCoy's 5A (1% l-Glutamine, 1% penicillin/streptomycin and 10% FBS). Human MCF-7 breast carcinoma cells were maintained in DMEM/High Glucose (1% l-Glutamine, 1% penicillin/streptomycin and 10% FBS and human MDA-MB-231 breast carcinoma cells were maintained in RPMI-1640 (1% l-Glutamine, 1% penicillin/streptomycin and 10% FBS).
Rabbit antibodies for the detection of Phospho-IKKα/β (Ser176/180), IκBα, Snail, Slug, XIAP, RelB, p52, Tubulin and HA-tag as well as mouse antibodies against Phospho-IκBα (Ser32/36) and HA-tag were purchased from Cell Signaling Technology (Massachusetts, USA). Mouse antibodies specific for IKKα, IKKβ, and SOD1 were purchased from BD Pharmingen. Rabbit antibodies against NF-κB p65 as well as mouse antibodies against NF-κB p50, Bcl-xL and PP2A-B56α, B56β, B56γ, B56δ, B56ε were purchased from Santa Cruz Biotechnology (Texas, USA). Rabbit antibody for the detection of SOD2 as well as mouse antibodies against PP2A-C subunit (clone 1D6) and 3-nitrotyrosine were purchased from Merck Millipore (Massachusetts, USA). Rabbit antibody against IKKβ was purchased from Invitrogen Thermo Fisher Scientific (Massachusetts, USA) and rabbit antibody against PP2A-C subunit was purchased from Proteintech Biotechnology (Illinois, USA). Rabbit antibody against SOD1 was purchased from Sigma Aldrich (Missouri, USA). Diethyldithiocarbamate (DDC), 4,5-dihydroxy-1,3-benzenedisulfonic acid (Tiron), sodium nitroprusside (SNP), ammonium tetrathiomolybdate (ATN), phorbol 12-myristate 13-acetate (PMA), N,N′-Dimethyl-9,9′-biacridinium dinitrate (Lucigenin), were all purchased from Sigma Aldrich (Missouri, USA). Okadaic acid, bisindolylmaleimide IV, 5,10,15,20-Tetrakis (4-sulfonatophenyl) porphyrinato Iron (III) Chloride (FeTPPS) were purchased from Merck Millipore (Massachusetts, USA). Peroxynitrite (ONOO−) and JSH-23 were purchased from Cayman Chemical (Michigan, United States).
pCEP4 empty vector was a generous gift from Professor David Marc Virshup from DUKE-NUS GMS, Singapore. NF-κB luciferase reporter vector and renilla luciferase vector were generous gifts from Professor Pieter Eichhorn from Curtin University, Australia. pCEP4-HA-B56γ3 (#14535) was purchased from Addgene. Scrambled siRNA and siRNAs targeting human SOD1 (J-0088364-09 and J-0088364-11) were purchased from Dharmacon Technologies. IKKβ siRNA (#VHS40301) was purchased from Invitrogen Life Technologies (Massachusetts, USA). The respective siRNAs targeting human PP2A-B56α (#SI02225839), B56β (#SI02659034), B56γ (#SI02663626), B56δ (#SI02653350) and B56ε (#SI02659041) were purchased from Qiagen.
Intracellular O2•- was determined independently either by a lucigenin-based chemiluminescence assay or by flow cytometry using O2•--sensitive fluorescent probe, dihydroethidium (DHE) (Thermo Fisher Scientific, Massachusetts, USA). For lucigenin-based assay, harvested cells were washed with 1X PBS and lysed in 450 μl of ATP lysis buffer (Sigma Aldrich, Missouri, USA). Upon cell lysis, lysates were immediately aspirated for chemiluminescence monitoring using the Berthold Sirius Luminometer (Bad Wildbad, Germany) at a rate of 0.6 s interval over 14.4 s. The luminescence emission was measured and recorded as relative light units (RLU). The average of RLUs was then calculated and subjected to normalization by respective sample protein concentration, determined by Coomassie Plus protein assay reagent (Pierce Biotechnology, Massachusetts, USA).
For flow cytometry-based detection of O2•- harvested cells were resuspended with plain medium containing 5 μM DHE and incubated for 15 min at 37 °C. Stained cells were then analysed by CytoFLEX LX Flow Cytometer (Beckman Coulter, California, USA) with excitation wavelength set at 595 nm and cell counter at 10,000 events. Subsequent data analysis was done using the CytExpert software.
Cells harvested were lysed with ice-cold co-immunoprecitation (co-IP) lysis buffer (50 mM Tris, pH 7.6, 150 mM NaCl, 0.5% Nonidet-P40, 1 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 20 μg/ml pepstatin A, 1 mM NaF, 1 mM Na3VO4 and 1 mM PMSF). 1.5 mg of cell lysate was pre-cleared using Protein-A agarose beads for an hour before overnight incubation with 3 μg of rabbit anti-IKKβ antibody. Protein A-agarose beads were then added to the mixture for 6 h to pull down the immune-complexes. Following which, protein A-agarose beads were washed thrice with co-IP lysis buffer without Nonidet-P-40. Protein samples mixed with Laemelli loading buffer were heated at 100 °C for 10 min. Samples and molecular weight protein markers (Bio-Rad, California, USA) were then subjected to 9% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). Resolved proteins were transferred onto a polyvinyl difluoride (PVDF) membrane using the wet transfer method. Membranes were incubated with blocking buffer for an hour before overnight incubation with antibodies specific for the protein of interest. Following which, membranes were washed thrice with TBS/Tween-20 (TBST) and incubated with the appropriate secondary antibody in TBST containing 1% fat-free milk on shaker for 1 h. Membranes were subsequently washed thrice with TBST before incubation with EZ-ECL chemiluminescent substrate (Biological Industries, Beit HaEmek, Israel). Protein intensities were detected and visualized using the iBright FL1000 imaging system (Invitrogen Thermo Fisher Scientific, Massachusetts, USA).
Cells were harvested and pelleted down following treatment. Stepwise lysis of cells and centrifugal isolation of nuclear and cytoplasmic protein fractions was performed according to manufacturer's recommendation using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Cat#78835; Thermo Fisher Scientific, Massachusetts, USA).
PLA was performed using the Duolink® In-Situ Red Starter Kit Mouse/Rabbit purchased from Sigma Aldrich (Missouri, USA). Treated cells on coverslips were fixed with 4% paraformaldehyde and permeabilized using 0.2% Triton-X. Apart from overnight primary antibody incubation, subsequent steps were performed according to manufacturer's protocol. Prepared samples were subsequently imaged for red signals using the Olympus Fluoview FV1000 confocal microscope (Tokyo, Japan).
Treated cells were harvested and resuspended in 1% FBS medium. Approximately 6 × 104 cells were seeded in a Boyden chamber insert (Corning, New York, USA) and suspended over a well containing medium supplemented with 1% l-glutamine and 10% FBS. Following 24-h incubation, non-migratory cells were removed from the upper surface of the chamber insert using a cotton tip swab moistened with medium. Migrated cells on the lower surface of the chamber insert were fixed with methanol and stained using crystal violet solution. The migrated cells were visualized and images were captured using the Olympus IX71 microscope (Tokyo, Japan). Cell counting was performed on four random microscope fields per sample using the Fiji Image J software.
Treated cells were harvested and resuspended in 1% FBS medium. Approximately 3 × 105 cells were seeded in a Matrigel-coated chamber insert (Corning, New York, USA) suspended over a well containing medium supplemented with 1% l-glutamine and 10% FBS. Following 24-h incubation, non-invading cells were removed from the upper surface of the chamber insert using a cotton tip swab moistened with medium. Invaded cells on the lower surface of the chamber insert were fixed with methanol and stained using crystal violet solution. The invaded cells were visualized and images were captured using the Olympus IX71 microscope (Tokyo, Japan). Cell counting was performed on four random microscope fields per sample using the Fiji Image J software.
Harvested cells were subjected to nuclear-cytoplasmic fractionation as per the abovementioned protocol. Isolated nuclear protein fractions were then applied to the ELISA plate provided in the kit and subsequent steps were performed according to manufacturer's instructions to assess NF-κB p65 binding activity (AbCam, Cat. #133112). The final absorbance readout was measured at 450 nm using the Spectrafluor Plus spectrophotometer (TECAN, Männedorf, Switzerland).
Cells transfected with NF-κB luciferase reporter vector and renilla luciferase vector were treated, 24-h post transfection, with the intended reagents in a triplicate format. Treated cells were then harvested per the protocol provided by the manufacturer (Promega, Cat. #E1910). The luminescence emission was detected and measured using the Varioskan LUX Multimode Microplate Reader (Thermo Fisher Scientific, Massachusetts, USA). The respective firefly and renilla luciferase luminescence activities were recorded as relative luminescence units (RLU). Fold change in activity was subsequently calculated after subtracting the reporter activity from the negative control.
Cells were subjected to intended treatments and supernatant was aspirated 24-h post treatment and transferred to a black 96-well optical-bottom plate. Matrix metalloproteinases (MMP) activity was then detected using a MMP activity kit (Cat. #112146) purchased from Abcam (Cambridge, United Kingdom).
Harvested cells post-treatment were transferred to a 96-well plate in a triplicate format and mixed with 4 mg/ml MTT (Sigma Aldrich, Missouri, USA). The mixture was incubated for an hour at 37 °C before centrifugation at 4200 rpm for 25 min. Supernatant was then discarded and the resulting formazan crystals were dissolved in a solution containing DMSO and Sorenson's glycine buffer (0.1 M glycine, 0.1 M NaCl, pH 10.5) for 30 min on a shaker. Cell viability was subsequently assessed by quantitating the resultant mixture at an absorbance wavelength of 570 nm using the Spectrafluor Plus spectrophotometer (TECAN, Männedorf, Switzerland).
Formalin-fixed, paraffin-embedded (FFPE) breast tissues from anonymised patients spotted as tissue microarray (TMA) (#30212) was obtained from Novus Biologicals (Colorado, USA). Immunohistochemistry staining of FFPE sections was performed using the BOND-MAX Automated Immunohistochemistry Biosystem (Leica Biosystems, Wetzlar, Germany) according to the following protocol. Briefly, tissues were deparaffinised and subjected to antigen retrieval and peroxidase blocking. Tissues were then washed and incubated with primary antibody for 8 min. Following which, tissues were washed and incubated with polymer for 8 min and DAB for 10 min prior to hematoxylin staining for 10 min. Images of the sections were photographed at 20X magnification. The cores which were missing or insufficient to score were excluded from the analysis. H scores were derived by multiplying the intensity grades (0: no expression; +1: mild; +2: moderate, +3: strong) by the extent of expression (in percentage). The cut-off for the low and high groups was determined per the median of the normal matched tissues.
Results were presented as mean + SD. One-way ANOVA was performed in cases where there were three or more comparisions while Student t-test (two-tailed) was employed for pairwise comparisions. Experiments were repeated three times unless otherwise stated. Statistical significance was set at P < 0.05.
Several images included in the manuscript have been adapted from the thesis titled, ‘Superoxide-mediated activation of NF-κB signalling via redox-mediated PP2A inactivation’, of Yee Yi Hui (first author) for the fulfilment of the degree of Doctor of Philosophy at National University of Singapore (NUS).
The authors declare no potential conflicts of interest.
1
2
4
5
6
7
9
10
11
12
13
14
15
16
17
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
37
38
39
40
41
42
43
44
45
47
48
49
50
51
52
53
54
55
57
58
59
60
61
62
The authors wish to thank David M. Virshup (DUKE-NUS, Singapore) for the generous gift of anti-B56α and B56β antibodies as well as the pCEP4 empty vector. We thank Pieter Eichhorn (Curtin University, Perth, Australia) for providing us the NF-κB luciferase reporter vector and renilla luciferase vector. The authors also extend their gratitude to Supriya Srivastava and Jie Qing Eu for providing technical support to this study, and Deepika Raman for her thorough and constructive feedback on the manuscript. This work is supported by grants from National Medical Research Council (NMRC) (NMRC/CIRG/1433/2015) and Ministry of Education (MOE2013-T2-2-130), Singapore to SP as well as grants from NMRC (NMRC/CSA-SI/0006/2016), National Research Foundation (NRF) Singapore, and Ministry of Education, Singapore, under its Research Centres of Excellence initiatives to GBC.
List of antibodies (species, company and catalogue no, dilution and application).

| Primary Antibody | Company | Catalogue no. | Dilution | Application |
|---|---|---|---|---|
| Phospho-IKKα/β (Ser176/180) | Cell Signalling Technology | #2687 | 1:1000 1:100 | WB IHC |
| Phospho-IκBα (Ser32/36) | Cell Signalling Technology | #9246 | 1:1000 | WB |
| IκBα | Cell Signalling Technology | #9242 | 1:1000 | WB |
| Snail | Cell Signalling Technology | #3879 | 1:1000 | WB |
| Slug | Cell Signalling Technology | #9585 | 1:1000 | WB |
| XIAP | Cell Signalling Technology | #2045 | 1:1000 | WB |
| RelB | Cell Signalling Technology | #4954 | 1:1000 | WB |
| p100/p52 | Cell Signalling Technology | #4882 | 1:1000 | WB |
| Tubulin | Cell Signalling Technology | #2125 | 1:1000 | WB |
| HA-Tag | Cell Signalling Technology | #3724 | 1:1000 | WB |
| HA-Tag | Cell Signalling Technology | #2367 | 1:1000 | WB |
| PARP | BD Pharmingen | 611039 | 1:1000 | WB |
| IKKβ | BD Pharmingen | 51–8120 GR | 1:1000 | WB |
| SOD1 | BD Pharmingen | 556360 | 1:1000 | WB |
| NF-κB p65 | Santa Cruz Biotechnology | SC-514451 | 1:500 | WB |
| NF-κB p50 | Santa Cruz Biotechnology | SC-8414 | 1:500 | WB |
| Bcl-xL | Santa Cruz Biotechnology | SC-8392 | 1:500 | WB |
| PP2A-B56α | Gift from DMV | NA | 1:500 | WB |
| PP2A-B56β | Gift from DMV | NA | 1:500 | WB |
| PP2A-B56γ | Santa Cruz Biotechnology | SC-374379 | 1:500 3 μg | WB IP |
| PP2A-B56δ | Santa Cruz Biotechnology | SC-81605 | 1:500 | WB |
| PP2A-B56ε | Santa Cruz Biotechnology | SC-376176 | 1:500 | WB |
| PP2A-C | Merck Millipore | 05–421 | 1:1000 | WB |
| 3-nitrotyrosine | Merck Millipore | 05–233 | 1:1000 | WB |
| IKKβ | Invitrogen Thermo Fisher Scientific | PA1-32139 | 3 μg | IP |
| PP2A-C | Proteintech | 13482-1-AP | 3 μg | IP |
| HA-Tag | Proteintech | 51064-2-AP | 3 μg | IP |
| IKKβ | Santa Cruz | SC-8014 | 1:100 | PLA |
| PP2A-C | Cell Signalling | #2038 | 1:100 | PLA |
| SOD1 | Sigma Aldrich | HPA001401 | 1:2000 | IHC |
Cell Signalling Technology (Massachusetts, USA), BD Pharmingen (California, USA), Santa Cruz Biotechnology (Texas, USA), Millipore (Massachusetts, USA), Proteintech (Illinois, USA), Invitrogen Thermo Fisher Scientific (Massachusetts, USA).
Rabbit primary antibodies against B56α and B56β were a generous gift from Prof David Virshup (Duke-NUS Medical School, Singapore).
Phospho-IKKα/β (Ser176/180) antibody recognizes IKKβ only when phosphorylated at Ser177/181.
 Sustained IKKβ phosphorylation and NF-κB activation by superoxide-induced peroxynitrite-mediated nitrotyrosine modification of B56γ3 and PP2A inactivation
Sustained IKKβ phosphorylation and NF-κB activation by superoxide-induced peroxynitrite-mediated nitrotyrosine modification of B56γ3 and PP2A inactivation
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
