 The First Bismuth Borosulfates Comprising Oxonium and a Tectosilicate‐Analogous Anion
The First Bismuth Borosulfates Comprising Oxonium and a Tectosilicate‐Analogous Anion
Angewandte Chemie (International Ed. in English)



- Altmetric
The first bismuth borosulfate (H3O)Bi[B(SO4)2]4 is only the second featuring a three‐dimensional anion, the first tectosilicate‐analogous borosulfate synthesised solvothermally without a precursor (from Bi(NO3)3⋅5 H2O and B(OH)3 in oleum); moreover, it is the first comprising two differently charged cations and crystallises in a new structure type in space group I4‾ (no. 82) (a=11.857(1), c=8.149(1) Å, 1947 refl., 111 param., wR2=0.037), confirmed by a second harmonic generation (SHG) measurement. The B(SO4)4 supertetrahedra are connected via all four sulfate tetrahedra resulting in a three‐dimensional anion with both H3O+ and Bi3+ cations in channels. Additionally, the crystal structure of a further bismuth borosulfate, Bi2[B2(SO4)6], is elucidated crystallising isotypically to the rare‐earth borosulfates R2[B2(SO4)6] in space group C2/c (No. 15) (a=13.568(2), b=11.490(2), c=11.106(2) Å, 3127 refl., 155 param., wR2=0.035). Moreover, the optical and thermal properties of both compounds are discussed.
The first tectosilicate‐analogous oxonium–bismuth borosulfate obtained from the reaction of oleum with bismuth nitrate and boric acid decomposes to the first bismuth borosulfate—a ring‐silicate‐analogous one—delivering exciting insights in the chemistry and the crystal structures of this still expanding material class.
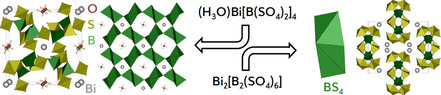
Crystalline borosulfates are a fast growing class of silicate‐analogous compounds since discovering the first representative K5[B(SO4)4] in 2012. [1] Meanwhile, over sixty compounds were characterised by mid of 2020.[ 2 , 3 , 4 , 5 , 6 ] Silicate‐analogous materials comprise tetrahedral basic building units as characteristic structural motif. The absence of an inversion centre within these tetrahedra often leads to interesting optical properties as they foster non‐centrosymmetric coordination environments around emitting cations and thus promote high luminescence probabilities or even non‐linear optical properties if the whole crystal structure lacks inversion symmetry.[ 7 , 8 , 9 ] Borosulfates may further be classified as silicate‐analogous in two ways: On the one hand, the anionic substructure so far consists exclusively of borate and sulfate tetrahedra. On the other hand, the boron atoms may be treated as coordinated by sulfate tetrahedra yielding supertetrahedra TX 4 like B(SO4)4. Consequently, borosulfates can be ranked by the dimensionality of their anion adapting Friedrich Liebau's classification of silicates. [10] There are either zero‐dimensional (0D) structures with non‐condensed supertetrahedra (T/X=1:4), 1D chains or 0D rings of TX 4 (normally showing a ratio of T/X=1:3), 2D layers and 3D frameworks (normally T/X=1:2). For the latter, the only reported compound so far is Li[B(SO4)2] [11] despite the huge number of reported borosulfates in the last eight years. Further, borosulfates feature simple cations like in Li[B(SO4)2] [11] or R 2[B2(SO4)6] (R=Y, La–Nd, Sm–Lu)[ 8 , 12 ] but also molecular cations as observed in H3O[B(SO4)2] [13] or (NH4)3[B(SO4)3]. [5] CsK4[B(SO4)4], Cs3Na2[B(SO4)4], and Cs3Li2[B(SO4)4] are the only borosulfates containing two different cations up to date and, apparently, here they all exhibit the same charge. [2]
In a solvothermal synthesis starting from Bi(NO3)3⋅5 H2O and B(OH)3 in oleum (65 % SO3) we obtained (H3O)Bi[B(SO4)2]4 as a colourless and almost phase‐pure powder containing single crystals (for further details, see the Supporting Information). Rietveld analysis revealed Bi2[B2(SO4)6] and Bi(HSO4)3 [15] as fractional side phases (see Figure S1 and Table S1 in the Supporting Information). According to our single‐crystal X‐ray diffraction (XRD) structure determination (H3O)Bi[B(SO4)2]4 crystallises in a new structure type in space group I (no. 82). Refinement details are summarised in Table S1. [38]
Figures 1 a and 1 b display the unit cell of the title compound. The anion consists of TX 4 supertetrahedra (T=B, X=SO4) sharing all four corners with neighbouring ones yielding a 3D network. Herein, Vierer and Achter rings perpendicular to c are found giving rise for channels along c as depicted in Figure 1 c; the Vierer ring channels host the bismuth, the Achter ring channels the oxonium cations. The formed network TX 2 resembles the composition of tectosilicates like SiO2[ 11 , 16 ] and features found in naturally occurring silicates: adjacent Vierer and Achter rings—however in a different ratio—are known for pure silica chabazite prepared via a template‐based synthesis with the sum formula SiO2. [17] The mineral harmotome Ba2Al4Si12O32⋅12H2O [14] exhibits a network of AlO4/SiO4 tetrahedra comprising channels of adjacent Vierer and Achter rings resembling the topology found for the B(SO4)4 supertetrahedra in (H3O)Bi[B(SO4)2]4 (see Figure 1 d).

![Crystal structure of (H3O)Bi[B(SO4)2]4: a) shows the borate (green) and sulfate (yellow) tetrahedra, b) shows the B(SO4)4 supertetrahedra neglecting the oxygen atoms (red) except the oxonium one; bismuth is depicted in grey and hydrogen in white; c) illustrates channels of supertetrahedra (dark green) forming Vierer and Achter rings along the [001] direction; the H3O+ and Bi3+ cations are located inside these channels; oxygen atoms are neglected except the oxonium one; d) depicts the anion of harmotome Ba2Al4Si12O32⋅12H2O
[14]
comprising AlO4/SiO4 tetrahedra (light blue) forming analogous Vierer and Achter rings.](/dataresources/secured/content-1765834313083-d73d51d3-ecbb-440d-ab9e-67c06df6642c/assets/ANIE-60-1503-g001.jpg)
Crystal structure of (H3O)Bi[B(SO4)2]4: a) shows the borate (green) and sulfate (yellow) tetrahedra, b) shows the B(SO4)4 supertetrahedra neglecting the oxygen atoms (red) except the oxonium one; bismuth is depicted in grey and hydrogen in white; c) illustrates channels of supertetrahedra (dark green) forming Vierer and Achter rings along the [001] direction; the H3O+ and Bi3+ cations are located inside these channels; oxygen atoms are neglected except the oxonium one; d) depicts the anion of harmotome Ba2Al4Si12O32⋅12H2O [14] comprising AlO4/SiO4 tetrahedra (light blue) forming analogous Vierer and Achter rings.
The trivalent bismuth ions are coordinated with distances ranging from 237.9(3) to 246.3(3) pm by eight oxygen atoms belonging to monodentate sulfate tetrahedra resulting in a distorted square antiprismatic coordination environment (Figure S2 in the Supporting Information). The H3O+ ions are equally distributed with an occupation of 50 % on site 4f along the channels (Figure 1 c); there, two different orientations regarding the hydrogen atoms due to the local symmetry are observed. The oxonium ions are held in place by medium strong hydrogen bonds [18] to two monodentate sulfate tetrahedra (Figure S3) with a donor–acceptor distance of 284(1) pm, longer than that reported for H3O[B(SO4)2] (272 pm). [13] The interatomic distances listed in Table S2 are in good agreement with the sum of the respective ionic radii. [19] Especially, the Bi–O distances do not indicate any influence of the lone pair of the Bi3+ ion. For the sulfate and borate tetrahedra, the deviation from tetrahedral symmetry was calculated by the method of Balić‐Žunić and Makovicky based on all ligands enclosing spheres on experimental data.[ 20 , 21 , 22 ] They can all be classified as regular [23] with deviations of 0.17 and 0.31 % for the sulfate and 0.34 % for the borate tetrahedra. The electrostatic reasonability of the crystal structure of (H3O)Bi[B(SO4)2]4 and all coordination numbers were confirmed by calculations based on the MAPLE concept (see Table S3–S4).[ 24 , 25 , 26 ] Hence, (H3O)Bi[B(SO4)2]4 is the first borosulfate containing two differently charged cations with their coordination environments differing distinctively and only the second borosulfate comprising a 3D anionic substructure. Further, it is the first tectosilicate‐analogous borosulfate synthesised directly without a precursor. Li[B(SO4)2] could only be obtained by thermal decomposition of Li[B(S2O7)2]. [11] We assume that three factors are crucial for the formation of (H3O)Bi[B(SO4)2]4: the high SO3 content—recently shown to be promoting the formation of S−O−S bonds and a higher degree of condensation [6] —, the moderate synthesis temperature favouring a higher degree of condensation,[ 8 , 11 , 16 ] and the presence of the oxonium cations balancing the large charge of the Bi3+ cations—the stabilisation of three‐dimensional borophosphates by H3O+ is known in the literature. [27] To prove the absence of an inversion centre and thus space group I , (H3O)Bi[B(SO4)2]4 was investigated with the powder second‐harmonic generation (SHG) method developed by Kurtz and Perry. [28] This method is commonly used as a first step to estimate the nonlinear optical properties of new materials or to detect the absence of an inversion centre in crystalline structures. [29] Table S5 (in the Supporting Information) shows the intensity of the SHG signal of a sample in comparison to reference compounds (Al2O3, quartz and glass capillary). The ratio of the sample signal and the reference signal provides an estimation of the effective SHG coefficient. The title compound (H3O)Bi[B(SO4)2]4 yielded a SHG intensity approximately twice that of quartz. The strong SHG signal indicates that the sample (H3O)Bi[B(SO4)2]4 clearly crystallises in a non‐centrosymmetric space group confirming the crystal structure determination by single‐crystal XRD. Moreover, (H3O)Bi[B(SO4)2]4 was investigated by FTIR spectroscopy (Figures 2 a, S4) and UV/Vis spectroscopy (Figure S5). In the former, the absorption bands between 1400 and 400 cm−1 indicate the presence of borate and sulfate tetrahedra. [30] Especially the section below 700 cm−1 shows coincidence with the results on R 2[B2(SO4)6]. [8] Here, the O−S−O and O−B−O bending and the B−O−S bridging vibration modes occur. In accordance with earlier results on borosulfates,[ 3 , 6 , 8 ] the bands between 840 and 1040 cm−1 can be assigned to B−O stretching vibrations and the ones above 1100 cm−1 to S−O stretching vibrations. The UV/Vis spectrum shows an absorption edge in the UV region related to the fundamental absorption of the sample overlapping with allowed s–p transitions of Bi3+ and is in line with the colourless powder. According to our thermal analyses, (H3O)Bi[B(SO4)2]4 is stable up to 180 °C before decomposition (Figure 2 b and S6). A chemically plausible decomposition process follows the reaction equations 1, 2, 3, 4, 5].
![a) Infrared spectra and b) thermogravimetric analyses of (H3O)Bi[B(SO4)2]4 and Bi2[B2(SO4)6].](/dataresources/secured/content-1765834313083-d73d51d3-ecbb-440d-ab9e-67c06df6642c/assets/ANIE-60-1503-g002.jpg)
a) Infrared spectra and b) thermogravimetric analyses of (H3O)Bi[B(SO4)2]4 and Bi2[B2(SO4)6].
The expected mass losses for this process agree well with the observed values (see Table S6). The formation of Bi2[B2(SO4)6] and Bi2O(SO4)2 was confirmed by variable‐temperature powder XRD (see Figure S7).
Often, bismuth compounds behave chemically similar to respective rare‐earth ones as both comprise similarly sized [19] trivalent cations and thus also adopt crystal structures found for the latter. [31] For instance, the tungstates Na5 R(WO4)4 crystallise isotypically with Na5Bi(WO4)4. [9] Hence, we were not astonished that we also discovered a pure bismuth borosulfate isotypic to the rare‐earth borosulfates R 2[B2(SO4)6]. [8] The bismuth borosulfate Bi2[B2(SO4)6] was synthesised solvothermally from as‐made Bi2[CO3]O2 (phase‐pure sample according to Figure S8 in the Supporting Information), boric acid, oleum (65 % SO3) and H2SO4 yielding a colourless phase‐pure powder containing single crystals (further details see Supporting Information). The crystal structure was elucidated by single‐crystal XRD and phase purity was confirmed by Rietveld analysis (Figure S9 and Table S7). Like R 2[B2(SO4)6], it adopts space group C2/c (no. 15, Table S7). The trivalent bismuth atoms are coordinated eight‐fold by terminal oxygen atoms with distances ranging between 233.9(2) and 257.3(2) pm in a similar way in Bi2[B2(SO4)6] as in (H3O)Bi[B(SO4)2]4. The resulting square antiprismatic coordination environment is less distorted in the former than in the latter. Again, no influence of the lone‐pair activity of the Bi3+ ion is indicated by the Bi–O distances. However, the method of Balić‐Žunić and Makovicky was used to calculate the distance of the bismuth atom from the centroid of the BiO8 polyhedron.[ 20 , 21 , 22 ] In Bi2[B2(SO4)6], the distance of 16 pm is significantly larger than the one in the isotypic R 2[B2(SO4)6] (see Figure S10) of 7±1 pm indicating an influence of the lone pair of the Bi3+ ion. On the other hand, the distance of 0.016 pm, approximately 0 pm, found for (H3O)Bi[B(SO4)2]4 confirms no influence of the lone pair of the Bi3+ ion, presumably due to the rigidity of the tectosilicate‐analogous anionic substructure surrounding the Bi3+ cation. Further, the weak coordination of the anion—the coordination strength of inorganic acids decreases with increasing condensation—prevents the Bi3+ lone pair from taking shape. The electrostatic reasonability of the crystal structure Bi2[B2(SO4)6] and all coordination numbers were confirmed by calculations based on the MAPLE concept (see Table S3, S4 and S8).[ 24 , 25 , 26 ]
Like the bismuth oxonium borosulfate, Bi2[B2(SO4)6] was investigated by FTIR spectroscopy (Figures 2 a and S4), UV/Vis spectroscopy (Figure S5), and thermogravimetry (Figure 2 b). The IR spectrum is in very good agreement with the spectra reported for the isotypic rare‐earth compounds R 2[B2(SO4)6]. [8] Similar to (H3O)Bi[B(SO4)2]4, the UV/Vis spectrum shows absorption in the UV region due to fundamental absorption and s–p transitions of Bi3+. Thermally, Bi2[B2(SO4)6] decomposes above 370 °C presumably to Bi2O3 and B2O3 accompanied by evaporation of SO3. [8] The experimental mass losses (see Table S9) point at Bi2(SO4)3 and Bi2O(SO4)2 as intermediates and are in agreement with earlier results of the thermal decomposition of the former. [32] The detailed investigation of the decomposition remains the objective of future research.
Crystal structures comprising two different cations frequently inherit specific features of their parent compounds, here meant in terms of oxonium borosulfate and bismuth borosulfate. This behaviour is well‐known as chemical twinning [33] or intergrowth [34] like in the mixed Na2 R[PO4][WO4] [35] where formally fragments of Na2PO4 are twinned with RWO4 fragments. Shifting the consideration on this contribution, the anion of H3O[B(SO4)2] is a chain of edge‐sharing supertetrahedra (1D anion) [13] while Bi2[B2(SO4)6] comprises non‐condensed dimers of edge sharing supertetrahedra (0D anion). In our title compound (H3O)Bi[B(SO4)2]4 the combination of Bi3+ and H3O+ results in a 3D network, obviously very different from both parent compounds—neither twinning nor intergrowth, it is something new. This demonstrates the high structural versatility of silicate‐analogous compounds like borosulfates very well as normally tailored solutions are found for specific situations. However, the environments of the cations in (H3O)Bi[B(SO4)2]4, H3O[B(SO4)2], and Bi2[B2(SO4)6] show similarities—the BiO8 polyhedra are very similar in (H3O)Bi[B(SO4)2]4 and Bi2[B2(SO4)6] and oxonium is coordinated via hydrogen bonds by two and three oxygen atoms in (H3O)Bi[B(SO4)2]4 and H3O[B(SO4)2], respectively. Consequently, the dimensionality of the anion dominates the overall structure of these borosulfates with (H3O)Bi[B(SO4)2]4 representing a new structure type providing two channels of different size for the cations of different size.
In this contribution, we reported the solvothermal syntheses, the crystal structure, as well as the optical and thermal properties of the first two bismuth borosulfates (H3O)Bi[B(SO4)2]4 and Bi2[B2(SO4)6]. The presence of oxonium ions agrees with the highly acidic character of borosulfate ions. Borosulfates can be considered salts of the complex acid H[B(HSO4)4], which was characterised as a super acid. [36] (H3O)Bi[B(SO4)2]4 is the second tectosilicate‐analogous borosulfate so far and the first one synthesised directly without a precursor. This is an obvious leap in borosulfate synthesis. Further, (H3O)Bi[B(SO4)2]4 is the first borosulfate comprising two differently charged cations.
Further work devoted to the question whether the Bi3+–R 3+ analogy can be used both ways are in train: with Bi2[B2(SO4)6] crystallising isotypically to R 2[B2(SO4)6] the unknown oxonium borosulfates (H3O)R[B(SO4)2]4 [37] may be expected to adopt the (H3O)Bi[B(SO4)2]4 structure type. As borosulfates are known to be weakly coordinating ligands, [8] this also merits a study on the luminescence properties of such new rare‐earth compounds since the dimensionality of the borosulfate anion should influence the coordination strength. (H3O)Bi[B(SO4)2]4 and Bi2[B2(SO4)6] or—even better—(H3O)R[B(SO4)2]4 and R 2[B2(SO4)6] would be suitable compounds to test this hypothesis investigating their optical properties.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for financial support under the project HO 4503/5‐1. Open access funding enabled and organized by Projekt DEAL.
References
1
1
3
4
5
7
7a
8
9
10
11
12
12
14
15
17
18
18
19
20
21
22
23
24
24
25
25
26
27
28
29
30
31
32
32a
32b
33
33a
33a
33b
34
36
37
38