 A new linear cyclin docking motif that mediates exclusively S‐phase CDK‐specific signaling
A new linear cyclin docking motif that mediates exclusively S‐phase CDK‐specific signaling
The EMBO Journal



- Altmetric
Cyclin‐dependent kinases (CDKs), the master regulators of cell division, are activated by different cyclins at different cell cycle stages. In addition to being activators of CDKs, cyclins recognize various linear motifs to target CDK activity to specific proteins. We uncovered a cyclin docking motif, NLxxxL, that contributes to phosphorylation‐dependent degradation of the CDK inhibitor Far1 at the G1/S stage in the yeast Saccharomyces cerevisiae. This motif is recognized exclusively by S‐phase CDK (S‐CDK) Clb5/6‐Cdc28 and is considerably more potent than the conventional RxL docking motif. The NLxxxL and RxL motifs were found to overlap in some target proteins, suggesting that cyclin docking motifs can evolve to switch from one to another for fine‐tuning of cell cycle events. Using time‐lapse fluorescence microscopy, we show how different docking connections temporally control phosphorylation‐driven target degradation. This also revealed a differential function of the phosphoadaptor protein Cks1, as Cks1 docking potentiated degron phosphorylation of RxL‐containing but not of NLxxxL‐containing substrates. The NLxxxL motif was found to govern S‐cyclin‐specificity in multiple yeast CDK targets including Fin1, Lif1, and Slx4, suggesting its wider importance.
The short linear motif NLxxxl present in several yeast cyclin‐dependent kinase substrates like Far1 and Slx4 targets them specifically to Clb5‐Cdk1 complexes active in S‐phase.
Introduction
Cyclin‐dependent protein kinases (CDKs) are central regulators executing hundreds of phosphorylation events that trigger, order, check, and finalize the complex process of eukaryotic cell division cycle (Morgan, 2007; Enserink & Kolodner, 2010). CDKs are unique among hundreds of other representatives of the eukaryotic protein kinase superfamily, as they use complex, sequential multi‐docking phosphorylation mechanisms; these involve interactions of a phosphoadaptor subunit Cks1 with primed phospho‐sites, as well as recognition of short linear motifs (SLiMs) in target proteins by cyclin docking pockets (Örd & Loog, 2019).
The initial view of cyclins, in which they function as periodically synthesized and degraded activators of CDK kinase subunits, each acting at their specific cell cycle stage, has been considerably broadened recently. First, it was found that different cyclins modulate the intrinsic active site specificity toward the phosphorylation motifs (Loog & Morgan, 2005; Kõivomägi et al, 2011a; Topacio et al, 2019). Second, for a long time it was known that a subset of CDK targets are tethered to CDK complexes via RxL docking motifs (a SLiM with the consensus sequence R/K‐x‐L‐φ or R/K‐x‐L‐x–φ) that bind to the hydrophobic patch (hp) in human cyclin A or E (Russo et al, 1996; Schulman et al, 1998; Takeda et al, 2001) or in S‐phase cyclin Clb5 in budding yeast (Wilmes et al, 2004; Loog & Morgan, 2005). However, recent studies have uncovered several other cyclin‐specific SLiMs that interact either with the hp or other areas on the cyclins (Bhaduri & Pryciak, 2011; Kõivomägi et al, 2011a; Bhaduri et al, 2015; Topacio et al, 2019; Örd et al, 2019a; Allan et al, 2020; Jackman et al, 2020). These studies widened the scope of the third general function of cyclins, namely in addition to being an activator and activity modulating factor for CDK subunit, cyclins also act as protein scaffolds that present various pocket configurations to dock different SLiMs in target proteins (Tatum & Endicott, 2020). A full set of highly specific and selective motifs have been defined for each of the four major cyclins in budding yeast: the LP, RxL, PxF, and LxF motifs for G1‐, S‐, G2‐, and M‐phase cyclins, respectively (Bhaduri & Pryciak, 2011; Kõivomägi et al, 2011a; Örd et al, 2019a; Örd et al, 2020). Further, recent studies have also revealed that human cyclins utilize a wider variety of docking motifs, as cyclin D and cyclin B1 were found to bind different helical docking motifs (Topacio et al, 2019; Allan et al, 2020; Jackman et al, 2020). Altogether, these findings define cyclins as versatile proteins with a plethora of docking options for SLiMs to achieve specific cell cycle signaling via CDK phosphorylation.
Such a complexity of substrate docking motifs highlights the uniqueness of CDK among protein kinases. Its grand task of temporally ordering hundreds, if not thousands, of individual phosphorylation events at precise activity thresholds during the cell cycle (Stern & Nurse, 1996; Coudreuse & Nurse, 2010; Swaffer et al, 2016) is quite different from the tasks performed by many other kinases and signaling enzymes, whose mode of action can be more simply described as binary switching between active and inactive state. In addition to the differential docking mechanisms provided by cyclins, a key to the combinatorial complexity of CDK substrate phosphorylation is conferred by the phosphoadaptor subunit Cks1 (Kõivomägi et al, 2011b; McGrath et al, 2013), which provides a functional feature to the CDK complex that is unique among protein kinases. The majority of CDK targets contain multiple phosphorylation sites in intrinsically disordered regions (Holt et al, 2009), and the cyclin‐CDK‐Cks1 complex functions as a scaffold with three fixed points of substrate contact (Fig 1A), which allows the process of multisite phosphorylation to proceed in an ordered manner (Kõivomägi et al, 2013; Örd et al, 2019b). The phosphorylated TP motifs bind to the Cks1 phospho‐pocket and facilitate the phosphorylation of secondary sites located C‐terminally from the pTP priming sites (Kõivomägi et al, 2013; McGrath et al, 2013). This mechanism enables three types of substrate sequences—pTP priming sites bound by Cks1, phospho‐acceptor sites recognized by the CDK active site, and diverse docking motifs recognized by cyclin—to be combined in “barcoded” linear patterns along the disordered targets. These patterns can be read by the CDK complexes to achieve a different input–output function of the net phosphorylation rate for a target (Örd et al, 2019b). In other words, the barcoded patterns act as timing tags that assign a target to a specific CDK activity threshold and hence a specific time point during the cell cycle. These findings created a unified model bringing together the CDK threshold model and cyclin specificity model: cyclin specificity and multisite phosphorylation patterns help to encode different execution orders and the CDK thresholds into the target proteins.


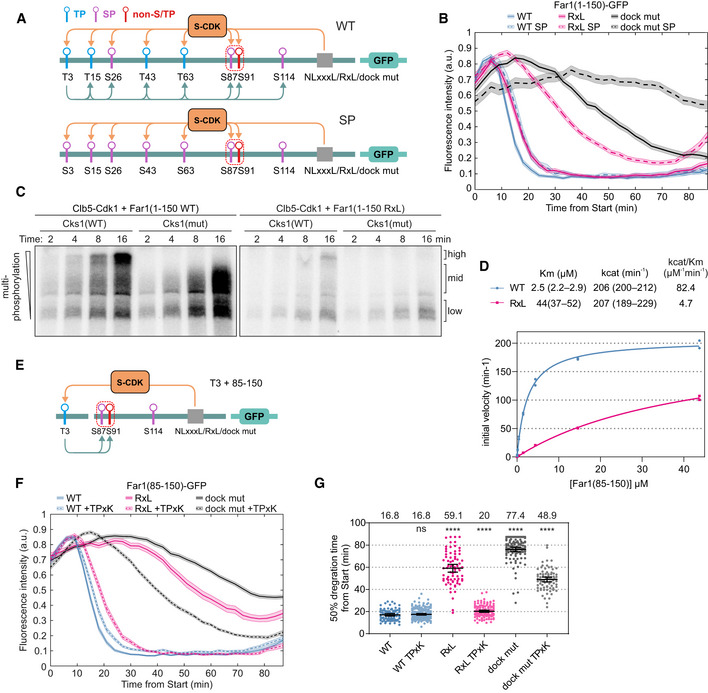
NLxxxL motif promotes S‐CDK‐mediated phosphorylation and degradation of N‐Far1
AScheme showing the major cyclin‐substrate interactions of Cdk1 complex.
BScheme showing the CDK phosphorylation sites and the potential Clb5 docking region in the disordered N terminus of Far1 (residues 1–150).
CCo‐immunoprecipitation of Cdc4 using either unphosphorylated or Clb5‐Cdk1‐phosphorylated Far1(85–150) as bait. The experiment was repeated twice, a representative example is shown.
DTime‐lapse fluorescence images showing the expression and degradation of wild‐type and S87A S91A mutant Far1(1–150)‐GFP and nuclear‐cytoplasmic shuttling of Whi5‐mCherry. Nuclear export of Whi5‐mCherry takes place at 0 min (Start point) and was used to synchronize the cells for quantitative analysis.
E, FMean ± SEM fluorescence intensities of the indicated Far1(1–150)‐GFP mutants over the cell cycle. The number of cells analyzed for each construct is noted in Table EV2.
GTime from Start to degradation of 50% of the indicated Far1‐GFP sensor in single cells. The bars show median ± 95% confidence intervals. The numbers above the plot show median values. ****P‐value < 0.0001, ***<0.001 and ns > 0.05 for pairwise comparisons with wild type using Mann–Whitney U‐test. The number of cells analyzed for each construct is noted in Table EV2.
H In vitro kinase assay showing the effect of N130A, L131A, L135A, and L136A mutations on Far1(1–150)‐GFP phosphorylation by Clb5‐Cdk1. WT/mut shows the decrease in phosphorylation rate of the mutant compared to wild type. The experiment was repeated twice, a representative example is shown.
Source data are available online for this figure.
Despite these recent advances, it has remained unclear how the two major elements of multisite substrate docking, the Cks1‐binding priming phosphorylation sites, and the cyclin‐binding SLiM (Fig 1A), are cooperating with or compensating for each other’s effects to define the CDK thresholds and timings of the cell cycle events. It is also not understood what is the possible sequence diversity, functional interrelationships and the balance between specificity and affinity of different SLiMs binding the hp of B‐type cyclins.
Here we report a Saccharomyces cerevisiae SLiM docking motif, with a consensus sequence NLxxxL, that promotes phosphorylation exclusively by S‐CDK (Clb5 cyclin bound to the yeast CDK Cdc28). This motif was found in several S‐phase CDK targets and, intriguingly, in multiple cases it overlapped with conventional RxL motifs. The NLxxxL‐mediated Clb5 docking enabled fast phosphorylation at the G1/S transition as well as early dephosphorylation in anaphase. The NLxxxL motif, when part of a CDK substrate, stands out among the known cyclin docking motifs with its considerably lower KM value. Finally, we also discovered an unexpected difference in the Cks1‐dependent phosphorylation behavior depending on whether the substrate utilized a weaker RxL motif versus a more efficient NLxxxL motif.
Results
NLxxxL motif promotes S‐CDK‐mediated phosphorylation of a di‐phosphodegron in Far1
An S‐CDK‐specific putative linear docking motif was initially mapped via truncations of the N‐terminal disordered region of Far1 within the segment of 130–140 of Far1 (Valk & Loog, manuscript in preparation). In the current study, the motif was further mapped using site‐directed mutagenesis and quantitative time‐lapse fluorescence microscopy. For this, we employed an approach using CDK threshold sensors, a set of variable GFP‐tagged substrate constructs that are degraded in response to phosphorylation at defined levels of CDK activity (Örd et al, 2019b). We based the sensors on a fragment comprised of the first 150 N‐terminal amino acids from Far1 (Far1(1–150)) (Fig 1B). This fragment contained Cdk1 phosphorylation site S87, which is necessary for cell cycle‐dependent degradation of Far1 (Gartner et al, 1998). Far1 is ubiquitinated by SCFCdc4 (Blondel et al, 2000), and S87–S91 matches the consensus for a Cdc4 di‐phosphodegron (Hao et al, 2007; Fig EV1A). To confirm that the Clb5‐Cdk1‐mediated phosphorylation of S87 and S91 is necessary for Far1‐Cdc4 interaction, we performed Cdc4 pull‐down experiments with truncated Far1(85–150). These experiments showed that Clb5‐dependent phosphorylation of Far1(85–150) and the presence of both S87 and S91 was necessary for interaction with Cdc4 (Fig 1C).
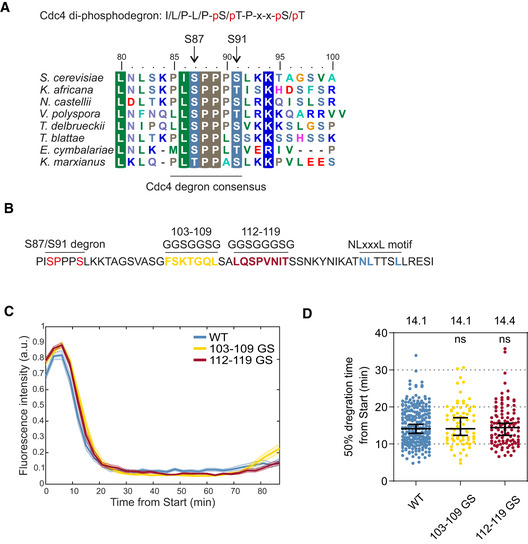
Mapping of the S87/S91 di‐phosphodegron and the linker region
Alignment of S. cerevisiae Far1(80–100) with homologs from other yeast species. Far1 segment 85–91 matches the consensus for Cdc4 di‐phosphodegron.
Sequence of Far1 positions 85–140 showing the linker sequence between the S87/S91 degron and the NLxxxL motif. To study the linker region, positions 103–109 and 112–119 were replaced with glycine‐serine linkers in two constructs that were analyzed in time‐lapse microscopy in panel “C”.
Mean ± SEM fluorescence intensities of the indicated Far1(1–150)‐GFP constructs after Start. For sample size, please see Table EV2.
Plot showing the 50% degradation timing of indicated Far1(1–150)‐GFP mutants in individual cells. The bars show median ± 95% CI. The numbers above the plot show median values for each construct. ns denotes P‐value > 0.05 for pairwise comparisons with wild type using Mann–Whitney U‐test. The number of cells analyzed is shown in Table EV2.
Source data are available online for this figure.
To estimate the effects of systematic alanine mutations within the docking motif on CDK‐dependent phosphorylation of the degron, we measured the dynamics of sensor degradation. Ubiquitination‐driven degradation of sensors with di‐phospho‐degrons is mediated by E3 ligase SCF‐Cdc4 and proteasome, that are constitutively active throughout the cell cycle (Zhou & Howley, 1998), allowing to measure the dynamics of sensor phosphorylation alone. Using a previously described live‐cell fluorescent microscopy protocol (Örd et al, 2019b), we followed the timing of phosphorylation‐dependent degradation of GFP‐tagged sensors (Fig 1D and E). For time point zero, we used the cell cycle Start, defined as the nuclear export of 50% of Whi5‐mCherry, the transcriptional inhibitor of early cell cycle genes (Doncic et al, 2011). We found that a construct based on the wild type Far1(1–150) was degraded rapidly, declining to 50% of its maximal levels by ~ 14 min from Start (Fig 1E). This is in good agreement with our previous observations of the timing of G1/S transition, marked by Sic1 degradation and accumulation of free Clb5‐Cdk1 complex (Venta et al, 2020). In contrast, a double‐alanine mutation of the di‐phosphodegron stabilized the sensor for the length of the cell cycle, which for budding yeast in rich media is about 90 min (Fig 1D and E).
We introduced alanine mutations within the identified docking region and tested a set of them in the context of the sensors (Fig 1F and G). Single mutants L131A and L135A showed a considerable delay in sensor degradation, the mutation N130A caused an intermediate effect with a ~ 22‐min half‐time, while L136A mutation, and a triple mutation of the motif TTS in the middle of the segment behaved like a wild type. We also introduced simultaneous alanine mutants into four amino acid segments flanking the 10 amino acid region initially mapped by truncations. The 4xAla mutations of the N‐terminal flanking sequence IKAT and the C‐terminal RESI individually did not cause a delay in degradation, and however, a minor 3‐min delay was detected with simultaneous mutation of both flanking regions (Fig 1F and G).
As the sequence of the segment did not bear any obvious resemblance to the conventional RxLxF motif, we wondered if a region between the docking motif and the degron phosphorylation sites would provide crucial additional contacts with the CDK complex. To test this, we replaced two segments in this linker region with Gly‐Ser stretches (Fig EV1B). However, no changes in degradation dynamics were observed (Fig EV1C and D). These results indicate that phosphorylation of the degron is controlled by the NLxxxL motif, and this motif fulfills the general criteria of a SLiM, defined as a recognition sequence of up to ten amino acids within a disordered segment (Davey et al, 2012).
To compare the in vivo data with the biochemical measurements of CDK specificity in vitro, we performed assays to determine the initial velocity of phosphorylation using purified CDK complexes and the sensor proteins, by 32P autoradiography of SDS–PAGE. The phosphorylation by Clb5‐Cdk1 was disrupted by mutations at the three key positions (N130A, L131A, L135A) and to a degree that correlated well with their degradation orders observed in vivo (Fig 1H). This indicates that NLxxxL is a docking motif for Clb5‐Cdk1, which is necessary for timely phosphorylation of the Far1 N terminus.
NLxxxL motif is specific for S‐phase cyclins Clb5 and Clb6
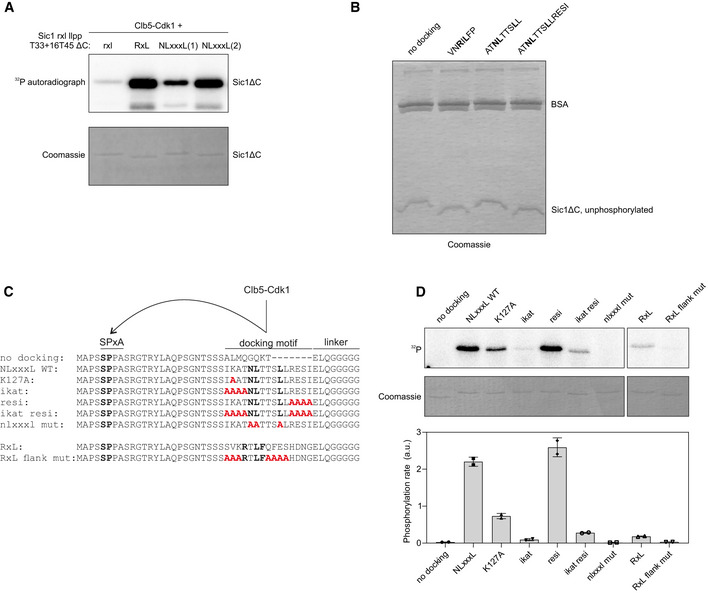
To analyze the cyclin specificity of the identified docking motif, we performed in vitro phosphorylation assays with eight cyclin‐Cdk1 complexes (Figs 2A and EV2A–C). For the four major complexes, we also tested their corresponding cyclin docking pocket mutants (Fig 2A). As controls, we used: (i) histone H1, a substrate that lacks cyclin docking motifs and relies only on a consensus phosphorylation site; and (ii) a non‐inhibitory truncated version of Cdk1 inhibitor Sic1 (Sic1ΔC), containing a conventional cyclin docking motif, RxL. The results show that the discovered docking motif was specific for both S‐phase complexes, Clb5‐ and Clb6‐Cdk1. The other closely related G2‐ and M‐CDK complexes showed very weak phosphorylation rate. The G1‐CDK complexes Cln1‐ and Cln2‐Cdk1 also showed considerable phosphorylation specificity, and however, this was neither dependent on the discovered motif in Far1, nor on the known hydrophobic substrate‐binding pocket on Cln2 (Bhaduri et al, 2015).
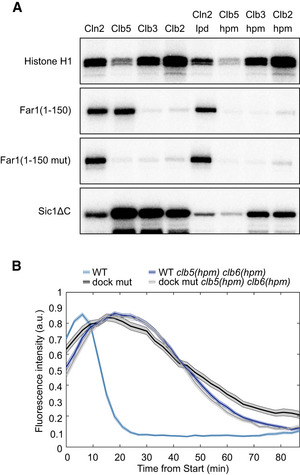
NLxxxL is a specific SLiM for docking by S‐phase Clb5/6‐Cdk1
Autoradiograph of a cyclin specificity analysis of the indicated substrates with wild‐type or docking site mutant cyclin‐Cdk1 complexes. Far1(1–150 mut) is L131A L135A L136A mutant, Sic1ΔC is the non‐inhibitory mutant of Sic1 (deletion of 216–284). The experiment was repeated twice, a representative example is shown.
Mean ± SEM fluorescence intensities of wild‐type and NLxxxL motif mutant Far1(1–150)‐GFP in either wild‐type or clb5(hpm) clb6(hpm) strain over the cell cycle. The number of cells analyzed is noted in Table EV2.
Source data are available online for this figure.
The Clb5 specificity and Clb5 interaction with the Far1 N terminus were dependent on the conserved hp docking pocket in the cyclin and the NLxxxL motif in Far1 (Figs 2A and EV2D). Also, in vitro phosphorylation experiments in the presence of competitor peptide with NLxxxL motif inhibited the rate of Clb5 phosphorylation of both wild‐type Far1(1–150 WT) and Far1(1–150 RxL), where NLxxxL motif is replaced with an RxL motif, further indicating that the NLxxxL motif binds to the hp (Fig EV2E and F, for Far1(1–150 RxL) see also Fig 3 later). Finally, we observed that NLxxxL motif is required for Clb5‐dependent phosphorylation of both Cdc4 di‐phosphodegron sites S87 and S91 (Fig EV2G).

NLxxxL motif is more effective than RxL motif in directing Clb5‐Cdk1 activity
Schemes of Far1(1–150)‐GFP constructs, either wild‐type or mutant where NLxxxL motif is replaced with RxL motif (VKRTLF), that are used in time‐lapse microscopy experiments in panel (C).
Sequence alignment showing the introduction of wild‐type or mutated RxL motif to Far1.
Plot showing the mean ± SEM fluorescence intensities of Far1(1–150)‐GFP constructs with the indicated docking site. In rxl mutant, the NLxxxL motif is replaced with a mutated non‐functional RxL motif (VKATAF). The number of cells analyzed is noted in Table EV2.
Kinetic characterization of Far1(1–150 WT)‐GFP and Far1(1–150 RxL)‐GFP phosphorylation by Clb5‐Cdk1. The data are from two experimental replicates.
Mean ± SEM fluorescence intensities of wild‐type or RxL‐dependent Far1(1–150)‐GFP in wild‐type or the indicated cyclin hpm mutant strains. The number of cells analyzed is noted in Table EV2.
Pheromone sensitivity halo assay with PGAL1‐3HA‐CLB5 sic1Δ strains carrying either wild‐type FAR1, far1Δ, far1(L135A), or far1(RxL). Different concentrations of α‐factor were pipetted on the paper disks. On glucose plates, α‐factor triggers cell cycle arrest. CLB5 overexpression from PGAL1 in sic1Δ strain causes lethality presumably by inhibition of replication licensing, which is rescued by the presence of α‐factor, which leads to Far1‐dependent inhibition of excess Clb5‐Cdk1 activity. The experiment was repeated twice, a representative example is shown.
Source data are available online for this figure.
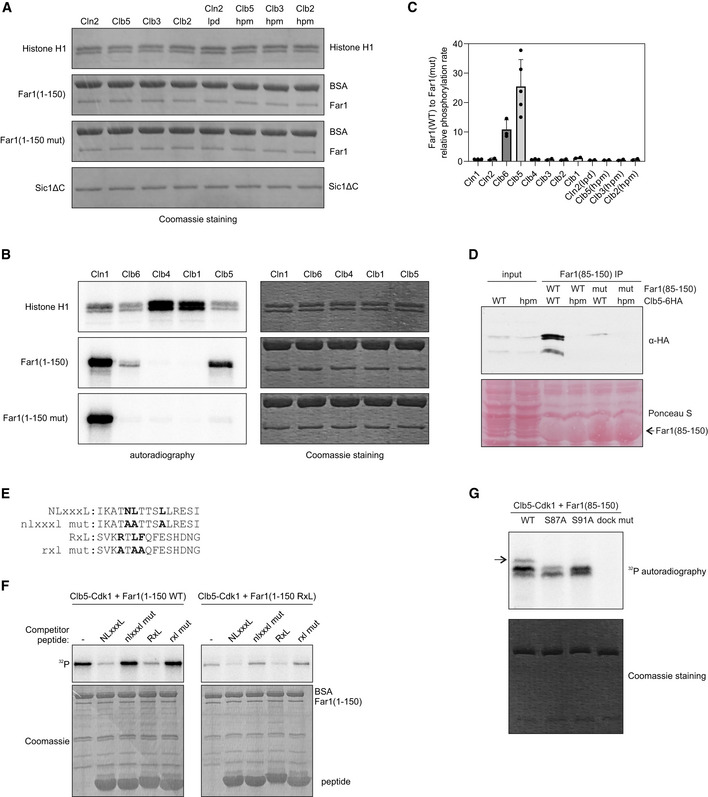
Cyclin hp‐dependency and S‐CDK specificity of the NLxxxL motif
Coomassie‐stained SDS–PAGE gels from kinase assays presented in Fig 2A.
Phosphorylation of histone H1, Far1(1–150), and Far1(1–150 mut) by Cln1‐, Clb6‐, Clb4‐, Clb1‐, and Clb5‐Cdk1 in vitro. In Far1(1–150 mut), the NLxxxL motif is mutated.
The relative phosphorylation rate of wild‐type Far1(1–150) compared to NLxxxL motif mutant Far1(1–150) by the indicated cyclin‐Cdk1 complex. The data are mean from at least two experiments, and the error bars show standard deviation. Individual experiment data points are also shown.
Co‐immunoprecipitation of either wild‐type or hpm Clb5 using either wild‐type or NLxxxL motif mutant Far1(85–150) as bait. The experiment was repeated twice, a representative example is shown.
Alignment of the competitor peptides used in Fig EV2F. The peptides were fused via ELGGGGG linker to GB1 domain.
In vitro phosphorylation experiment showing the phosphorylation of Far1(1–150) with either NLxxxL (WT) or RxL motif (RxL) by Clb5‐Cdk1 in the presence of 210 µM competitor peptides with the indicated motifs. The experiment was repeated twice, a representative example is shown. Sequences of the competitor peptides are shown in Fig EV2E.
The indicated Far1(85–150) variant was phosphorylated with Clb5‐Cdk1, and the multisite phosphorylation was studied using Phos‐tag SDS–PAGE autoradiography. The arrow points to the product with phosphorylation at both S87 and S91.
Source data are available online for this figure.
To confirm that the effects of NLxxxL motif mutation observed in microscopy (Fig 1F) were indeed due to the hp‐mediated docking of the substrate to Clb5 and Clb6, and not due to changes in counteracting phosphatase specificity in the case of the mutated NLxxxL motif, we performed a microscopy experiment with strains where S‐phase cyclins were replaced with versions containing mutated docking pockets, the Clb5(hpm) and Clb6(hpm) (Fig 2B). As the degradation curves for wild‐type and NLxxxL mutant sensor in clb5(hpm) clb6(hpm) strain were nearly identical, we can conclude that phosphatase specificity toward NLxxxL mutant was not changed. This also provides in vivo evidence for the Clb5/6‐specificity of the NLxxxL motif.
We have previously shown that conventional RxLxF cyclin docking motifs usually enhance the specificity of targets via binding into the cyclin hp pockets of different cyclins, showing a trend of compensation for the gradually weakening specificity toward the histone H1‐based consensus phosphorylation peptide (k cat/K M: Clb2>Clb3>Clb5), such that the docking effect is strong for Clb5, intermediate for Clb3, and mild for Clb2 (Kõivomägi et al, 2011a). More recently, we have shown that the hp pockets of different Clbs recognize distinct non‐RxL motifs, such as LxF for Clb2 and PxxPxF for Clb3 (Örd et al, 2020). In this respect, the S‐CDK‐specific motif described here falls into the latter category, being specific for only a particular cyclin despite using the hydrophobic docking pocket conserved in multiple Clb cyclins.
Substitution of the NLxxxL motif with an RxL results in partial loss of function
Having found that NLxxxL is an hp‐binding motif like RxL, we asked if the two motifs have any functional differences other than the S‐CDK specificity of NLxxxL. For this, we replaced the NLxxxL motif in the Far1(1‐150)‐GFP sensor with an RxL motif from Sic1 (Fig 3A and B, and Appendix Fig S1A). This substitution caused a minor 3‐min delay in degradation, whereas a sensor with a mutated RxL motif showed a 30‐min delay (Fig 3C, Appendix Fig S1B), comparable to mutations in the NLxxxL determinants (Fig 1G). To directly analyze the phosphorylation kinetics of Far1(1–150) with the different docking motifs, we performed Michaelis–Menten steady‐state kinetic analysis with Clb5‐Cdk1. The K M value for Far1(1–150) with the NLxxxL motif was around 1.5 µM, and substitution of NLxxxL with RxL increased K M to 10 µM (Fig 3D). This results in around 13‐fold higher specificity (ratio of k cat/K M values) of the substrate with NLxxxL compared to the one with RxL. Interestingly, however, this large difference in specificity manifests only in a minor difference in degradation timing (Fig 3C). This result implies that early accumulation of CDK activity around G1/S has very high fold change, as the activity of Clb5‐Cdk1 seems to increase by roughly 10‐fold in the 3‐min period around G1/S. This suggests that extensive changes in specificity are needed to precisely order phosphorylation events at the onset of S‐phase due to the drastic increase in Cdk1 activity mediated by the activation of S‐Cdk1 (Örd et al, 2019b). Further, mutation of Clb5 hp caused a considerable delay in Far1(1–150 RxL)‐GFP degradation (Fig 3E, Appendix Fig S1C). Far1(1‐150 WT)‐GFP with NLxxxL motif, however, was not delayed by clb5(hpm) mutation, indicating that specific docking of Clb6‐Cdk1 fulfilled the minimal CDK threshold for phosphorylation of the Far1(1–150 WT) (Fig 3E, Appendix Fig S1C).
To further analyze the functional differences of the two motifs, we tested the importance of the docking on the double‐negative feedback loop between Far1 and Clb5‐Cdk1, by using a halo assay for pheromone sensitivity combined with overexpression of CLB5. Overexpression of CLB5 causes lethality in sic1Δ background (Jacobson et al, 2000), presumably due to inhibition of replication origin licensing by Clb5‐Cdk1 activity (Lengronne & Schwob, 2002). Activation of Far1 inhibitory activity toward CDK by pheromone, however, rescues the effect of CLB5 overexpression in sic1Δ cells, presumably by inhibition of the excess Clb5‐Cdk1 activity, thus enabling the cells to grow only in the presence of pheromone (Fig 3F). Disruption of the NLxxxL docking by mutation of N130A, L131A, or L135A decreases the ability of the CLB5 overexpressing cells to grow in the presence of pheromone, as the cells barely grow only in a specific concentration range of diffusing pheromone (Fig 3F, Appendix Fig S1D). This could be because of the inability of Clb5‐Cdk1 to degrade Far1 in these cells. Furthermore, substitution of the NLxxxL motif with RxL motif results in a similar phenotype as L135A mutation, showing that RxL cannot substitute for NLxxxL motif in this case.
The contribution of Cks1 to the degron phosphorylation depends on the cyclin docking motif
In our previous studies of multisite phosphorylation of a number of key Cdk1 targets (Sic1, Cdc6, Swe1, Ndd1, etc.), we have described a general mechanism of Cks1‐mediated N‐to‐C‐terminally directed sequential phosphorylation that can have varying degrees of processivity (Kõivomägi et al, 2011b, 2013; McGrath et al, 2013; Örd et al, 2019a). To study how Cks1 docking affected phosphorylation of Far1(1–150), we mutated the TP sites to SP (Fig 4A), as only phospho‐threonine bind to Cks1 (Kõivomägi et al, 2013; McGrath et al, 2013) and analyzed degradation timing using microscopy. Disconnection of Cks1 docking by the T‐to‐S mutations caused a ~5‐min delay in sensor degradation (Fig 4B, Appendix Fig S2A), indicating that Cks1 promotes the degron phosphorylation. Interestingly, the delay in degradation caused by mutation of Cks1 binding sites was much greater in Far1(1–150) constructs in which the NLxxxL motif was replaced with an RxL motif or which had no cyclin docking motif (Fig 4B, Appendix Fig S2A). This indicates that the Cks1‐dependence of the substrate phosphorylation also depends on the efficiency of cyclin docking motif.
The Cks1 dependency is affected by the affinity of the cyclin docking motif
Schemes of the Far1(1–150)‐GFP constructs used in (B). The constructs contain either wild‐type phosphorylation sites or have all TP sites mutated to SP sites (SP mutant). The predicted Cks1 docking connections are shown with the arrows below the scheme.
The degradation of Far1(1–150)‐GFP with either wild‐type or all serine‐based phosphorylation sites and the indicated docking motifs was measured in time‐lapse microscopy. Plot shows mean ± SEM of GFP fluorescence intensities from Start. The number of cells is shown in Table EV2.
Phos‐tag SDS–PAGE autoradiographs showing the Cks1 dependency of Clb5‐Cdk1‐mediated multisite phosphorylation of Far1(1–150) with either NLxxxL (WT) motif or RxL motif. The experiment was performed twice, a representative example is shown.
Plot showing the kinetics of Clb5‐Cdk1‐mediated phosphorylation of Far1(85–150) with either wild‐type (NLxxxL) or RxL docking motif. The data are from two replicates.
Scheme of Far1(85–150)‐GFP with either NLxxxL, RxL, or mutated cyclin docking motif and with or without T3 primer site introduced upstream of the S87 S91 degron.
Mean ± SEM GFP fluorescence intensities of the indicated Far1(1–85) constructs, synchronized at the time of Start. The number of cells is noted in Table EV2.
Plot showing the timing of 50% degradation of the indicated Far1(85‐150) constructs in single cells. The numbers above the plot show median values. The bars show median ± 95% CI. **** and ns indicate P‐values < 0.0001 and > 0.05 by Mann–Whitney test for pairwise comparisons with WT. The number of cells is noted in Table EV2.
Source data are available online for this figure.
Elimination of both cyclin and Cks1 docking led to the stabilization of the sensor to a similar extent as mutation of the degron (Figs 1E and 4B), showing that the docking mechanisms are essential for phosphorylation of the degron sites. This supports a previously suggested mechanism by which helper networks of Cks1 binding sites and cyclin docking motifs can enhance phosphorylation of output degron sites to assign the timing of the phosphorylation switch to a specific point in the cell cycle (Örd et al, 2019b).
Analysis of the phosphorylation reactions using Phos‐tag SDS–PAGE revealed that Far1(1–150) can be efficiently multi‐phosphorylated by Clb5‐Cdk1 and that the appearance of highly phosphorylated forms is dependent on Cks1 for both NLxxxL and RxL substrates (Fig 4C, Appendix Fig S2B and C). Although the substitution of NLxxxL motif with RxL results in a significant decrease in total phosphorylation rate, it has a lesser impact on the phosphorylation pattern compared to mutation of the Cks1‐binding sites (Fig 4C, Appendix Fig S2B and C). Here again, despite a relatively large difference in overall phosphorylation signal (Fig 4C), the high fold change of CDK accumulation during the early stages (10–20 min after Start) allows us to see only a ~ 2–3 min difference in degradation timing between NLxxxL and RxL versions. In the case of Far1(1–150 RxL SP), both higher affinity cyclin docking and Cks1‐mediated connections are lost, and this results in decreased accumulation of multi‐phosphorylated forms and greatly delayed sensor degradation (Fig 4B and C, Appendix Fig S2A–C).
To better understand the role of Cks1‐dependent route in context of the two very different docking motifs, we created a Cks1‐independent minimal system by deleting the region N‐terminal of the degron (positions 1–84). Kinetic analysis of Far1(85–150), a protein lacking 5 N‐terminal phosphorylation sites, and therefore also the Cks1 binding sites, revealed a slight increase in K M compared to Far1(1–150) with a NLxxxL motif, and a stronger increase for the RxL motif (Figs 3D and 4D). However, strikingly, the k cat values were found to be considerably higher compared to the 1–150 version and, importantly, were now almost equal for the NLxxxL and RxL motifs. This suggests that the Cks1‐dependent N‐terminal route may have dual function. While acting as a phospho‐priming guided sequential path that promotes processivity and degron phosphorylation, it also could act as a diversional path that, due to a larger number of steps, slowing down the net k cat of the process.
Finally, we tested how introduction of a single optimal Cks1 priming site (based on Far1 T3) affects the degradation dynamics of substrates with different cyclin docking motifs (Fig 4E). The deletion of positions 1‐85 had only a minor effect in case of NLxxxL docking motif, and addition of T3 priming site did not improve the degradation (Fig 4F and G). For reporters with the RxL motif or lacking any docking motif at all, the major delay in degradation was rescued by introduction of just this single Cks1 priming site (Fig 4F and G). These results show that via NLxxxL docking, Clb5‐Cdk1 is capable of efficiently phosphorylating the degron consisting of a minimal consensus (S87) and a non‐proline site (S91), whereas with RxL docking the required thresholds are not reached. Interestingly, this difference between the motifs could make some S‐phase targets dependent on priming by other kinases, such as G1‐CDK.
Modularity of the Far1 NLxxxL motif
In order to test if the identified linear docking motif is modular, and transferrable between CDK targets without losing its function, we replaced an RxL motif with a 9 amino acid segment (ATNLTTSLL) containing the motif from Far1 into a modified version of Sic1ΔC (Fig 5A and B), whose timely phosphorylation is dependent on cyclin docking (Örd et al, 2019b). Unexpectedly, degradation of this Sic1ΔC‐GFP‐based construct was very inefficient, reaching its half maximum only slightly earlier than the control sensor with no docking site at all, at ~ 55–60 min (Fig 5C and D). The construct with intact RxL motif showed degradation half‐life much earlier, at around ~ 30 min. However, when we introduced a longer version of the NLxxxL motif that also included the 4 amino acids from the C‐terminal flanking segment (ATNLTTSLLRESI) (Fig 5B), a striking effect on degradation was observed, shifting the half‐time to ~ 23 min after the Start. Interestingly, the C‐terminal flanking residues (RESI) from the core motif NLxxxL did not affect the degradation of the sensor in Far1 context, while it was required for fast degradation within the Sic1‐based construct. This latter difference was corroborated by in vitro phosphorylation assays (Fig EV3A). We speculate that although the modular replacement of the docking motif from one substrate into the other was successful, there are apparently other weaker specificity elements in the local sequence context that may become more important if the module is replaced into a foreign sequence context.

NLxxxL motif can substitute RxL in targeting Clb5‐Cdk1 activity for degradation of Sic1
Schemes of the CDK threshold sensors based on Sic1 used in panels (C, D). The sensors contain the non‐inhibitory domain of Sic1 (1–215), have mutated RxL and LP docking sites and insertion of 4 residues between T33 and T45. The docking sites are inserted to the position of Sic1 RxL motif.
Alignment of the Sic1 segment with the different introduced docking motifs.
Plot showing the mean ± SEM fluorescence intensities of the Sic1‐based threshold sensors shown in (A) containing the indicated cyclin docking motif. The sample size for each strain is noted in Table EV2.
The time from Start to the fluorescence levels of the threshold sensors with indicated docking motifs decreasing to 50% of peak level was measured in single cells. The black lines show median ± 95% CI. The numbers above the plot show median degradation timings of the indicated sensors. The number of cells is noted in Table EV2.
In vitro phosphorylation kinetics of Sic1(T33 + 16T45 rxl llpp ΔC) with the indicated cyclin docking motif by Clb5‐Cdk1. The data are from two replicate experiments.
Analysis of multisite phosphorylation of Sic1(T33 + 16T45 rxl llpp ΔC) with different docking motifs using Phos‐tag SDS–PAGE autoradiography. The experiment was performed twice, a representative example is shown.
The fraction of maximally phosphorylated form from total phosphorylation signal was quantified from Phos‐tag gels as shown in panel (F).
Source data are available online for this figure.
NLxxxL is a linear and modular docking motif
Autoradiograph and Coomassie‐stained gel of phosphorylation reactions of the indicated Sic1ΔC variant and Clb5‐Cdk1.
Coomassie‐stained Phos‐tag SDS–PAGE gel of the phosphorylation experiment in Fig 5F.
Sic1‐based minimal model substrates used to study the specificity determinants of NLxxxL and RxL motifs. Fifteen amino acid segments with the docking motifs were introduced 20 amino acids downstream of a minimal consensus site SP. The motifs are followed by a linker and fused to GB1 domain.
In vitro phosphorylation assay with Clb5‐Cdk1 using the Sic1‐based minimal model substrates described in panel “C”. The data are from two independent experiments. The error bars show standard deviation of the mean from two experiments.
Source data are available online for this figure.
The Sic1‐based sensor with the extended NLxxxL module was degraded even more rapidly than the construct with a native RxL motif from Sic1 (Fig 5C and D). However, in an in vitro phosphorylation assay we observed similar kinetics for the substrates with either docking motifs (Fig 5E). On the other hand, a high‐resolution Phos‐tag SDS–PAGE assay providing the quantitative profiling of multi‐phosphorylated species showed that the total signal of the most highly phosphorylated form was higher in case of the extended NLxxxL substrate (Figs 5F and EV3B). Furthermore, the relative quantitative pattern of multiply phosphorylated species was more shifted toward higher bands in case of NLxxxL compared to RxL (Fig 5G). This result explains the faster degradation timing of the extended NLxxxL substrate, as efficient multisite phosphorylation of Sic1 is essential for Sic1 degradation (Kõivomägi et al, 2011b).
To better understand the impact of local context and flanking residues, we constructed minimal model substrate based on the N terminus of Sic1, where NLxxxL and RxL motifs were introduced into a foreign context for both motifs (Fig EV3C). As in Far1‐based constructs, the NLxxxL motif promoted phosphorylation of a minimal consensus site much more efficiently than the RxL motif (Fig EV3D). In this particular context, mutation of the IKAT sequence N‐terminal of NLxxxL decreased the phosphorylation rate, with the lysine in −3 position having a moderate effect (Fig EV3D). Mutation of the C‐terminal flanking residues (RESI), however, had no effect. Analogously, in case of RxL motifs, the residues N‐terminal of the core motif have been found to interact with cyclin (Lowe et al, 2002). Also, we observed that the RxL motif is dependent on its flanking residues for specificity (Fig EV3D). This indicates that both NLxxxL and RxL have a more defined core motif, but also require favorable flanking residues, which can be more variable. In the Far1 context, the IKAT and RESI sequences had only a minor effect presumably due to a wider optimal context.
Homology of the NLxxxL and RxL motifs
We performed a search for potential NLxxxL motifs from the intrinsically disordered proteins in yeast and found that the consensus sequence is present in 300 proteins, 50 of which have been previously found to be Cdk1 targets (Table EV1). Based on this motif prediction, we found that Lif1, a DNA ligase IV complex component functioning in DNA repair that is phosphorylated in S‐phase (Matsuzaki et al, 2012), and Slx4, an endonuclease processing recombination intermediates (Cussiol et al, 2017), are specific targets of Clb5‐Cdk1 (Figs 6A and B, and EV4A). Furthermore, their phosphorylation was dependent on a functional hp in Clb5 and an NLxxxL motif in each substrate (Fig 6B and C). This shows that NLxxxL also controls the phosphorylation of other Cdk1 targets.
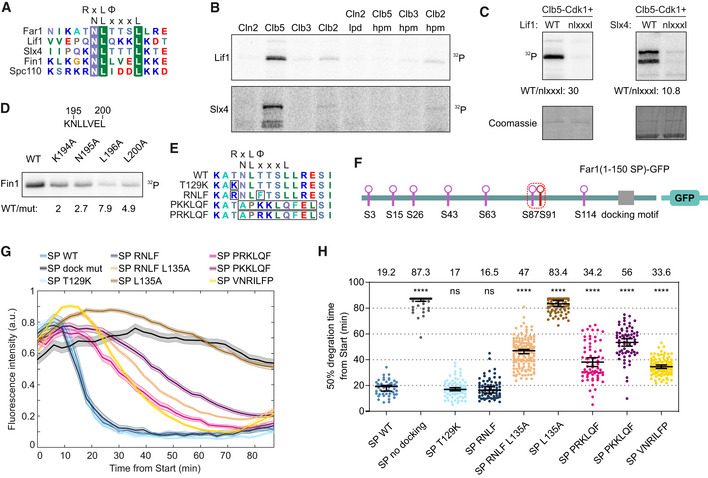
NLxxxL motif is present in other S‐CDK targets and is homologous with RxL motif
An alignment of predicted NLxxxL motifs from Far1, Lif1, Slx4, Fin1, and Spc110. In case of Fin1 and Spc110, the motif overlaps with an RxL motif.
Autoradiograph showing the cyclin specificity and docking dependency of Lif1 and Slx4 phosphorylation. The experiments were repeated twice, representative examples are shown.
NLxxxL is necessary for efficient phosphorylation of Lif1 and Slx4 by Clb5‐Cdk1, as shown by the in vitro kinase assay. The experiments were repeated twice, representative examples are shown.
Autoradiograph of Clb5‐Cdk1‐mediated phosphorylation of indicated Fin1 mutants. The experiments were repeated twice, representative examples are shown.
Alignment showing the introduction of RxL motif determinants to Far1 NLxxxL motif in mutants analyzed in time‐lapse microscopy in panels (G, H).
Scheme of Far1(1–150 SP)‐GFP sensors used for time‐lapse microscopy‐based analysis of different docking motif variants. All TP sites have been mutated to SP sites.
The effect of different docking motifs on the degradation of Far1(1–150 SP)‐GFP, where all TP sites are mutated to SP. Plot shows mean ± SEM of fluorescence intensities in the cell cycle. For the sample sizes, please see Table EV2.
The time from Start to the degradation of 50% of Far1(1–150 SP)‐GFP with the indicated docking motif was measured in single cells. The numbers above the plot show median values for the sensors, and the black lines show median ± 95% CI. **** and ns denote P‐value < 0.0001 and > 0.05, respectively, for pairwise comparisons with SP WT using Mann–Whitney U‐test. For the sample sizes, please see Table EV2.
Source data are available online for this figure.

Conservation of NLxxxL motifs in Fin1, Far1, and Lif1 and mapping of the Clb5 docking interface
ACoomassie‐stained gels for phosphorylation assay presented in Fig 6B.
B–DAlignment of Fin1 (B), Far1 (C), and Lif1 (D) homologs in yeasts showing the conservation and overlap of cyclin docking motifs. The match to either RxL or NLxxxL motif consensus is presented on the right. The RxL consensus was defined as R/K‐x‐L‐Φ or R/K‐x‐L‐x‐Φ.
ECoomassie‐stained gels of phosphorylation assays presented in Fig 6D.
FComputational structural model of Clb5 with RxL peptide bound to the hydrophobic patch. The RxL interaction is based on alignment of Clb5 model with crystal structure of cyclin A in complex with Cdc6 RxL peptide (2CCI).
GPhosphorylation of Far1(1–150), Lif1, and Sic1ΔC by different Clb5‐Cdk1 mutants was measured under initial velocity conditions. The plots show the mean of phosphorylation rate by the indicated Clb5 mutant compared to wild‐type Clb5‐Cdk1. The data are from two independent experiments, and the error bars show standard deviation.
Source data are available online for this figure.
Surprisingly, we found that predicted NLxxxL motifs overlap with previously described RxL motifs in the Clb5‐specific targets Fin1 and Spc110 (Fig 6A). This gives rise to a hypothesis that the two motifs are homologous and have evolved slightly different binding modes to the cyclin hp, as the key residues in the interaction are different. Interestingly, an alignment of the Fin1 194KNLLVEL motif within yeast species reveals that the motif earlier thought to function as an RxL motif could also be an NLxxxL motif, as the NLxxxL determinants are conserved, whereas the R/K of RxL has been lost in some species (Fig EV4B). This indicates that the NLxxxL motifs could be derived from RxL motifs, as they can overlap, but by losing the RxL determinants a highly Clb5‐specific docking motif can emerge. Whereas Fin1 homologs have uniform conservation of the NLxxxL consensus, Far1 and Lif1 homologs show higher diversity, with some homologs having either NLxxxL or RxL motif consensus sequences and some are not carrying any known docking site in the region (Fig EV4C and D). To test the functionality of the overlapping motifs in Fin1, we introduced single mutations to the docking region in Fin1 and analyzed phosphorylation of these substrates by Clb5‐Cdk1 in vitro. Mutation of K194, a determinant of RxL motif, caused only a twofold decrease in phosphorylation rate, whereas mutation of N195 and L200, the NLxxxL determinants, resulted in a greater decrease (Figs 6D and EV4E). Mutation of L196, however, had the greatest effect, likely because L196 is a determinant for both RxL and NLxxxL, indicating that both motifs have retained their functionality in Fin1.
To better understand the interaction of the two motifs with cyclin, we analyzed more precisely how mutations in the hp and its vicinity affect phosphorylation of an RxL‐dependent substrate, Sic1ΔC, and two NLxxxL‐dependent substrates, Lif1 and Far1. From the tested mutations, single mutation of the Clb5 hp residues L201A and W204A caused the largest decrease in phosphorylation of all tested substrates (Fig EV4F and G). Mutation of Clb5 T269 and D270 led to a considerable decrease in phosphorylation rate of Far1(1–150) and Lif1, whereas the effect on Sic1ΔC phosphorylation was minor (Fig EV4G), indicating that these positions could interact specifically with the NLxxxL motif.
To understand the overlap of the two motifs better, we introduced the RxL motif determinants to Far1 NLxxxL motif in Far1(1–150) mutant, where all TP sites have been mutated to SP sites (Fig 6E and F). Mutation of the threonines to serines increases the CDK threshold, making timely phosphorylation more dependent on cyclin docking (Fig 4B). The addition of the RxL determinants into the NLxxxL motif, resulting in overlapping motifs KNLTTSL and RNLFTSL, did not significantly affect the timing of sensor degradation (Fig 6G and H). Mutation of L135 in the RNLFTSL motif (RNLFTSA) greatly delayed the degradation, but not to the same extent as L135A substitution in wild‐type background, indicating that the introduced RNLF motif is functional, but much less efficient in promoting Far1(1–150 SP)‐GFP degradation than the NLxxxL motif (Fig 6G and H). Furthermore, when the NLxxxL motif was entirely replaced by conventional RxL motifs PKKLQF, PRKLQF, and VNRILFP, the degradation was also delayed compared to the wild type (Fig 6G and H), confirming the stronger phosphorylation potentiation by the NLxxxL motif. Interestingly, the degradation timing of the sensors with different RxL motifs varied by over 20 min. This highlights how sequence variations around RxL cyclin docking motif can affect the docking specificity and the timing of phosphorylation throughout the cell cycle.
Clb5‐specific docking motifs facilitate anaphase‐specific dephosphorylation
Docking motifs that are exclusively specific for S‐CDK could be important for assigning the timing of dephosphorylation to early anaphase, as Clb5 is degraded before the metaphase–anaphase transition, while Clb2 is degraded in late anaphase (Lu et al, 2014). To further analyze the temporal order of cyclin degradation, we performed time‐lapse fluorescence microscopy with strains expressing B‐type cyclins tagged with Citrine. For a reference, the 50% nuclear import time point of Whi5‐mCherry was used, which we determined to take place at about 14 min after anaphase onset detected by movement of Spc42‐GFP‐tagged spindle pole bodies (SPBs) (Fig EV5A). The data confirmed that Clb5 degradation is complete at the onset of anaphase, while Clb2 and Clb1 are degraded over 20 min later (Fig 7A and B). The quantitative model of CDK function states that as the CDK activity rises during the cell cycle, different activity thresholds are reached (Stern & Nurse, 1996; Coudreuse & Nurse, 2010). In metaphase–anaphase, the sequential degradation of cyclins leads to a decrease in total CDK activity and ordered dephosphorylation of targets (Touati et al, 2018). Without cyclin specificity, the phosphorylation switches would occur in the last‐in‐first‐out (LIFO) order, as the highest CDK thresholds met during the accumulation are also the first to be switched off in anaphase (Fig 7C). However, different docking specificity and degradation profiles of cyclins might influence the order of dephosphorylation during mitotic exit, creating the possibility for first‐in‐first‐out (FIFO) switches (Fig 7C).
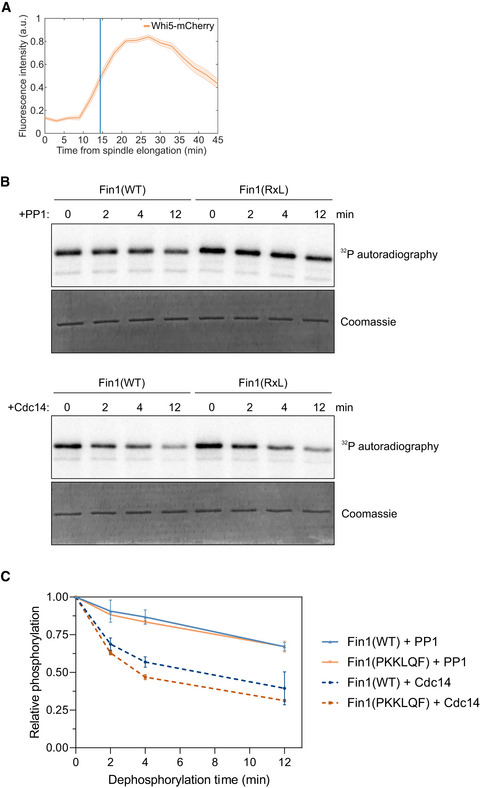
Nuclear import of Whi5 and phosphatase activity on Fin1(WT) and Fin1(PKKLQF)
The nuclear fluorescence levels of Whi5‐mCherry after spindle elongation were measured in single cells using time‐lapse microscopy. The plot shows mean ± SEM levels from a group of cells synchronized at the time of anaphase onset detected by movement of Spc42‐EGFP. The data are from 95 cells.
Dephosphorylation of Fin1(WT) and Fin1(PKKLQF) by Glc7 (PP1) and Cdc14. Fin1 was phosphorylated with Clb2‐Cdk1 before addition of phosphatase and Sic1 to inhibit Clb2‐Cdk1.
Quantified data from the dephosphorylation experiment in panel “B”. Data are from at least two replicates, and error bars show standard deviation.
Source data are available online for this figure.
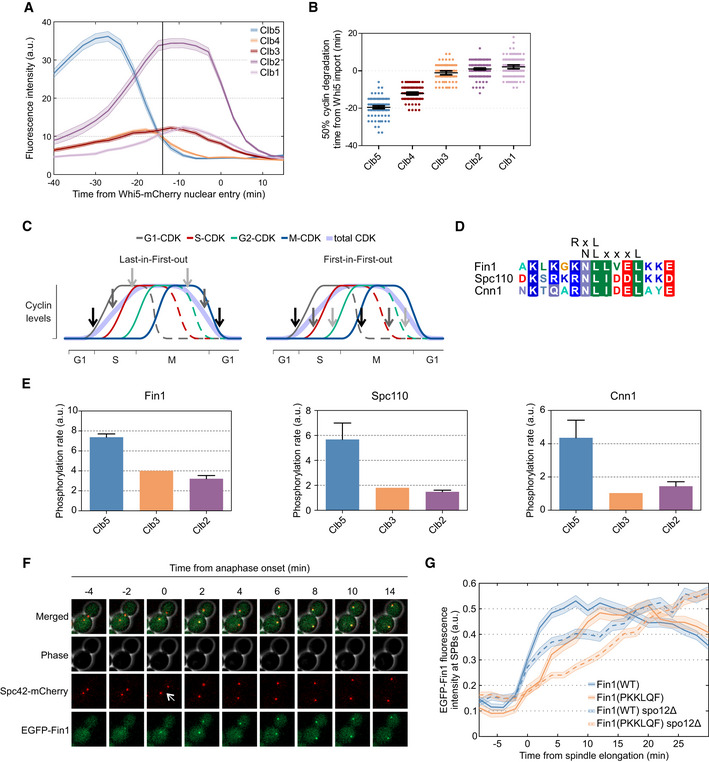
Exclusively Clb5‐specific docking motifs improve substrate dephosphorylation in anaphase
Time‐lapse microscopy was used to measure cyclin–citrine levels during mitotic exit in unperturbed cell cycles. The cells were synchronized by the nuclear import of 50% of Whi5‐mCherry at mitotic exit. The plot shows mean ± SEM fluorescence intensities averaged over a population of cells. The vertical line at −14 min marks the estimated time of anaphase entry. Sample sizes are shown in Table EV2.
Cyclin–citrine degradation relative to the nuclear import of 50% Whi5‐mCherry was followed in single cells. Plot shows the time from nuclear import of 50% Whi5‐mCherry to the degradation of 50% of the indicated cyclin in individual cells. The error bars show 95% CI of the mean. The number of cells is noted in Table EV2.
Simplified cell cycle model showing the expression profiles of 4 cyclins and the activation and inactivation of 3 CDK‐dependent switches A, B, C. On the left plot, the switches are controlled in the last‐in‐first‐out order, and on the right, the first‐in‐first‐out order.
Sequence alignment of predicted Clb5‐specific motifs in Fin1, Spc110, and Cnn1.
The effect of cyclin specificity on the phosphorylation of Fin1, Cnn1, and Spc110 was studied in a kinase assay. The plots show the phosphorylation by Clb5‐, Clb3‐, and Clb2‐Cdk1, the error bars show standard deviation (n = 2).
Images from a time‐lapse microscopy experiment of an exemplar cell expressing Spc42‐mCherry for detection of SPBs and EGFP‐Fin1. Cells were imaged every 2 min, the white arrow indicates onset of anaphase.
The accumulation of EGFP‐Fin1 to SPBs in anaphase was measured in experiments described in (F). The plot shows mean EGFP fluorescence intensities at SPBs around the time of anaphase onset. The error bars are ± SEM. The number of cells analyzed is shown in Table EV2.
Source data are available online for this figure.
We found that a group of Cdk1 targets that are dephosphorylated in anaphase, Fin1, Spc110, and Cnn1 (Woodbury & Morgan, 2007; Bock et al, 2012; Liang et al, 2013) contain Clb5‐specific NLxxxL motifs (Fig 7D). We analyzed the cyclin specificity of phosphorylation of these substrates and confirmed that, as predicted, Fin1, Spc110, and Cnn1 are most efficiently phosphorylated by Clb5‐Cdk1, when compared with Clb3‐ and Clb2‐Cdk1 (Fig 7E). Fin1 is an intermediate filament protein that is phosphorylated by Clb5‐Cdk1 and localizes to the nucleoplasm in metaphase, and following dephosphorylation at the onset of anaphase, is localized to the SPBs and the spindle (Woodbury & Morgan, 2007). We set out to analyze if the exclusively Clb5‐specific NLxxxL motif determined the timing of Fin1 dephosphorylation. For this, we replaced the motif in Fin1 (GKNLLVEL) with an RxL motif that promotes phosphorylation also by Clb3‐ and Clb2‐Cdk1 (PKKLQF). To study the dephosphorylation dynamics of Fin1 in an unperturbed cell cycle, we measured the accumulation of GFP‐Fin1 to the SPBs relative to the onset of anaphase, as detected by Spc42‐mCherry tagged SPBs (Fig 7F). Wild‐type Fin1 started accumulating at the SPBs at the time of spindle elongation (Fig 7F and G), as published previously (Woodbury & Morgan, 2007)). Fin1 with a PKKLQF RxL motif, however, showed a delay in SPB localization, suggesting that Fin1(PKKLQF) is dephosphorylated later. Fin1 interacts with phosphatases Cdc14 and PP1 (Bokros et al, 2016). Furthermore, Fin1 is dephosphorylated by Cdc14 upon activation of FEAR network (Woodbury & Morgan, 2007). To test whether the replacement of NLxxxL motif with RxL motif affects targeting of Fin1 by these phosphatases, we measured the dephosphorylation of Fin1 by Cdc14 and PP1 (Glc7) in vitro (Fig EV5B and C). These experiments show that both Fin1(WT) and Fin1(PKKLQF) are targeted by Cdc14 and PP1, and a slightly faster dephosphorylation of Fin1(PKKLQF) compared to wild‐type Fin1 was detected with Cdc14. Next, we tested how the kinase docking specificity affects the Fin1 SPB recruitment in a spo12Δ strain, where there is no release of Cdc14 in early anaphase (Stegmeier et al, 2002). Interestingly, wild‐type Fin1 was partially recruited to the SPBs at the start of spindle elongation with similar dynamics as in wild‐type strain, followed by slower gradual accumulation, indicating that the initial SPB localization of Fin1 is not Cdc14‐dependent (Fig 7G). SPB accumulation of Fin1(PKKLQF) in spo12Δ background, however, was delayed and gradual throughout anaphase (Fig 7G).
This result demonstrates that different cyclin docking motifs may be used to assign different Cdk1 substrate dephosphorylation timings in anaphase. Importantly, previous studies have shown that while dephosphorylation of Fin1 is dependent on Clb5 degradation, dephosphorylation of Orc6 is not and occurs in later anaphase (Touati et al, 2018). While both Fin1 and Orc6 contain RxL motifs, the Orc6 RxL motif (RRKLAF) matches the consensus for conventional RxL motif promoting phosphorylation by Clb5‐ as well as Clb3‐ and Clb2‐Cdk1. Therefore, the pattern of specificity for different cyclin docking motifs might add another level of complexity to the dephosphorylation timing of CDK substrates in mitotic exit and facilitate mixed LIFO and FIFO switching orders (Fig 7C).
Discussion
Our work sheds further light on the ability of cyclins to function as versatile targeting scaffolds that can bind a wide range of short linear motifs in CDK substrate proteins. In fact, it has been a longstanding enigma why evolution has chosen large proteins, with an energetically costly synthesis and degradation cycle, to temporally activate CDK kinase domains. By comparison, there are plenty of alternative and less costly ways to temporally and reversibly activate kinases by upstream signals, such as via phosphorylation or small second messenger molecules. Similar to cyclins and CDKs, the regulatory subunits of PP2A phosphatases have been recently found to regulate the substrate targeting of the phosphatase complex (Hertz et al, 2016; Kruse et al, 2020).
Recent studies have revealed that cyclins have both common and exclusive substrate docking interactions. For example, RxLxF motifs promote phosphorylation by S‐, G2‐ and M‐Cdk1. Such motifs can establish a LIFO switching order, which could be necessary, for example, to avoid re‐replication by keeping the proteins controlling replication licensing phosphorylated and inactivated from the start of S‐phase till the inhibition of Cdk1 activity in late mitosis. Alternatively, there are exclusively cyclin‐specific motifs, such as NLxxxL presented in this study, and LP, PxF, LxF for G1‐, G2‐, and M‐CDK, respectively (Bhaduri & Pryciak, 2011; Kõivomägi et al, 2011a; Örd et al, 2019a, 2019b). The motifs with cyclin‐exclusive recognition provide greater flexibility in temporal ordering of Cdk1 phosphorylation events.
In addition to triggering cell cycle events by Cdk1‐mediated phosphorylation during the accumulation of cyclins, the temporal ordering of late mitotic events is also governed by dephosphorylation on specific targets (Kataria et al, 2018). As Clb5 is degraded prior to anaphase and the NLxxxL motif was predicted in several proteins that are dephosphorylated in early anaphase, the S‐CDK‐specific docking may have evolved to promote earlier dephosphorylation and achieve more complexity of CDK function via an FIFO switching order.
An important element revealed by this study was the relationship between the Cks1‐dependent sequential phosphorylation route and the cyclin docking mechanisms. In the case of more efficient docking via NLxxxL, the Cks1 path was non‐essential for fast degradation, whereas when docking is weaker, as seen with an RxL motif in the Far1 context, the Cks1 route became essential. Strikingly, Far1(1–150), a target with Cks1‐dependent route, showed lower net k cat values than the minimal substrate Far1(85–150) lacking the Cks1‐dependent priming sites. This suggests that the N‐terminal Cks1 docking domain may have a dual function: While providing extra docking specificity to empower processive phosphorylation, it also acts as a diversional route, making the degron phosphorylation more difficult. The reason for this could be the necessity to compromise between efficient phosphorylation and filtering the erroneous noisy peaks of kinase signals. Such diversional function of Cks1‐driven multisite phosphorylation route is also reminiscent to our recent finding revealing that within the CDK‐Sic1 inhibitor complex, two competing Cks1‐dependent diversional routes and their various kinase inputs control the degron phosphorylation (Venta et al, 2020).
In conclusion, based on these and other recent findings, we predict that cyclins will accommodate even more SLiMs with various binding modes to facilitate substrate and inhibitor protein binding with a wide range of affinities. Cyclin surfaces functioning as scaffolds for a wide variety of SLiMs broadens our view of CDK function and opens new insights on how the order of CDK‐controlled events is achieved and how the CDK thresholds are robustly read in context of noisy biomolecular signaling context.
Materials and Methods
Yeast strains
Saccharomyces cerevisiae strains were of W303 background and are described in Table EV2. Gene deletions and mutations, promoter substitutions, and tagging were carried out using PCR‐based homologous recombination (Longtine et al, 1998; Janke et al, 2004). All gene modifications were confirmed by DNA sequencing. Far1(1–150) constructs were cloned into pRS306 vector containing SIC1 promoter (1–420 base pairs upstream of SIC1 gene) and C‐terminal EGFP. The vectors were linearized and integrated to URA3 locus. The transformants were selected for single‐copy integration by fluorescence intensity.
Time‐lapse fluorescence microscopy
Cells were grown at 30°C in synthetic complete media with 2% glucose (SC) to OD 0.2–0.6 before the experiment. Cells were then pipetted onto 0.8‐mm cover glass and covered with a 1‐mm thick 1.5% SC/glucose agarose pad (NuSieve™ GTG™ Agarose, Lonza). Cells were incubated under the agarose pad for 1 h before the start of the experiment. Imaging was executed using a Zeiss Observer Z1 microscope with a 63×/1.4NA oil immersion objective and Axiocam 506 mono camera (Zeiss), using 3 × 3 binning. The sample was kept at 30°C using Tempcontrol 37–2 digital (PeCon). Cells were imaged every 3 min, expect for experiments with GFP‐Fin1 Spc42‐mCherry, where images were taken every 2 min. The experiments were 8 h long and contained up to 12 positions that were followed using an automated stage and ZEN software (Zeiss). Focus was kept using Definite Focus. Colibri 470 LED module with exposure time of 15 ms was used for excitation of EGFP‐tagged proteins. Excitement of cyclins fused with yeCitrine was performed using a Colibri 505 LED module for 500 ms. Whi5‐mCherry was excited using Colibri 540–580 LED module for 750 ms. All Colibri modules were used at 25% power. Filter set 61 HE (Zeiss) was used for imaging EGFP and mCherry, and filter set 72 HE (Zeiss) was used for yeCitrine.
Image segmentation, cell tracking, and quantification of fluorescence signals were performed using MATLAB (The MathWorks, Inc.) as described in (Doncic et al, 2013). All plots with microscopy data contain data from at least two experiments, and the exact number of cells analyzed from each strain is presented in Table EV2.
Protein purification
Plasmids used for recombinant protein expression are listed in Table EV3. Cdk1 substrate proteins were purified from Escherichia coli. Far1(1–150)‐EGFP, Sic1(1–215), Fin1, Cnn1, and Spc110 were N‐terminally tagged with 6xHis tag and were expressed in BL21RP cells. Expression of Far1(1–150)‐EGFP, Cnn1, and Spc110 was induced with 0.3 mM IPTG at 16°C, whereas Sic1(1–215) and the GB1‐6xHis constructs were induced with 1 mM IPTG at 37°C. The proteins were purified using immobilized cobalt affinity chromatography and were eluted with imidazole. Fin1‐6xHis was expressed and purified as described previously (Woodbury & Morgan, 2007). 6xHis‐Cdc14 was expressed in BL21RP cells with 0.125 mM IPTG at 23°C and was purified using nickel affinity chromatography. GST‐Glc7 was expressed in BL21RP cells and was purified as described previously (Rojas et al, 2014).
Yeast cyclin‐Cdk1 complexes were purified from S. cerevisiae cells where the tagged cyclin was overexpressed from GAL1 promoter. Clb6, Clb5, Clb4, Clb3, Clb2, and Clb1 were purified using the tandem affinity purification method (Puig et al, 2001; Ubersax et al, 2003). Cln2 with N‐terminal 3HA tag was purified as described (McCusker et al, 2007). Overexpressed GST‐Cln1 was purified from yeast lysate using Glutathione Sepharose (GE Healthcare). Yeast cells were lysed using Mixer Mill MM 400 (Retch). TAP‐tagged mutated Clb5‐Cdk1 complexes used in Fig EV4G were bound to IgG Sepharose (GE Healthcare), washed extensively, and were released from the IgG Sepharose by TEV‐mediated cleavage of the TAP tag. Cks1 was expressed in E. coli BL21RP and purified as described (Reynard et al, 2000).
Kinase assay
The phosphorylation reactions were performed at room temperature in buffer containing 50 mM Hepes‐KOH, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 20 mM imidazole, 2% glycerol, 0.2 mg/ml BSA, 500 nM Cks1, and 500 µM ATP [(with added [γ‐32P]‐ATP (Hartmann Analytic)]. Substrate protein concentrations were 500 nM (in the linear [S] versus v0 range, several‐fold below the estimated KM value) unless noted otherwise. The concentrations of kinase complexes were around 0.2 nM. The kinase assays were performed under conditions below 10% of initial substrate turnover. Reactions were stopped at two time points (8 and 16 min) with SDS–PAGE sample buffer and separated using SDS–PAGE. For multisite phosphorylation analysis of Far1, Phos‐tag SDS–PAGE with 8% acrylamide and 50 µM Phos‐tag was used. For Sic1 multisite phosphorylation analysis, 10% acrylamide SDS–PAGE gels supplemented with 100 µM Phos‐tag was used.
For analysis of Fin1 dephosphorylation, 0.5 µM Fin1(WT) and Fin1(PKKLQF) were first phosphorylated with 0.3 nM Clb2‐Cdk1 for 10 min. Then, a mixture of 1 µM full‐length Sic1 and either Cdc14 or Glc7 was added. Reactions with Glc7 were supplemented with 2 mM MnCl2. The dephosphorylation reaction was stopped at 10 s (0 min), 2, 4, and 12 min by pipetting aliquots into SDS–PAGE sample buffer.
γ‐32P phosphorylation signals were detected using an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare Life Sciences). Signals were quantified using ImageQuant TL (Amersham Biosciences), and GraphPad Prism 8 was used for data analysis. All kinase assays were performed in at least two replicate experiments.
Co‐immunoprecipitation and Western blot
Yeast cells of w303, MO578, and MO579 for Cdc4, Clb5(WT), and Clb5(hpm) co‐immunoprecipitation, respectively, were grown in 50 ml YPD to OD 0.8. Cells were collected and snap‐frozen. 6xHis‐Far1(85–150)‐GFP was expressed in BL21RP cells at 37°C with 1 mM IPTG. 6xHis‐Far1(85–150)‐GFP was bound to Chelating Sepharose. For Cdc4 co‐immunoprecipitation, 6xHis‐Far1(85–150)‐GFP was phosphorylated with Clb5‐Cdk1 for 1 h at room temperature. Yeast cells were lysed by bead beating in IP buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% NP40, 1 mM DTT). The cleared lysate was mixed with the beads with 6xHis‐Far1(85–150)‐GFP, and the mixture was incubated at 4°C for 1 h. Then, the beads were washed thoroughly using IP buffer and the proteins were eluted with SDS–PAGE sample buffer. The proteins were separated using SDS–PAGE and were transferred to a nitrocellulose membrane using Pierce G2 Fast Blotter (Thermo Scientific). For detection of Cdc4, goat polyclonal IgG antibody (yN‐19, sc‐6714) and HRP‐conjugated anti‐goat IgG antibody (1:7500) (31402, Invitrogen) were used. Anti‐HA.11 epitope tag antibody (1:500) (clone 16B12, BioLegend Cat. No. 901501) and HRP‐conjugated anti‐mouse IgG antibody (1:7,500) from Labas, Estonia were used to detect Clb5‐6HA.
Pheromone sensitivity assay
Yeast cultures were grown in YPD to stationary phase. 10 µl of the culture was mixed with 0.5% agar and plated on YPD or YPG plate. Then, filter paper disks were placed on the plate and 1 µl of solution containing 0, 20, 2 µg, or 0.2 µg of α‐factor in DMSO was pipetted on the disks. The plates were incubated at 30°C for 1 or 2 days for YPD or YPG plates, respectively.
Bioinformatics and statistical analysis
Potential NLxxxL motifs were searched from the intrinsically disordered regions (defined by positions with IUPRED score over 0.3) of S. cerevisiae proteome using SlimSearch 4 (Krystkowiak & Davey, 2017). All statistical tests were performed using GraphPad Prism 8. Mann–Whitney U‐test is used for statistical comparison of two populations of cells which are independent. No statistical methods were used to predetermine sample size.
Author contributions
IF, MÖ, and ML directed the study. IF, LB, FM, EV, and MÖ cloned the constructs, made the yeast strains, and purified the proteins. IF, LB, FM, and MÖ performed the experiments. MÖ and ML wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank P. Pryciak for comments on the manuscript and J. Mihhejev for excellent technical assistance. The work was funded by ERC Consolidator Grant 649124, Centre of Excellence for “Molecular Cell Technologies” TK143, and Estonian Science Agency grant PRG550 to M.L.
Data availability
This study includes no data deposited in external repositories.