 Polymeric nanoparticle vaccines to combat emerging and pandemic threats
Polymeric nanoparticle vaccines to combat emerging and pandemic threats
Biomaterials


- Altmetric
Subunit vaccines are more advantageous than live attenuated vaccines in terms of safety and scale-up manufacture. However, this often comes as a trade-off to their efficacy. Over the years, polymeric nanoparticles have been developed to improve vaccine potency, by engineering their physicochemical properties to incorporate multiple immunological cues to mimic pathogenic microbes and viruses. This review covers recent advances in polymeric nanostructures developed toward particulate vaccines. It focuses on the impact of microbe mimicry (e.g. size, charge, hydrophobicity, and surface chemistry) on modulation of the nanoparticles’ delivery, trafficking, and targeting antigen-presenting cells to elicit potent humoral and cellular immune responses. This review also provides up-to-date progresses on rational designs of a wide variety of polymeric nanostructures that are loaded with antigens and immunostimulatory molecules, ranging from particles, micelles, nanogels, and polymersomes to advanced core-shell structures where polymeric particles are coated with lipids, cell membranes, or proteins.

Introduction
Emerging diseases
Over the past decades, there have been a constant stream of new pathogens emerged from microbial reservoirs including those in animals or vectors and then spilt over into human populations, causing emerging infectious diseases. Globalization and climate change have contributed to amplify the emergence and transmission of the pathogens, posing pandemic threats to global human wellness [[1], [2], [3]]. A notable example is the ongoing pandemic coronavirus disease 2019 (COVID-19) caused by a novel severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) originated from bats [4]. SARS-CoV-2 emerged in Wuhan city of China in late 2019 [5] and has since infected over forty million individuals spread in 235 countries, areas or territories, and claimed the lives of more than one million people to date [6] along with widespread social and economic disruption [7]. SARS-CoV-2 shares genome sequence homology with severe acute respiratory syndrome coronavirus (SARS-CoV, ~79%) and Middle East respiratory syndrome coronavirus (MERS-CoV, ~50%) [8] that caused SARS [9] and MERS [10] outbreaks in China and Saudi Arabia in 2003 and 2012, respectively. Other emerging infectious diseases also occurred, including the emergence of Zika in Brazil in 2016 [11], the Ebola epidemic in West Africa in 2014 [12], the H1N1 swine influenza pandemic from Mexico in 2009 [13], the Dengue outbreaks in the Americas in 2000 [14] and the acquired immunodeficiency syndrome (AIDS) pandemic caused by human immunodeficiency virus (HIV) in the United States in 1980s [15]. Although most outbreaks are caused by viruses, perpetual bacterial and parasitic infections, such as methicillin-resistant Staphylococcus aureus (MRSA) [16], multidrug-resistant tuberculosis [17] and malaria [18], represent considerable risks to human health as well with the potential for an explosive growth of cases. Despite different causative agents, infectious diseases share commonalities; unpredictability of the re-emergence of known diseases and of emergence of new diseases from previously unrecognized pathogens, high morbidity and profound socio-economic impacts. Therefore, there is an urgent need to develop effective prophylactic measures for prevention and preparedness of both re-emerging and new emerging diseases which are listed as “Disease X″ in the top ten priority diseases by the World Health Organization [19].
Vaccine development and its challenges
Vaccination has proven to play critical roles in disease prevention improving global human health [20], as it has greatly reduced the burden of former epidemics like smallpox [21], polio [22], and measles [23] to name a few. Vaccination aims to achieve long-term protective immunity capable of rapid and effective reactivation upon subsequent microbial exposure. Designing a vaccine involves the selection of antigens, adjuvants, delivery strategies, and scalable manufacturing systems. Antigens derived from pathogens are the targets of protective immunity within the body, while adjuvants are sometimes required to stimulate the immune response toward co-delivered antigens by enhancing antigen presentation and/or by providing co-stimulation signals. Carriers can be used to deliver antigens to antigen presenting cells (APCs) which in turn determines the relative magnitude and quality of the resultant immune responses, whether adjuvants are needed, as well as the suitability of particular routes to administer the vaccine, and the requirement for a prime–boost vaccination regimen to improve protective immunity and its durability. It is desirable to develop a carrier that can be readily: (i) modularized with a broad antigen repertoire to boost immune responses, (ii) served as plug-and-play platform technologies adaptable to address multiple pathogens in parallel, and (iii) mass produced for enabling rapid responses to re-emerging and emerging disease epidemic.
Traditionally, vaccines have been developed empirically based on weakened or inactivated forms of the pathogens. Although the technology is mature, these live attenuated and inactivated whole cell/virus which act as carriers themselves may pose safety concerns emanated from inadequate inactivation processes and potential reversion to their pathogenic forms [24]. To improve safety, subunits vaccines have been designed and developed to only contain minimal antigenic element of pathogens, in lieu of the entire pathogens, based on selected protein, peptide or polysaccharide antigens [25]. Nucleic acid vaccines involving DNA or RNA encoding antigens have also been developed, which recently gain popularity due to their relatively rapid and ease of large scale manufacture [26]. These antigens or nucleic acid encoding the antigens can be encapsulated in or display on to a carrier platform, including nonreplicating viral vectors, protein-based nanoparticles, or synthetic nanoparticles, to provide stability and targeting ability to APCs [27]. However, the improved safety of subunit vaccines comes as a trade-off to their efficacy. Therefore, development of subunit vaccine formulations that could bridge the gap between safety and efficacy is an ongoing quest.
To date, there are no safe and effective vaccines yet to combat emerging diseases. In general, vaccine development takes years of research before they can be licensed for human use due to multifaceted challenges. First, high genetic diversity of especially zoonotic pathogens that are capable of cross-species transmission make it difficult for vaccine design. For example, influenza virus that tends to undergo antigenic drift/shift, as well as HIV and hepatitis C virus with their genetic plasticity represent a moving target for antigen selection and engineering. Second, there is a lack of well-established immune correlates of protection, including the type, quantity and quality of immune responses that are adequate to confer protection in humans [28]. Nevertheless, it is generally known that neutralizing-antibody responses are required to bind pathogens in a manner that block infections, while cell-mediated immune responses eliminate the infected cells and limit the spread of the pathogens [29,30]. To demonstrate vaccine efficacy and durability, it is critical to generate these dual arms of adaptive immunity especially against intracellular pathogens in suitable animal models and in clinical trials. Fourth, vaccine safety should also be evaluated rigorously. Studies on SARS, MERS and dengue have raised concerns about antibody-dependent enhancement (ADE) of the infections, which can occur when non-neutralizing antibodies or antibodies at sub-neutralizing levels contribute to exacerbating the disease severity [31,32]. Lastly, vaccine manufacture requires consistent batch-to-batch reproducibility in a scale that is deployable to large populations. Cold chain requirement for some vaccine candidates exhibits additional logistical and fiscal barriers for global vaccine distribution especially in low-income, tropical countries.
Polymeric nanostructures as vaccine platforms
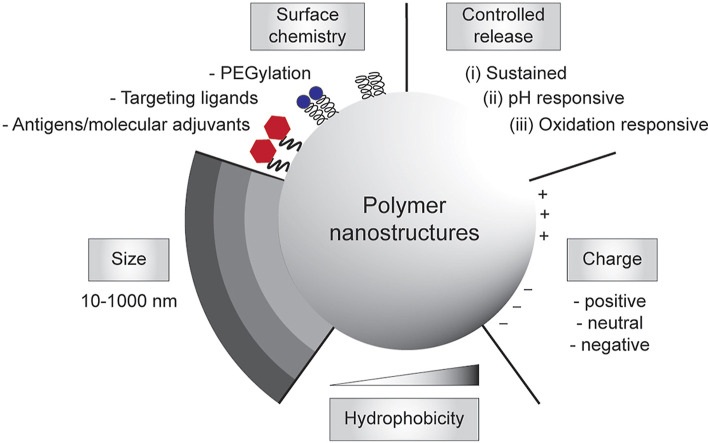
The need to achieve control over humoral and cell-mediated immune responses has catalyzed the research and development toward rational vaccine design. In the last decade, there has been a considerable progress in the rational design and synthesis of nanoparticles for vaccine development [[33], [34], [35], [36], [37]]. In particular, polymer-based nanostructures have been developed toward particulate vaccines against emerging infectious diseases owing to their tailorability, biocompatibility and biodegradability [[38], [39], [40]]. Many of vaccine-based polymeric nanostructures have also been utilized to fight against non-infectious diseases like cancers that also cause major problems in global human health, which paves the way to be potentially developed against emerging diseases as well. Additionally, different polymeric nanostructures can be synthesized, ranging from particles, nanogels, and micelles to polymersomes and core-shell particles, using vastly available materials, including natural polymers that are abundant in nature [41], synthetic polymers that can be facilely manufactured [42], or biopolymers that are amenable to large-scale production in microbial cell factories [43,44]. Due to their rich chemistry, polymeric nanostructures can be modularized to adopt immunological cues by mimicking biophysical and biochemical characteristics of pathogens to elicit robust and protective immune responses upon vaccination (Fig. 1 ).
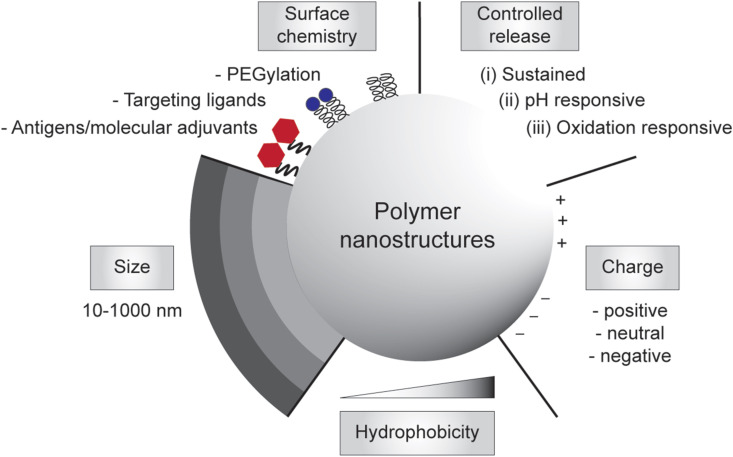
Engineering polymeric nanostructures for vaccine development. The physicochemical properties of the particulate polymers can be engineered to enhance desirable immune responses, including size, surface chemistry, controlled release, charges, and hydrophobicity.
The aim of this review is to highlight the key research progresses in immunology-guided design of polymeric nanostructures in efforts to develop particulate vaccines. As the fields of immunology and materials engineering evolve rapidly [45,46], we limit the review to only recently published studies. Here we will start describing strategies to develop polymeric particles that aim to modulate innate and adaptive immune responses. Next, polymeric nanoparticles will be categorized by their structures, including particles, nanogels, micelles, polymersomes and core-shell particles, and methods to incorporate antigens and/or molecular adjuvants along with their immunological properties in model animals.
Immunological basis for particulate vaccine delivery
Vaccination aims to provide long-term protection against infectious microbes and viruses and, in the context of cancer, target inhibition of tumor development by triggering specific anti-tumor immune responses. The immune system is enormously complex with a distributed network of immune cells and lymphoid organs, which can be generally classified into innate and adaptive components. The innate immune system provides immediate defense at entry sites of infections like mucosal surfaces or the skin, mediated by complement proteins and cellular effectors such as macrophages and dendritic cells (DCs) [47]. In contrast, the adaptive immune system comprised of B cells and T cells is slower to develop against the infection, but it can differentiate into long-term immunological memory that recognizes specific antigens from the pathogen and provides rapid protection on re-exposure to the same antigens [48]. Most of the activities of the immune system take place in the lymph nodes (LNs) where APCs, B cells and T cells interact with each other in close proximity. Therefore, many nanoparticles have been developed to target LNs aiming to improve the immunological outcomes [49].
Particulate delivery to the lymph nodes (LNs)
Particulate vaccines are usually administered parenterally through the muscle, subcutaneous (s.c.), or intradermal (i.d.) tissues. Following injections, there are two trafficking routes of particulate vaccines to the LNs: (i) by diffusion or convection through the afferent lymphatics; or (ii) by cell-mediated transport from the peripheral tissue interstitium.
Passive drainage of polymeric nanoparticles into LNs are influenced by size [49], rigidity [50], charge [51] and composition [52] of the nanoparticles. Nanoparticles with hydrodynamic diameters of 20–200 nm were shown to passively drain into and accumulate at LNs after s.c. injection [[53], [54], [55], [56], [57]], in which their transport kinetics are size-dependent [54]. Rigid nanoparticles were demonstrated difficult to be passively migrated to LNs, as the number of 25 nm-sized rigid polystyrene nanoparticles that were drained to LNs after i.d. injection were 10-fold less than similarly sized flexible dextran particles [50]. The chemical properties of the nanoparticles determine their interactions with the extracellular matrix (ECM) that composes the lymphatic tissues [58]. Hydrogel-based polymeric nanoparticles engineered with poly(ethylene glycol) (PEG) were demonstrated to enhance their passive drainage to the LNs by blocking interactions with the ECM [52], although it should be noted that dense PEGylation of nanoparticles using high molecular weight PEG could prevent subsequent nanoparticle uptake by APCs [59]. In addition, carbohydrate pullulan nanogels (<100 nm) were shown to passively migrate to LNs after s.c. injection facilitated by their uncharged hydrophilic surfaces that prevented nonspecific binding in ECM during the transport [51]. For particles with hydrodynamic diameters of 500–2000 nm, Manolova et al. showed that polystyrene particles of those sizes were internalized by DCs at the injection site and then transported to the LNs by DC-mediated trafficking [55].
Once nanoparticles have arrived into LNs, they could be distributed into different areas within the LNs, in which the nanoparticle size was found to influence the cellular distribution [45,55]. Further, the nanoparticles may be internalized by APCs (e.g. macrophages or DCs) that line the subcapsular sinus or medullary sinuses within the LNs [34,60]. DCs are often targeted by antigen-carrying nanoparticles, because DCs constitute a diverse subsets that plays major roles in initiating and regulating innate and adaptive immune responses [61]. To enhance the internalization of nanoparticles by DCs, nanoparticles can be surface-functionalized with specific ligands that recognize receptors expressed on the surface of DCs [62]. For example, PEGylated poly(lactic-co-glycolic acid) (PLGA) nanoparticles were functionalized with a monoclonal antibody targeted to a DC cell surface receptor CD40, which demonstrated enhanced DC uptake used for anti-tumor vaccines [63]. Another study also used PLGA nanoparticles but were coated with chitosan to enhance DC uptake, because (i) cationic properties of chitosan improved nanoparticle internalization by DCs through electrostatic interaction with the DCs’ membranes, and (ii) chitosan promotes clathrin-mediated endocytosis [64]. Liu et al. surface-engineered PLGA nanoparticles with hyaluronic acid to facilitate CD44 receptor-mediated endocytosis in DCs and enhance the internalization by DCs [65], as hyaluronic acid has a specific affinity towards CD44 which is the major cell receptor for HA highly expressed on DCs [66].
Antigenic cargoes carried in polymeric particles can be digested by DCs, and the resultant antigen peptide fragments are presented on the surfaces of DCs in the context of major histocompatibility complexes (MHCs). There are two pathways for antigen presentation (Fig. 2 ): (i) the class II MHC (MHC II) antigen presentation pathway for CD4+ T cell activation, and (ii) the class I (MHC I) antigen cross presentation pathway for CD8+ T cell activation, which both have significant roles in induction of humoral and cellular immunities.
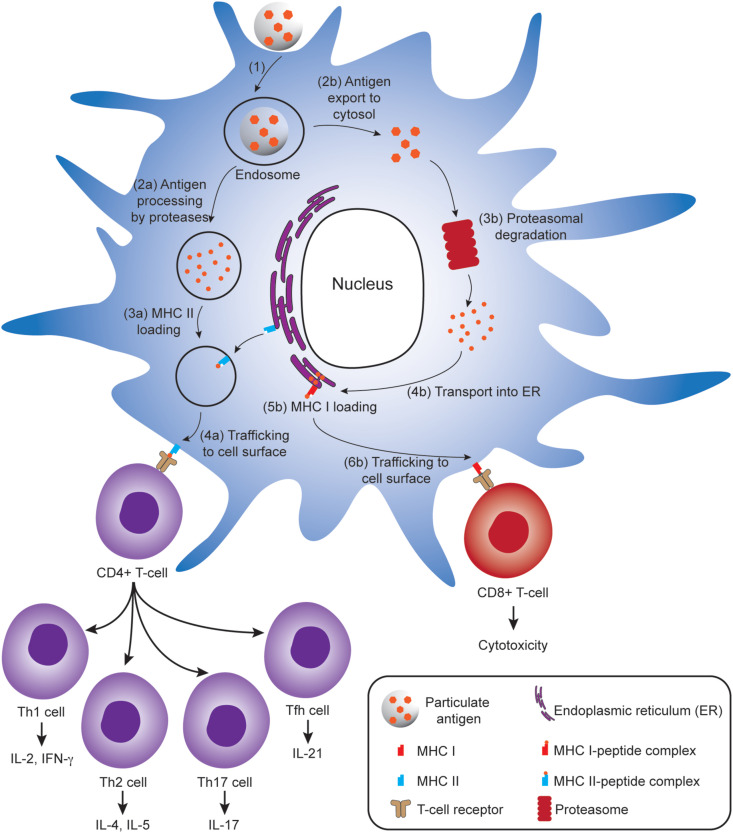
Antigen processing and presentation in a dendritic cell (DC) that lead to activation of CD4+ T-cells (left) and CD8+ T-cells (right). (1) Antigen-loaded particles are internalized by DC through cell uptake pathways such as phagocytosis or endocytosis. To activate CD4+ T cells, (2a) particulate antigens are processed to peptide fragments by proteases in endosomes, (3a) subsequently loaded onto MHC II molecules, (4a) and the formed MHC II–peptide complex is then trafficked to the cell surface where it presents the antigen to CD4+ T cells bearing cognate T cell receptors (TCRs). To activate CD8+ T cells (following the cytosolic pathway as depicted), (2b) the antigens are translocated to cytosol and (3b) subsequently processed by proteasome. (4b) The resulting peptide fragments are transported to endoplasmic reticulum (ER), (5b) then loaded onto MHC I molecules in ER, followed by (6b) trafficking of the formed MHC I–peptide complex to cell surface where they can interact with CD8+ T cells bearing cognate TCRs. Activated CD4+ T cells can differentiate into T helper type 1 (Th1), Th2, Th17 and T follicular helper (Tfh) cells producing various cytokines for signaling B cell activation, whereas activated CD8+ T cells can kill infected/mutated cells.
Activation of CD4+ T cells
After processing the antigenic cargo into antigen fragments in the endolysosomal compartment, DCs present the antigen fragment bound to MHC II molecules on their surfaces to activate CD4+ T cells (Fig. 2). CD4+ T cells, also known as helper T cells, can differentiate into different subclasses, including T helper type 1 (Th1), Th2 and Th17 cells [67], as well as T follicular helper (Tfh) cells [68]. Th1 cells can release cytokines such as interferon-γ (IFN-γ) and interleukin-2 (IL-2) to assist CD8+ cytotoxic T cell (CTL) activation facilitating cell-mediated immune response. Th1 also play an important role in the development of memory T cells for long-term protective immune response. Th2 cells secrete cytokines like IL-4, IL-5, IL-10 and IL-13, which support humoral immune response. Th2 cells help to activate B cells to secrete antibodies against specific antigens that link to most extracellular pathogens including bacteria and parasites [69]. Both Th1 and Th2 cell lineages are critical for the proliferation of B cells and immunoglobulin class switching. For example, immunoglobulin G2a (IgG2a) production and IgG1/IgE class switching are regulated by Th1 cytokine IFN-γ and the Th2 cytokine IL-4, respectively. Tfh cells also mediate humoral immunity through interaction with B lymphocytes in the germinal centre, which assist B cells in dividing into long-lived plasma cells that produce high-affinity antibodies or memory B cells for long-term protection. Th17 cells secretes cytokines like IL-17 to promote inflammation in response to infections [70].
Antigen delivery to MHC II loading pathway has been demonstrated to be influenced by size, shape, and charge of the polymeric particles. It is clear that the delivery of antigens using particles with different particle sizes could result in different immune response (Table 1 ) [[71], [72], [73], [74], [75], [76], [77], [78], [79], [80], [81]]. For example, polystyrene (PS) particles conjugated with Ovalbumin peptide (Ova) having sizes in a narrow nano-sized range (20, 40, 49, 67, 93, 101, and 123 nm) were demonstrated to elicit distinct T cell responses, with 40–49 nm particles induced highest level of antigen-specific Th1 cytokine IFN-γ, while 93–123 nm particles mainly generating Th2 cytokine IL-4 [71]. However, when comparing nano- and micro-sized particles, there is no clear consensus which particle size is more effective at inducing strong immune response (Table 1). Some studies reported that smaller particles were more effective at inducing strong immune responses than the larger counterparts [72,73,76,77], whereas other studies reported the opposite [[79], [80], [81]]. For example, 40-nm Ova-conjugated PS particles induced higher production of IFN-γ and IgG as compared to other particle sizes (20, 100, 200, 500, 1000, and 2000 nm) [72]. In contrast, 1000-nm poly(lactide-co-glycolic acid) (PLGA) particles loaded with bovine serum albumin generated stronger antibody responses (IgG, IgG1 and IgG2a) than those elicited by 200- and 500-nm particles [79]. The differences in the resultant immune responses were likely due to the differences in the material compositions, administration routes, size distribution of the particles, antigen loading methods, and/or antigen doses [82,83].

| Materials | Diameter (nm) | Administrationroute | Measured immune response | Comments | Ref. |
|---|---|---|---|---|---|
| PS particles conjugated with Ova | 20, 40, 49, 67, 93, 101, 123 | i.d. | IFN-γ,IL-4,IgG1 | •40–49-nm particles induced highest level of IFN‐γ | [71] |
| •93–123-nm particles mainly generated IL‐4 | |||||
| •IgG1 production was observed across the particle size range | |||||
| PS particles conjugated with Ova | 20, 40, 100, 200, 500, 1000, 2000 | i.d. | IFN-γ,IgG | •40-nm particles induced strongest immune response | [72] |
| PS particles conjugated with HA or Ova–HEL | 40, 200, 1000 | s.c. | Tfh, IgG1, IgG2c | •200-nm particles conjugated with HA generated strongest antibody responses | [73] |
| •1000-nm particles failed to induce an antibody response | |||||
| •200-nm conjugated with Ova–HEL enabled more sustained antigen presentation by DCs, leading to the enhanced production of Tfh cells | |||||
| PPS particles conjugated with Ova | 30 and 200 | i.n. | IFN-γ, TNF-α, IL-2, IgA, IgG, IgG1, IgG2c | •200-nm particles induced higher levels of IFN-γ, TNF-α, and IL-2 in lung | [74] |
| •200-nm particles generated higher titers of IgG, IgA, and IgG2c, but a similar level of IgG1 induced by both groups | |||||
| PS particles conjugated with Ova | 193 and 521 | s.c. | IFN-γ,IL-4, IgG,IgG1, and IgG2a | •193-nm particles elicited a higher amount of IFN-γ, IgG, | [75] |
| IgG1, and IgG2a, but a similar level of IL-4 induced by both 193- and 521-nm particles | |||||
| •Overall, 193-nm particles induced stronger Th1 and Th2 immune responses than 521-nm particles | |||||
| PVA-grafted PLG particles loaded with TT | 100, 500, 1500 | p.o., i.p., i.n. | IgG, IgA | •100-nm particles elicited higher antibody titers than those of 500-nm after p.o. or i.p. injection | [76] |
| •1500-nm particles failed to induce an antibody response | |||||
| •100- and 500-nm particles were seen to induce equal levels of antibody responses upon i.n. administration | |||||
| PLGA particles co-loaded with Ova and CpG ODN | 300, 1200, 6700, and 17400 | i.p. | IgG2a, IgG1 | •300-nm particles induced strongest antibody response with highest IgG2a/IgG1 ratio (Th1 biased immunity) | [77] |
| PLA particles loaded with HbsAg | 200–600, 2000–8000 | i.m. | IFN-γ,IL-4, IgG | •2000–8000-nm particles induced higher IgG response than those of 200–600-nm particles | [78] |
| •200–600-nmm particles promoted IFN-γ secretion (Th1 biased response), while 2000–8000 nm particles mainly driving IL-4 release (Th2 biased response) | |||||
| PLGA particles loaded with BSA | 200, 500, 1000 | i.n., p.o., s.c. | IgG, IgG1, IgG2a | •1000-nm particles generated stronger antibody responses than those elicited by 200- and 500-nm particles | [79] |
| •500-nm particles elicited stronger antibody responses than 200-nm particles following i.n. administration | |||||
| •200- and 500-nm particles induced similar immune responses upon p.o. and s.c. administration | |||||
| PLGA/lecithin core-shell particles loaded with HPV 16 L1 pentamer | 300, 1000, 3000 | i.m. | IFN-γ,IL-4, IgG, IgG1, IgG2a Tfh | •1000- or 300-nm particles induced higher production levels of cytokines and antibodies than 3000-nm particles | [80] |
| •Balanced IgG1/IgG2a ratio was seen across the particle size range | |||||
| •1000-nm particles provided a more sustained antigen release than 300-nm particles, leading to the enhanced Tfh cell response | |||||
| PLGA particles mixed with Ova or H5N1 antigen | 538.5, 972.5, 2126, 4934 | i.m. | IFN-γ,IL-4, IgG, IgG1, IgG2a, IgG2b | •972.5-nm particles elicited strongest immune responses | [81] |
Abbreviation: i.d., intradermal; i.m., intramuscular; i.n., intranasal; i.p., intraperitoneal; p.o., oral; s.c., subcutaneous; DCs, dendritic cells; IFN-γ, interferon gamma; IL-4, interleukin 4; BSA, bovine serum albumin; CpG ODN, CpG oligodeoxynucleotides; HA, hemagglutinin; HbsAg, hepatitis B surface antigen; HPV, human papilloma virus; HEL, hen egg lysozyme; Ova, ovalbumin; TT, tetanus toxoid; PS, poly(styrene); PLA, poly(lactide); PLGA, poly(d,l-lactide-co-glycolide); PPS, poly(propylene sulfide); PVA, poly(vinyl alcohol); PLG, poly(lactide-co-glycolide); Tfh, T follicular helper.
The shape of polymeric particles may also play a role in the induction of T helper cell responses. Spherical polystyrene particles induced higher Th1 responses as measured through IgG1:IgG2a ratio, whereas rod-shaped particles of the same composition led to Th2-biased immune response during in vivo immunization study [75]. It was also found that cell uptake of rod-shape polystyrene particles was shown higher and more specific compared to spherical particles [84]. Rough or budding polystyrene particles were demonstrated to be more effective at inducing the inflammasome compared to the smooth counterparts [85]. Although this is a part of the innate immune system, it shows that there is a clear role for surface textures interacting with the immune system.
The surface charge of polymeric nanoparticles has also shown to affect which type of T helper response is induced. Cationic particles steered the immune system towards a Th1 type response, while anionic particles showed a more balanced Th1/Th2 immune response [86,87]. Previous studies conducted using cationic synthetic polymer-based particles such as polyethyleneimine confirmed these results and showed high Th1 responses following vaccination [88,89].
Activation of CD8+ T cells
CD8+ cytotoxic T cells are required to act synergistically with humoral immunity to eliminate cells infected by, for example, rotavirus, polio and influenza [33]. Strong CD8+T cells are also needed to kill mutated cells caused by cancers. CD8+ T cells recognize and respond to peptide antigen fragments cross presented by MHC I molecules expressed on DCs [90]. Following cell uptake in DCs, there are two main pathways for antigen processing that lead to CD8+ T cell response: (i) the vacuolar pathway and (ii) the cytosolic pathway [91]. In the vacuolar pathway, the antigens can be degraded within endolysosomal compartments following cell uptake in DCs caused by the acidic environment and proteases, in which the processed antigens can be directly loaded onto recycling MHC I in the phagosome. In the cytosolic pathway (Fig. 2), the processed antigens are translocated to the cytosol for further processing by proteasome before loading onto MHC I. Antigen-loaded polymeric nanoparticle have been demonstrated of inducing CD8+ T cell response.
Surface charges of polymeric nanoparticles play significant role in inducing CD8+ T cell responses. Cationic poly(ethylenimine) nanoparticles (PEI) were shown able to facilitate the loaded cargoes to escape from endosomes through a “proton-sponge” mechanism, enabling direct delivery to the cytosol where they can be processed by the MHC I loading pathway to trigger CD8+ T cell responses [92]. High-density amines within PEI absorb protons that are pumped by ATPase enzyme into the developing endosomes, causing more protons to be transferred into the endosomes to reach the desired pH along with an influx of counter ions to balance, which ultimately causes osmotic swelling and rupture of the endosome – a strategy that has been used for gene delivery [93].
CD8+ T cell responses can also be achieved by engineering stimuli-responsive polymeric nanoparticles to facilitate controlled release of encapsulated antigens in the endolysosomal compartment [[94], [95], [96], [97]]. The endosomal-lysosomal system is known to undergo acidification (to pH ~5.5, which is lower than the neutral pH in cytoplasm) to enable acidophilic hydrolases and other enzymes within the system to function [98]. Such conditions may produce an environment favorable to oxidation reactions as well. Taking advantages of these conditions, pH-responsive and oxidation-sensitive polymeric nanoparticles were developed to promote antigen release upon changes in pH [[94], [95], [96]] and in oxidative environment [99], respectively, resulting in antigen processing that induced CTL responses.
Polymeric nanoparticles designed to mimic viruses or bacteria in size [100] and surface antigenicity [[101], [102], [103], [104]] have been reported to elicit CD8+ T cell responses. Sub-200 nm polymer nanoparticles coated directly with cancer-cell membranes promoted cross-presentation the entire tumor antigen repertoire by DCs inducing expansion of tumor-antigen specific CD4+ and CD8+ T cells [101,102]. Other polymeric nanoparticles of similar sizes but synthetically displayed antigens with controlled antigen orientation have also been reported to enhance antigen cross-presentation [103,104].
Particulate pattern recognition receptor (PRRs) agonists
The present of molecular components unique to pathogens, like bacterial lipopolysaccharides, viral double-stranded RNA, or fungal cell wall components, provides “danger signals” to the innate immune system [105]. These pathogen components can be recognized by pattern recognition receptors (PRRs) expressed on many innate immune cells, including macrophages and DCs. The binding of danger signals to PRRs instructs the immune cells to produce pro-inflammatory cytokines and chemokines and to upregulate CD80 and CD86 maturation markers, inducing antigen-specific adaptive immune responses and functional differentiation of T cells [34,45]. Several PRRs located at different cellular domains have been intensively investigated, including (i) Toll-like receptors (TLRs) consisting of TLR3, TLR7, TLR8 and TLR9 located at the endosome, as well as TLR1, TLR2, TLR4 and TLR6 located at the cell membrane; (ii) nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) comprising NOD1 and NOD2 located at cytoplasm; (iii) retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) located in the cytosol; and (iv) stimulator of interferon genes (STING) located at the endoplasmic reticulum.
The hydrophobicity of polymeric nanoparticles served as danger signal [106], with increasing degree of hydrophobicity resulting in higher levels of cellular internalization and CD86 expression in DCs [107,108]. In addition to their intrinsic danger signal, polymeric nanoparticles have been designed to incorporate synthetic PRR agonists to enhance the pathogen-like qualities of nanoparticle vaccines, thus improving the strength and quality of adaptive immune responses. Synthetic PRR agonists can be encapsulated into polymeric nanoparticles driven by electrostatic, hydrophilic, or hydrophobic interactions (Fig. 3 A(a)), including polyinosinic:polycytidylic acid (poly(I:C)) (TLR3 agonist) [109], oligodeoxynucleotides with unmethylated CpG motif (CpG-ODN) (TLR9 agonist) [110], cyclic dinucleotide (CDN) [97] and cyclic diguanylate monophosphate (cdGMP) [96] (STING agonist), CL235 (NOD1 agonist) [111], CL365 (NOD2 agonist) [111], monophosphoryl lipid A (MPLA) (TLR4 agonist) [112], and imidazoquinoline compounds like imiquimod [113] and gardiquimod [114] (TLR7 agonist) as well as resiquimod [115](TLR7/8 agonist). Polymeric nanoparticles can also be used to display synthetic PRR agonists on their surfaces (Fig. 3A(b)): (i) CpG-ODN [112] or 5′-triphosphorylated stem-loop RNA (SLR) (RIG-I agonist) [116] through electrostatic interaction; (ii) biotinylated CpG-ODN bound via affinity interaction with avidin functionalized nanoparticles [117]; and (iii) CpG-ODN that was covalently conjugated through pyridyl disulfide-thiophosphate coupling [118,119]. Such polymeric particulate PRR agonists have been developed to target different TLRs that are located at cell membrane and endosome (Fig. 3B). It is necessary after the cell uptake that the associated agonists escape from the endosome into the cytosol to bind RLRs, NLRs or STING (Fig. 3B).
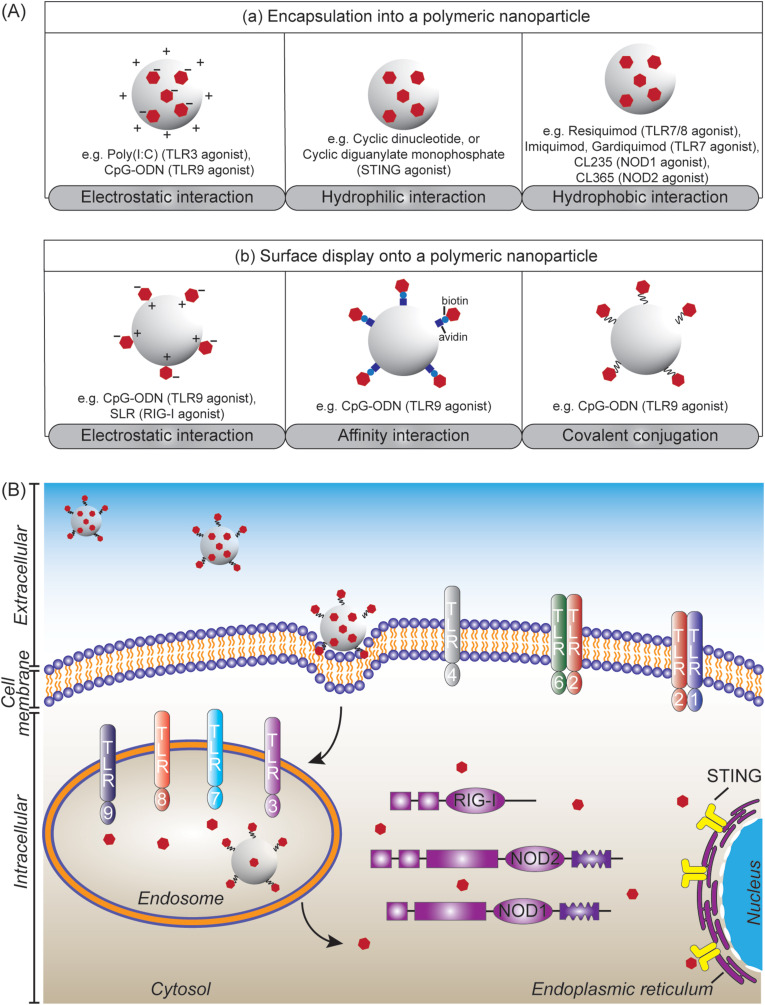
(A) Modes of presentation of pattern-recognition receptor (PRR) agonists (depicted as red-filled hexagon) to polymeric nanoparticles: (a) by encapsulation and (b) by surface display, achieved through physical and chemical interactions. (B) Polymeric nanoparticles incorporated with PRR agonists target PRRs that are located at different cellular domains to shape adaptive immune responses. The polymeric nanoparticles can target TLR1, TLR2, TLR4, and TLR6 at cell membrane. Cellular uptake of the nanoparticles leads to signalling endosomal TLRs including TLR3, TLR7, TLR8, and TLR9. Escaping the endosomes is required to target RIG-I, NOD1 and NOD2 at cytosol, as well as STING at endoplasmic reticulum. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The inclusion of molecular adjuvants within nanoparticles, as opposed to free molecular adjuvants without nanoparticles, would prevent their systemic distribution that could trigger undesirable reactogenicity and cytotoxicity. In addition, the nanoparticles facilitate the molecular adjuvants to be protected from degradation [120], enabling slow release, hence, prolonging exposure (i.e. depot effects) [121], be delivered to and internalized by DCs [63,109,122], and be translocated to intended cell compartments through stimuli-responsive releases [96,121]. Activation of PRRs using particulate PRR agonists has been demonstrated to shape adaptive immune responses. For example, most TLR activation results in stimulation of Th1-based adaptive immune responses, while endosomal TLR (i.e. TLR3, TLR7, TLR8, and TLR9) signalling enhances antigen cross-presentation and elicits CD8+ T cell responses [45]. Mice immunized with dextran particles encapsulating STING agonist resulted in 50-fold enhancement of type I interferon (IFN) including IFN-β, as compared to the free STING agonist [123]. Type I IFN has been reported to enhance DC activation [124] and T cell priming [125], as well as inducing B cell activation and Ig class switching [126]. As a result, the increased IFN-β in mice immunized with particulate STING was followed by 104-fold increase in the antibody titers and Th1-biased responses, hence, CD8+ T cell responses [123].
Polymer based delivery vehicles and their immunological properties
There are various polymeric structures that have been developed as vaccine delivery systems, including solid polymeric nanoparticles, micelles, nanogels, polymersomes, and core-shell nanoparticles (Fig. 4 A). A variety of antigens can be encapsulated into the core or displayed onto the surface of those polymeric structures or both (Fig. 4B) resulting in various antigen-loaded polymeric structures with a wide range of sizes (Fig. 4C). These include antigen-encoding nucleic acids (RNA or DNA), as well as antigens based on lipids, carbohydrates, peptides and proteins, or even bacterial lysates or inactivated viruses (Table 2 ). In addition, molecular adjuvant targeting PRR can be incorporated along with antigens in an individual polymeric structures in order to mimic pathogens and enable their codelivery to the same APCs, aiming for enhancing specific immune responses [96]. Therefore, the design and utility of polymeric structures have been an area of intense and expanding research in efforts to control emerging infectious diseases caused by influenza virus [127], Middle East Respiratory Syndrome Coronavirus [96], Zika virus [128], Ebola virus [129], Dengue virus [130], human immunodeficiency virus [92], Mycobacterium tuberculosis [131], etc. (Table 2).
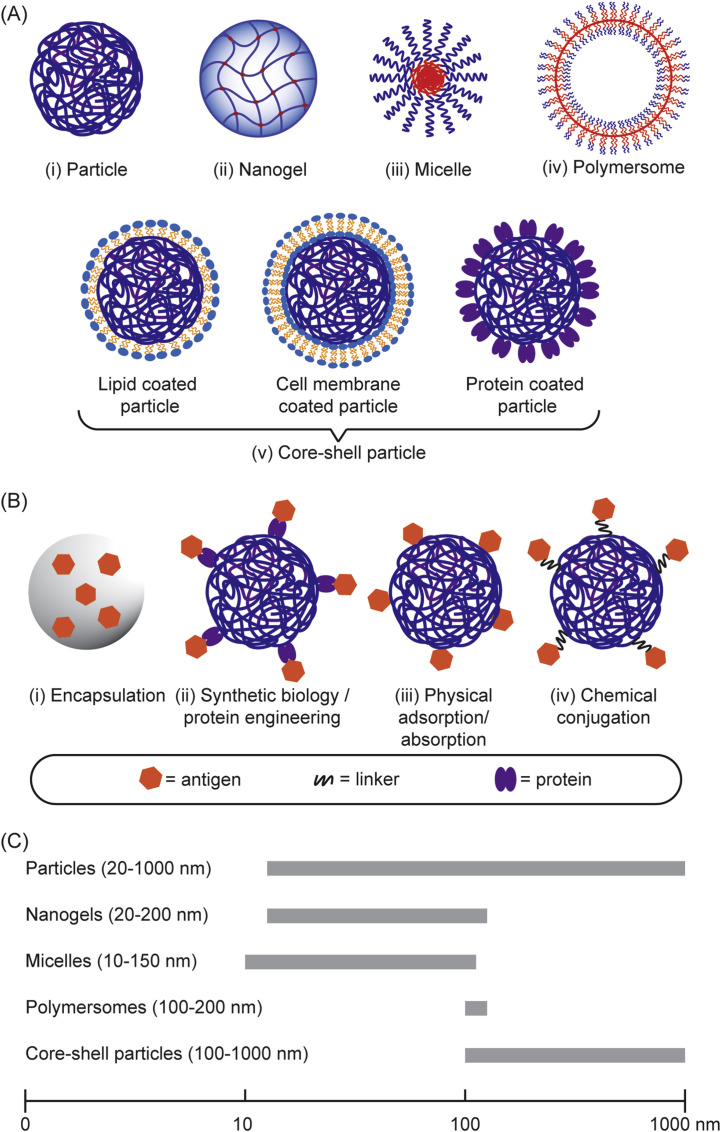
(A) Different structural features of particulate polymers used for vaccine development. (B) Assembly/loading of antigens into the particulate polymers. (C) The size range of the polymeric nanostructures used in vaccine development.
| Vaccine platform | Antigen | Adjuvant | Admin route | Organism model | Humoral response | T cell response | Other attributes | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| CD4+ Th cells | CD8+ T cells | ||||||||
| Viral Infection | |||||||||
| H5N1 avian influenza virus | |||||||||
| PLGA nanoparticle | HA | PLGA nanoparticle encapsulated MPL and R837 | s.c. | Mouse | IgG1, IgG2a, IgG2b, and virus neutralizing antibody titer | Th1 | Strong response | 100% survival and maintained body weight were achieved for 14-days post-infection. The antigen-specific memory CD4+ T cells was persistent for 1.5-year post-vaccination. | [132] |
| PA nanoparticle | H5 HA trimer | Pentablock copolymer hydrogel | s.c. | Mouse | Sustained virus neutralizing antibody titer for up to 70 days | N/E | N/E | Significant reduction of viral load in the lungs of mice, with maintained body weight similar to healthy, noninfected mice | [143] |
| H1N1 swine influenza virus | |||||||||
| PS-core protein-shell nanoparticle | HA | – | i.v. | Mouse | IgG, IgG1, IgG2a and HA inhibition titers | Th1 | Strong response with CTL activity | Protection against H1N1 virus for up to 16 days post-infection as indicated from high survival rate and minimum body-weight loss | [104] |
| pABOL nanoparticle | saRNA encoding for HA | – | i.m. | Mouse | HA IgG, HA inhibition, and virus neutralization titers | N/E | N/E | The mice were protected against the H1N1 challenge as reflected from 8% decrease of the body weight as compared to naïve mice which lost >25% of body weight | [127] |
| Middle East Respiratory Syndrome Coronavirus (MERS-CoV) | |||||||||
| Hollow-core PLGA-shell nanoparticle coated with lipid | Receptor binding domain protein | cdGMP | s.c. | Mouse | IgG, IgG1, IgG2a (for 300 days post-vaccination), with the virus neutralizing titer | Balanced Th1/Th2, with persistent central memory CD4+ T cell responses (for 28-day) | Strong and peptide specific responses, with CTL activity | Significant reduction of virus load titers in the lungs after the challenge, with 100% survival for 24-days post-infection | [96] |
| Zika virus (ZIKV) | |||||||||
| Chitosan/γ-PGA nanoparticle | Inactivated Zika virus | – | s.c. | Mouse | IgG, IgG1, IgG2a | Balanced Th1/Th2 | Strong response | The produced antibody was able to neutralize Zika virus infection | [128] |
| Ebola virus (EBOV) | |||||||||
| PAA-PPI dendrimers and lipid-PEG nanoparticle | VEEV replicon RNA for the EBOV glycoprotein | – | i.m. | Mouse | IgG | Th1 | Strong response with the production of IFN-γ and IL2 | After 23-days post-infection, the mice immunized with a single dose of the polymeric vaccines achieved 100% survival rate | [129] |
| Dengue virus (DENV) | |||||||||
| PLGA nanoparticle | Tetravalent E protein | – | s.c. | Mouse | IgG, IgG1, IgG2a, and virus neutralizing antibody titer | Balanced Th1/Th2 | N/E | A balanced serotype-specific antibody response was stimulated to each DENV serotype (DENV1, DENV2, DENV3 and DENV4) | [130] |
| Human immunodeficiency virus (HIV) | |||||||||
| Mannosylated PEI nanoparticle | Plasmid DNA encoding 15 protein antigens | – | t.d. | Human (Phase I, Phase I/II) | – | Th1 | Strong response with the production of IFN-γ and IL2, and CTL activity | HIV-specific precursor/memory T cells with high proliferation capacity was expanded in a dose-dependent manner at week 48 post-immunization | [144,145] |
| PLGA microparticle | Plasmid DNA encoding Gag and V2-deleted gp140 Env | MF59 during boosting | i.m. | Human (Phase I) | Strong neutralization antibody against the homologous HIV, but minimal neutralization breadth against the heterologous HIV | Th1 | Minimal response | Polyfunctional CD4+ T cell responses were elicited, comprising of IFN-γ (most dominant), IL2, TNF-α and IL-4 | [135] |
| PEI-cyclodextrin nanoparticle | mRNA for gp120 glyco-protein | – | i.n. | Mouse | IgG, IgG1, IgG2a, and secreted IgA in distal mucosa | Balanced Th1/Th2/Th17 | Strong response with cytotoxic T-lymphocyte activity | The nanoparticles could facilitate antigen delivery through intra- and paracellular pathways, inducing both systemic and mucosal immune response | [92] |
| PLGA nanoparticle | HIV-1 p24-Nef fusion peptide | Recombinant FLiC protein (TLR5 agonist) | i.d. | Mouse | IgG, IgG1 and IgG2a | Th1 and Th2 | Strong response with cytotoxic T-lymphocyte activity | Lowering the immunization dose significantly increased the Th1 cytokine and slightly decreased the humoral response | [136] |
| CS/DS, or CS/HA nanoparticles | PCS5 peptide antigen | Poly(I:C) (TLR3 agonist) | i.m. | Mouse | IgG | Th1 | Strong response | Central and effector memory CD4+ and CD8+ T cells were generated | [146] |
| Hepatitis B Virus (HBV) | |||||||||
| CS/γ-PGA nanogels | HBsAg | – | i.m. | Mouse | IgG | Th2 | Strong response with induction of effector memory CD8+ T cells | Single dose vaccination of cationic CS/γ-PGA nanogels cleared HBsAg and restored IgG production after plasmid challenge | [147] |
| CS nanoparticle | HBsAg | – | i.p. | Mouse | IgG, IgG1, IgG2a, IgG2b, IgG3 titers in serum, spleen and bone marrow | Th1 and Th2 | N/E | The humoral and cellular response were durable up to 30-weeks after single-dose vaccination, with increased in BAFF-R + B cells, CD138+ plasma cells, and Tfh cells | [133] |
| PLA-core lipid-shell microparticle | HBsAg | – | i.m. | Mouse | IgM, IgG, IgG1 and IgG2a | Th1 | Strong response with CTL activity | Granzyme B, the effector of cytotoxic T cell, was also produced | [148] |
| Hepatitis C Virus (HCV) | |||||||||
| PHB-core protein-shell particle | HCV core protein | CFA or Emulsigen | s.c. | Mouse | IgG1 and IgG2c | Th1 | N/E | Strong cytokine profiles, including IFN-γ, TNF-α, IL-17A, IL-2, IL-6, and IL-10 as compared to the respective soluble controls | [149] |
| PHB-core protein-shell particle | HCV core protein | Alum | i.m. | Mouse | IgG | Th1 | N/E | Reduction of virus load titer in ovaries after the challenge | [150] |
| Bacterial infection | |||||||||
| Mycobacterium tuberculosis | |||||||||
| PEG–PPS micelle | Mycolid acid (MA) – a lipid antigen | – | i.n. | Mouse | Anti-CD1b antibody titers | Th1 | N/E | The nanoparticles were primarily taken up by alveolar macrophages and DCs in the lung | [151] |
| PHB-core protein-shell particle | Mycobacterial fusion peptides Ag85B–TB10.4–Rv2660c | DDAB | s.c. | Mouse | Antigen-specific antibody titers, dominated by IgG1 | Th1, Th2 and Th17 | N/E | Strong production of cytokines, IFN-γ, TNF-α, IL-17A, IL-2, IL-6, IL-10, as compared to the respective soluble controls | [131] |
| Mycobacterium paratuberculosis | |||||||||
| PA nanoparticle | M. paratuberculosis culture filtrate | – | s.c. | Mouse | N/E | Th1 | Strong responses both post-vaccination and post-challenge | The bacterial load in spleen, liver, small intestine and mesenteric lymph node was reduced. | [152] |
| Mycobacterium bovis | |||||||||
| PHB-core protein-shell particle | Mycobacterial fusion proteins, Ag85A–ESAT-6 | Emulsigen | s.c. | Mouse | IgG | Th1 and Th17 | N/E | Reduction of bacterial count in the spleen and the lung after the challenge | [153] |
| Staphylococcus aureus (resistant to the antibiotic, methicillin) | |||||||||
| PLGA-core red blood cell-shell nanoparticle | α-hemolysin (Hla) protein | – | s.c. | Mouse | Anti-Hla IgG titer for up to 35 days, with germinal center formed | N/E | N/E | Minimum skin lesion area and reduced bacterial load in the skin. Reduced bacterial load was also observed in major organs (heart, kidney, spleen, lung and liver) after the challenge | [154] |
| PLGA-core red blood cell-shell nanoparticle | Combination of α-toxin, PVL, and γ-toxin | – | s.c. | Mouse | Anti-α-toxin, anti-PVL, and anti γ-toxin, with germinal center formed | N/E | N/E | Minimum skin lesion area, and reduced bacterial load at heart, lung and kidney after the challenge | [155] |
| Pseudomonas aeruginosa | |||||||||
| PLGA-core macrophage-shell nanoparticle | Combination of FliC, OprM, OprE and SSB | – | s.c. and i.n. | Mouse | IgG, with germinal center formed. | N/E | N/E | After i.n. injection, both systemic and mucosal immunities were elicited. Reduced bacterial load in lung for both s.c. or i.n. vaccination. | [138] |
| PHB-core protein-shell particle | Fusion antigenic epitopes AlgE, OprF and OprI | Alum | s.c. | Mouse | IgG1 and IgG2c, with opsonophagocytic antibody titer | Th1 | N/E | Without the adjuvant, Th1 immune response can be induced | [156] |
| Streptococcus pneumoniae | |||||||||
| cCHP nanogel | PspA | – | i.n. | Rhesus Macaque | PspA-specific bronchoalveolar fluid IgG and nasal wash IgA antibodies, with neutralizing antibody titer | Th2 and Th17 | N/E | The mice injected intraperitoneally with the pooled sera of macaques nasally immunized with the nanogels were protected from the challenge for at least 2 weeks | [157] |
| PHB-core protein-shell particle | Ply and 19F CPS | – | s.c. | Mouse | IgG with the dominant IgG1 and IgG2b, and opsonophagocy-tic antibody titer | Th2 | N/E | The IgG was persistent for up to 6 months and recognized Ply in whole cell lysates of six different S. pneumoniae serotypes | [158] |
| PHB-core protein-shell particle | PsaA | – | s.c. | Mouse | IgG with the dominant IgG1 and IgG2b | Th2 | N/E | The elicited IgG recognized PsaA in whole cell lysate of seven different serotypes of S. pneumoniae | [159] |
| Neisseria meningtidis | |||||||||
| PHB-core protein-shell particle | NadA and MenC | – | s.c. | Mouse | IgG with the dominant IgG1 and IgG2b | Th1 and Th17 | N/E | The serum exhibited bactericidal activity | [160] |
| Parasitic infection | |||||||||
| Plasmodium yoelii | |||||||||
| PS nanoparticle | MSP 4/5 | – | i.d. | Mouse | IgG, IgG1, IgG2a, IgG2b | Th1 and Th2 | N/E | Moderate survival rate of the immunized mice against the blood-stage malaria infection was demonstrated | [161] |
| Plasmodium falciparum | |||||||||
| PLGA nanoparticle | Pfs25 | – | s.c. | Rhesus macaque | IgG | Th1 | Strong response | •The T cell response increased the antibodies' avidity •The numbers of Pfs25-specific plasmablasts, circulating memory B cells, and plasma cells in the bone marrow were increased. | [162] |
| Plasmodium vivax | |||||||||
| PLGA-core lipid-shell nanoparticle | VMP001 | MPLA | s.c. | Mouse | IgG, IgG1, IgG2b, IgG2c, IgG3, with germinal center formed | Balanced Th1/Th2 | N/E | The antibodies had high avidity that could agglutinate live sporozoites 6-month after the vaccination | [163] |
Abbreviation: N/E, not evaluated.
1. Vaccine platform: cCHP, cholesteryl group-bearing pullulan; CS, chitosan; DS, dextran sulfate; HA, hyaluronic acid; PA, poly(anhydride); PAA-PPI, poly(amido amine)-poly(propylenimine); pABOL, poly(N,N-cystaminebis(acrylamide)-co-4-amino-1-butanol); PAS, poly(acrylic starch); PEG, poly(ethylene glycol); PHB, poly(3-hydroxybutyric acid); PLA, poly(lactic acid); PLGA, poly(lactic-co-glycolic acid); PS, poly(styrene).
2. Antigens: CPS, capsular polysaccharide; ESAT-6, early secreted antigenic target 6-kDa protein; Flagellin, FliC; HA, hemagglutinin; HBsAg, hepatitis B surface antigen; MenC, capsular polysaccharide from serogroup C; MSP 4/5, Recombinant merozoite surface protein 4/5; M2e, matrix protein 2 ectodomain; NadA, Neisseria adhesin A; NP, nucleoprotein; OprM and OprE, Outer membrane proteins; PA, polymerase protein; PCS5, protease cleavage site 5; Pfs25, a glycophosphotidylinositol-linked protein expressed on the ookinetes surface; Ply, Pneumolysin; PsaA, Pneumococcal surface adhesin A protein; PspA, Pneumococcal surface protein A; PVL, Panton–Valentine leucocidin; saRNA, self-amplifying RNA; SSB, single-stranded DNA binding protein; VEEV replicon RNA, Venezuelan equine encephalitis virus replicon RNA; VMP001, Vivax malaria protein – a recombinant antigen derived from the circumsporozoite protein.
3. Adjuvant: cdGMP, cyclic diguanylate monophosphate; CFA: complete Freund's adjuvant; CpG ODN, CpG oligodeoxynucleotide; DDAB, dimethyl dioctadecyl ammonium bromide; MPLA, monophosphoryl lipid A; poly(I:C), polyinosinic:polycytidylic acid.
4. Route of administration: i.d., intradermal; i.m., intramuscular; i.n., intranasal; i.t., intratracheal; p.o., oral; s.c., subcutaneous; t.d., transdermal.
Loading of these antigens can be carried out during particle synthesis (pre-loading) or after the particles have been formed (post-loading) via various ways depending on the polymer type and properties of the antigens (e.g. hydrophobicity or charges). In pre-loading methods, antigen loading is achieved through emulsification [132], polymerization [133] or ionic complexation [127], which result in the antigen encapsulation within polymeric matrices upon particle formation (Fig. 4B(i)). Another way for pre-loading of antigens is through synthetic biology or protein engineering using biopolymeric particles (Fig. 4B(ii)), in which the peptide or protein-based antigens are fused to the protein that mediates the self-assembly of biopolymeric particles, leading to the particle formation displaying the fusion proteins (Fig. 4A(v, right)) [42,44]. In post-loading methods, preformed particles are used to adsorb antigens to the particle surfaces driven by hydrophobic forces [134], electrostatic interactions [135], or covalent conjugation through thiol-maleimide group reaction [96] or NHS (N-hydroxysuccinimide)/EDC (carbodiimide) crosslinking reaction [136] (Fig. 4B(iii)). In addition, polymeric particles coated with cell membranes derived from red blood cells [137] or macrophages [138] (Fig. 4A(v, middle)) have been explored to enable absorption of biological toxin-based antigens due to their natural affinity towards the cell membranes (Fig. 4B(iii)).
Overall, the pre-loading methods facilitate simple manufacturing as particle synthesis and antigen loading occur simultaneously, and ultimately protect the loaded cargo avoiding burst release. However, organic solvents, elevated temperature or shearing forces that are utilized during particle synthesis may adversely affect the structural integrity of the loaded protein antigens. Also, cytotoxic components such as cationic surfactants that are sometimes used for chemical-based particle synthesis [139] should be removed carefully after antigen-loaded particles are formed, as their harsh removal may pose adverse effects on the antigen functionalities. It should also be noted that different modes of antigen loading have been demonstrated to result in different immune responses [[140], [141], [142]]. For example, lipid-coated polymeric nanoparticles containing either encapsulated ovalbumin antigens (OVA) or both encapsulated and physically adsorbed OVA elicited significantly higher OVA-specific immune responses (IgG, IFN-γ, Th1 dominant immune response) as well as higher numbers of activated B cells, CD4+ T cells, CD8+ T cells, and central memory and effector T cells than the OVA-adsorbed nanoparticles and free OVA [140]. It was proposed that physically adsorbed OVA was undergone initial burst release after injection, whereas encapsulating OVA in the nanoparticles created an antigen depot at the injection site by providing more sustained antigen release, hence, ensuring long-term antigen exposure, as well as facilitated delivery to cytosols following DCs uptake where cross-presentation occurred.
This section discusses the synthesis of the polymeric nanostructures, including the modes of loading/assembly of antigens and/or molecular adjuvants into the particulate polymers, as well as the immune responses associated with their deliveries in animal models.
Polymer particles
Natural polymer particles
Natural polymers, like chitosan and alginate, are attractive sources to synthesize particle-based vaccine delivery vehicles (Table 2) due to their abundance in nature, biocompatibility, biodegradability, and mucoadhesive characteristics [164]. Chitosan, also known as β(1–4)-linked 2-acetamido-2-deoxy-β-d-glucose (N-acetyl glucosamine), is a cationic polysaccharide, whereas alginate is an anionic polysaccharide composed of β(1–4)-linked d-mannuronic acid and α-l-guluronic acid units. Particulate formulation loaded with antigens using either chitosan or alginate is generally synthesized based on ionotropic gelation and self assembly of the polyelectrolytes without the use of organic solvents or high shear stress, thus preserving immunogenicity of the antigens [146,165].
As chitosan is only soluble in acidic media with pH < 5 due to its high density of acetylated monomers, chitosan can be deacytelated, and its increasing degree of deacetylation is correlated to the increasing cationic density and hence solubility [41]. Another way to increase solubility of chitosan is by modifying its amino groups, forming trimethyl chitosan (TMC) [166], chitosan lactate [167], or oleyl-carboxymethy-chitosan (OCMCS) [168]. This cationic property of chitosan makes it easier to encapsulate anionic antigens [146,169] and nucleic acids [167], and other negatively charged biopolymers [170]. Additionally, due to their mucoadhesiveness, chitosan based nanoparticles have been used for mucosal delivery [166,[168], [169], [170], [171]], which demonstrated enhanced DC uptake as well as promoted the opening of tight epithelial cell junctions in the mucosal tissues for paracellular transport of the antigen [172]. For example, intranasal administration of TMC nanoparticles containing lipopeptide antigens against group A Streptococcus (GAS) showed enhanced humoral immunity in mice, producing antibodies with high opsonic activity against clinical GAS strains [166]. In addition to ionic complexation for antigen loading, antigens can be chemically conjugated to chitosan aiming for providing more stable bonds and thus preventing premature antigen release – a strategy recently pursued for vaccine development against HIV [146]. In this study, the HIV protease cleavage site peptide antigen (PCS5) (used to cleave specific proteins critical for the HIV virion maturation [173]) was either (i) entrapped in the chitosan/dextran sulfate (DS) complexes (Fig. 5 A), or (ii) conjugated to cationic chitosan and then complexed with anionic dextran sulfate (DS) and poly(I:C) (TLR3 agonist, a molecular adjuvant) to form nanoparticles (Fig. 5B). PCS5 was also conjugated with hyaluronic acid (HA) through cleavable bonds prior to complexation with chitosan and poly(I:C) (Fig. 5C), which can be cleaved in the presence of glutathione (a cysteine-glutamic acid-glycine tripeptide present in the cytosol of cells) promoting antigen release [146]. Intramuscular immunization of mice with each nanoparticle formulation generated significant anti-PCS5 IgG titers for up to 16 weeks as compared to unimmunized mice (Fig. 5D). Of note, the nanoparticles with conjugated PCS5 elicited high proportions of memory CD4+ and CD8+ T cells, as well as IL-2- and TNF-α-producing CD4+ and CD8+ T cells [146]. Chitosan based nanoparticles have also been developed as antitumor vaccines due to their abilities to elicit significant adjuvant effect by stimulating innate immune responses [174,175]. This was achieved by encapsulating tumor cell lysates as the antigens generated from B16 melanoma cells and modifying the nanoparticle surfaces with mannose for DC targeting [176]. It was demonstrated that the nanoparticles promoted DC maturation and augmented anti-tumoral cellular immune responses in mice.
![Polysaccharide nanoparticles for vaccine development against human immunodeficiency virus (HIV). (A–C) Three nanoparticle vaccine formulations formed based on complexation between oppositely charged polysaccharides containing HIV protease cleavage site peptide antigen (PCS5) and/or molecular adjuvant poly(I:C): (A) complexes between chitosan (CS) and dextran sulfate (DS) entrapping PCS5 (diameter, ~119 nm); (B) complexes between PCS5-conjugated CS and DS entrapping poly(I:C) (diameter, ~141 nm); and (C) complexes between CS and PCS5-conjugated hyaluronic acid (HA) entrapping poly(I:C) (diameter, ~211 nm). (D) Humoral immune responses in mice following intramuscular injection with each of the nanoparticle groups (50 mice per group) as compared to that in nontreated naïve mice. Arrows indicate the time of vaccination. Reproduced with permission from Ref. [146]. Copyright (2019) American Chemical Society.](/dataresources/secured/content-1765799170241-d106e8c1-f09f-4b76-8335-3dc3d2fa44f7/assets/gr5_lrg.jpg)
Polysaccharide nanoparticles for vaccine development against human immunodeficiency virus (HIV). (A–C) Three nanoparticle vaccine formulations formed based on complexation between oppositely charged polysaccharides containing HIV protease cleavage site peptide antigen (PCS5) and/or molecular adjuvant poly(I:C): (A) complexes between chitosan (CS) and dextran sulfate (DS) entrapping PCS5 (diameter, ~119 nm); (B) complexes between PCS5-conjugated CS and DS entrapping poly(I:C) (diameter, ~141 nm); and (C) complexes between CS and PCS5-conjugated hyaluronic acid (HA) entrapping poly(I:C) (diameter, ~211 nm). (D) Humoral immune responses in mice following intramuscular injection with each of the nanoparticle groups (50 mice per group) as compared to that in nontreated naïve mice. Arrows indicate the time of vaccination. Reproduced with permission from Ref. [146]. Copyright (2019) American Chemical Society.
Sodium alginate is the form that is typically used in biomedical applications and is water soluble salt of alginic acid. Alginate is commonly used: (i) for protection of encapsulated antigens from degradative environments in stomach due to acid and protease upon oral delivery; and/or (ii) for controlled release of the loaded antigens, expected to prolong antigen presentation to APCs [94,165,177]. Calcium-alginate nanoparticles were used to encapsulate the attenuated Androctonus australis hector insect toxins to develop vaccines against scorpion envenomation [165]. Rabbit immunized with the nanoparticles showed increased of specific IgG titer and IgG1/IgG2a isotype balance, as well as 100% protective efficacy of all immunized rabbits [165]. For cancer immunotherapy applications, alginate nanoparticles that displayed mannose and model antigen ovalbumin (OVA) were developed [94]. OVA was conjugated to the alginate using pH-sensitive Schiff base bond. While mannose facilitated DC targeting and uptake, the stimuli-responsive nanoparticles promoted controlled antigen release in endosomes and subsequent cytosol delivery. Transdermal administration of the nanoparticles in mice led to DC maturation and CTL activation, and ultimately the inhibition of E.G7 tumor growth in mice [94].
Synthetic polymer particles
Synthetic polymers that are commonly used in biomedical applications include poly(lactic acid) (PLA), poly(glycolic acid) (PGA) and poly(lactic-co-glycolic acid) (PLGA) (recently reviewed in Ref. [42]). Other synthetic polymers that have also been used to develop particulate vaccines are poly(ethylene imine) (PEI), poly(N,N-cystaminebis(acrylamide)-co-4-amino-1-butanol) (pABOL), poly(ε-caprolactone) (PCL) and poly(anhydride) (PAN) will be discussed in this review (Table 2).
PEI is a cationic polymer available in linear or branched form with low or high molecular weight. PEI is capable of forming complexes with nucleic acids through electrostatic interactions (i.e. polyplexes). Increasing concentrations of PEI relative to nucleic acids has been demonstrated to increase the diameter of the resultant polyplexes, ranging from 80 to 450 nm [178]. Polyplexes possess high transfection efficiency, as PEI is able to induce proton-sponge effect [179]. PEI is rich in secondary and/or tertiary amines that could absorb protons during endocytic trafficking, causing more and more protons and associated counter ions to be transported into the endosomes. The increased ion concentration eventually results in osmotic swelling and rupture of the endosomes, allowing direct entry of the polyplexes into the cytosol [93]. In addition, immunostimulatory property of PEI promoted DC trafficking to LNs and induced non-pro-inflammatory cytokine responses [180]. Therefore, PEI complexes with nucleic acids have been developed for deliveries of nucleic acid vaccines, including antigen-encoding DNA [180], messenger RNA (mRNA) [92,181], and self-amplifying replicon RNA (repRNA) [182]. To improve vaccine delivery, PEI can be functionalized with, for example, cyclodextrin [92,181] known able to enhance permeation via nasal epithelium [183]. It has been demonstrated that mice immunized intranasally with polyplexes that were loaded with mRNA encoding HIV gp120 antigen were able to passage across the nasal epithelial barrier and ultimately generated strong mucosal and systemic immune responses in a balanced Th1/Th2/Th17 profile [92]. Mannose can also be used to functionalize PEI enabling DC targeting. Mannosylated PEI complexed with plasmid DNA that encode fifteen different HIV antigens in a single plasmid, which resulted in nanoparticles with sizes ranging from 70 to 300 nm have been developed as therapeutic nanoparticle vaccines for HIV [184,185]. These PEI nanoparticle vaccines so-called DermaVir completed clinical trials Phase I (NCT00712530) [145], Phase I/II (NCT00270205) [144] and Phase II (NCT00711230), and are now under development towards clinical trial Phase III. DermaVir can be applied topically using DermaPrep devices that disrupts stratum corneum layers of the skin to allow nanoparticle penetration and subsequent endocytosis by Langerhans cells (LCs) residing below the stratum corneum layer, leading to maturation of LCs to DCs where antigen expression and cross-presentation occur [185]. Clinical trials of DermaVir demonstrated elicitation of HIV-specific effector CD4+ and CD8+ T cells expressing IFN-γ and IL2 and the dose-dependent expansion of HIV-specific precursor/memory T cells with high proliferation capacity, which were manifested as significant reduction of HIV RNA (up to 70%) as compared to placebo [144,145,185].
The facile loading of nucleic acids within PEI through the formation of polyplexes has motivated the development of other cationic polymers such as pABOL to achieve a more effective delivery of the nucleic acids [127,186]. pABOL could serve as a proton sponge due to its high density of tertiary amines. Unlike PEI, pABOL also contains multiple disulfide linkages at the polymer backbone which can undergo rapid cleavage by glutathione present in the cytosol of cells, leading to biodegradation and intracellular cargo release [186]. pABOL was demonstrated to enable the delivery and expression of self-amplifying RNA (saRNA) encoding firefly luciferase in mice at significantly higher levels than PEI (Fig. 6 ) [127]. Furthermore, a library of pABOL with molecular weight ranging from 5 to 167 kDa was synthesized and developed to encapsulate influenza hemagglutinin (HA) antigen-encoding saRNA, thereby forming nanoparticles with diameters between 100 and 400 nm [127]. Intramuscular immunization of mice with 8 kDa pABOL nanoparticles containing 1 μg HA-encoding saRNA was able to generate high levels of HA-specific IgG (~40,000 ng/mL), HA inhibition titer (~500) and virus neutralization titer (IC50 ~2700) as compared to the PEI and 100 kDa pABOL containing the same dose of saRNA (Fig. 6). Following intranasal challenge with A/California/07/2009 flu virus, mice immunized with the 8 kDa pABOL-saRNA nanoparticles were completely protected with only 8% weight loss at peak viremia, whereas all non-immunized mice died within five days [127].
![Polymeric nanoparticle vaccines against H1N1 influenza virus. The nanoparticles are composed of a complex between linear, cationic polymer poly(N,N-cystaminebis(acrylamide)-co-4-amino-1-butanol (pABOL) and self-amplifying RNA (saRNA) encoding hemagglutinin (HA) antigen from the H1N1 A/California/07/2009 strain. (A) Synthesis of pABOL through aza-Michael polyaddition of 4-amino-1-butanol to N,N-cystaminebis(acrylamide) catalyzed by triethylamine (a), and its subsequent ionic complexation with saRNA, and high transfection efficiency of the formed nanoparticles as compared to poly(ethyleneimine) (PEI) based nanoparticles (b). (B) Typical TEM image of pABOL-saRNA nanoparticles stained with 2% uranyl acetate (scale bar: 100 nm). (C) Immunogenicity of the nanoparticles at different pABOL molecular weights and saRNA doses after intramuscular vaccination of mice: HA-specific IgG titer (a), HA inhibition (HAI) titer of Cal/09 flu virus (b), and neutralization IC50 against Cal/09 flu virus (c). Reproduced with permission from Refs. [127]. Copyright (2020) American Chemical Society.](/dataresources/secured/content-1765799170241-d106e8c1-f09f-4b76-8335-3dc3d2fa44f7/assets/gr6_lrg.jpg)
Polymeric nanoparticle vaccines against H1N1 influenza virus. The nanoparticles are composed of a complex between linear, cationic polymer poly(N,N-cystaminebis(acrylamide)-co-4-amino-1-butanol (pABOL) and self-amplifying RNA (saRNA) encoding hemagglutinin (HA) antigen from the H1N1 A/California/07/2009 strain. (A) Synthesis of pABOL through aza-Michael polyaddition of 4-amino-1-butanol to N,N-cystaminebis(acrylamide) catalyzed by triethylamine (a), and its subsequent ionic complexation with saRNA, and high transfection efficiency of the formed nanoparticles as compared to poly(ethyleneimine) (PEI) based nanoparticles (b). (B) Typical TEM image of pABOL-saRNA nanoparticles stained with 2% uranyl acetate (scale bar: 100 nm). (C) Immunogenicity of the nanoparticles at different pABOL molecular weights and saRNA doses after intramuscular vaccination of mice: HA-specific IgG titer (a), HA inhibition (HAI) titer of Cal/09 flu virus (b), and neutralization IC50 against Cal/09 flu virus (c). Reproduced with permission from Refs. [127]. Copyright (2020) American Chemical Society.
Other synthetic polymer nanoparticles, PCL [103,104,187,188] and PAN [152,189,190], have been synthesized using emulsion methods for vaccine development due to their biocompatibility and biodegradability. Antigens are generally encapsulated within the polymers, taking advantage of the sustained release properties of the polymer nanoparticles, hence promoting sustained antigen availability. Hepatitis B surface antigen was encapsulated into sub-100 nm PCL nanoparticles which were then used as a single-dose oral vaccine [187]. Mice vaccinated orally with the PCL nanoparticles generated significant antigen-specific antibody titers without booster dose for both systemic and mucosal immunity [187]. PAN nanoparticles with diameters of 200 nm were loaded with protein antigens obtained from culture filtrates of Mycobacterium avium subsp. paratuberculosis (MAP) lysates [152]. Subcutaneous administration of the PAN nanoparticles in mice had significantly higher frequency of antigen-specific CD4+ and CD8+ T cells, and conferred improved protection against intraperitoneal MAP challenge, compared with non-immunized mice and Mycopar® (containing whole inactivated MAP)-immunized mice [152].
Polymer nanogels (hydrogel nanoparticles)
Nanogels are three-dimensional spherical-like polymer chains with open network structures (i.e. porosity) [191]. The polymers are characterized by specific chemical groups available within the polymers that drive the polymer network formation through chemical or physical crosslinking. While the former results in strong and rigid connections, the latter tends to give more degradable structures due to the nature of the weaker electrostatic, host–guest, or hydrophobic/hydrophilic interactions. Nanogels possess the characteristics of both hydrogels (i.e. capable of swelling via incorporation of a large amount of water or biological fluids) and nanoparticles (i.e. large specific surface area for payload delivery, enhanced bioavailability of the payloads, and multiple surface functionalization) [192]. There are several ways to form nanogels, including (i) self-assembly [60,121,[193], [194], [195], [196], [197]]; (ii) emulsion polymerization [198,199]; and (iii) particle replication in nonwetting template (PRINT) technology [200]. Synthetic polymers [121,193,195,200], polysaccharides [60,[196], [197], [198], [199]] and their combination [201] have been developed to form nanogels for vaccine applications.
Nanogels formed from synthetic polymers, including poly(ethylene glycol) [200], poly(tri(ethylene glycol)methyl ether methacrylate)–block–poly(pentafluorophenyl methacrylate) (P(mTEGMA)x-b-P(PFPMA)y) [121,193], and poly(hydroxy methacrylate) (PHEMA) [195], have been used for the delivery of molecular adjuvants [121,193] or antigens [195] or both [200]. Functionalization of PHEMA with hydrophobic moieties like pyridine (Pyr) increased the hydrophobicity of the formed Pyr-PHEMA nanogels which, in turn, initiated an innate immune response through TLR2 [195]. This intrinsic adjuvant property of the nanogels is beneficial for development of particulate vaccines as immune responses to antigens can be enhanced without any exogenous adjuvant. The capacity of the Pyr-PHEMA nanogels encapsulating model antigen OVA to modulate humoral immune responses was compared to soluble OVA and OVA-loaded PLGA nanoparticles supplemented with the alum adjuvant [195]. In this study, mice deficient in TLR5 were used as the model animals known to develop metabolic syndrome with poor immune responses. It was demonstrated that the nanogels enhanced trafficking in the draining LNs and expression of MHC II on CD169+ macrophages and CD11c + dendritic cells in mice compared to the soluble OVA. Moreover, the nanogels stimulated TLR2 and induced an increase in GL7+FAS + CD19+ germinal center B cells in mice compared to alum-supplemented PLGA nanoparticles [195]. These germinal center B cells have undergone somatic hypermutation and affinity maturation, along with production of OVA-specific IgG titers.
Nanogels can also be formed using polysaccharides like dextran [198,199], alginate [197], and cholesterol-bearing pullulan and dextran [60,196]. Most polysaccharide based nanogels have been recently developed in efforts to achieve effective intracellular delivery potential for cancer vaccines [60,[196], [197], [198], [199]]. Shen et al. synthesized and developed sub-200 nm alginate nanogels as an oral DNA vaccine by loading with Tat peptide and gp100 encoding plasmid DNA through electrostatic interactions [197]. Tat peptide is a cluster of basic amino acids within the HIV1 transactivation protein, which was found responsible for enabling the protein penetration through cell membranes [202], and has been used to functionalize nanoparticles for mediating intracellular delivery of the nanoparticles [203]. On the other hand, gp100 encoding plasmid DNA prevented metastasis and relapsed melanoma as well as inhibited tumor growth [204]. The in vitro delivery of alginate nanogels containing Tat peptide showed that alginate assisted in mucus penetration, while the Tat peptide facilitated passage through intestinal epithelial cell layer [197]. Further, oral vaccination of mice with the Tat peptide and gp100 plasmid DNA-encapsulated alginate nanogels enhanced secretion of IFN-γ and activation of CTL, led to B16F10 tumor inhibition growth.
Polymer micelles
Amphiphilic polymers are able to undergo self-assembly in an aqueous solution, forming particulate micellar nanosystems with diameters ranging from 13 nm [205] to 150 nm [206]. The micelles consist of a hydrophobic polymer core conjugated to a hydrophilic polymer constituting the shell, which have been utilized either as an adjuvant [207] or as a nanocarrier for antigens [151,206,[208], [209], [210]] as well as for co-delivery of molecular adjuvants and antigens [[211], [212], [213], [214]]. The hydrophilic polymer shells are mostly formed using PEG to provide colloidal stability, as well as to reduce retention at the injection site and improve lymphatic trafficking to the draining LNs [215]. To enhance cellular internalization, functionalized micelles can be synthesized (i) by conjugating amphiphilic polymer precursors with positively-charged polyarginine [205], or (ii) by combining two different amphiphilic block copolymers as the micelle precursor, e.g. poly(ε-caprolactone) (PCL)–PEG and PCL–poly(ethyleneimine) (PEI), in which PEI is used to impart positive surface charges to the formed micelles [216]. While hydrophobic cargoes can be easily entrapped into the micelle core [151,205,209,212], it is necessary to conjugate hydrophilic payloads with hydrophobic moieties, such as palmitic acid [211], to enable their encapsulation within the core during micelle formation (pre-loading). Another way to incorporate hydrophilic cargoes is by noncovalent conjugation onto the surfaces of preformed micelles (post-loading) [205,208,210,214].
The antigen-delivery systems based on polymeric micelles have been developed against bacterial pathogens [151,209]. Shang et al. loaded mycolic acid, one of the lipid components of Mycobacterium tuberculosis, into PEG–PPS micelles [151]. In contrast to free mycolic acid, intranasal immunization of mice using the micelles induced potent CD1b-restricted, mycolic acid-specific T cell responses in the lungs of mice.
Polymeric micelles have also been used for anti-tumor vaccines due to their sub-100 nm sizes which would facilitate passive targeting to the LNs [208,[211], [212], [213], [214]]. LNs are critical targets of cancer vaccines as antigen presentation and initiation of T-cell mediated immune responses occur mainly at these sites [217]. Li et al. prepared sub-60 nm polymer micelle nanocarriers as an anti-tumor vaccine with the OVA peptide displayed on the surfaces and the TLR7 agonist CL264 encapsulated within the micelles [214]. Mice immunized with these micelles generated balanced Th1 and Th2 immune responses and were protected against challenge with E.G7 cells expressing OVA.
Polymersomes (aqueous-core polymeric nanoparticles)
Polymersomes are a nanostructured polymeric vesicles with sizes ranging from 100 to 200 nm, and consisting of an aqueous core encapsulated by a polymeric bilayer or complicated interdigitated membrane structures [218]. The aqueous interior allows facile loading of hydrophilic payloads including antigens and molecular adjuvants without the need for prior chemical modifications, while the polymeric bilayer can retain hydrophobic molecular adjuvants. Such nanostructure is reminiscent of liposomes but is made based on the self-assembly of amphiphilic block copolymers instead of lipids. PEG is usually used as the hydrophilic block to prevent nonspecific interactions with cells or proteins, whereas the hydrophobic block includes poly(propylene sulfide) (PPS) [99,[219], [220], [221]] and poly(dicholorophosphazene) (PDCP) [95]. In general, synthesizing polymersomes requires the accessible hydrophilic block fraction (f hydrophilic) to be about 35 ± 10% of the block copolymer (as compared to micelles that are obtained at f hydrophilic > 0.50) [222], but this also depends on the molecular weight, concentration, and geometry of the polymers [218]. Similar to liposome preparation methods, polymersomes can be synthesized using methods like thin film rehydration [99,[219], [220], [221]] and flash nanoprecipitation [223]. For some block copolymers that cannot be directly hydrated as in lipids due to high glass-transition temperature of the hydrophobic block [95,224], such block copolymers can be dissolved in an organic solvent, followed by gradual addition of water to induce the formation of polymersomes with subsequent dialysis to remove the organic solvent.
To enhance antigen presentation and adjuvant stimulation mediated by efficient endosomal escape, stimuli-responsive polymersomes have been developed as a nanocarrier for dual-delivery of antigen and molecular adjuvants [95,97,99]. Oxidation-sensitive polymersomes were prepared based on PPS due to its ability to undergo oxidative conversion from a hydrophobe to a hydrophile (i.e. poly(propylene sulfoxide) and poly(propylene sulfone)) [225]. These polymersomes were used as a vaccine against Mycobacteria tuberculosis by encapsulating Ag85B/p25 antigens in the aqueous core and incorporating molecular adjuvant CL075 (TLR8 agonist) in the polymer layer [99]. CL075 is systematically toxic, and therefore its encapsulation would protect surrounding biological environment upon injection. On the other hand, oxidation-sensitive polymersomes permitted antigen release in an oxidative environment within diverse endosomal compartments where both MHC-I and MHC-II antigen presentation can occur [219,220]. In vitro study of the polymersome trafficking in bone-marrow derived DCs suggested that (i) the polymersomes released encapsulated cargoes like TLR agonists in endosomes, which is beneficial for promoting their interactions with endosome-resident TLRs (e.g. TLR3, TLR7, TLR8 and TLR9) and the subsequent TLR activation enabling improved immune responses, and (ii) the polymersomes facilitated endosomal escape of the encapsulated antigens to the cytosols, which is desirable for antigen processing for MHC I presentation [219]. Mice immunized with these 125-nm polymersomes increased the production of IL-12p70 by four orders of magnitude enhancing Th1-polarized immune responses as compared to the live Bacillus Calmette–Guèrin (BCG) vaccine [99].
The pH-responsive polymersomes were synthesized based on PDCP grafting with diisopropylethylenediamine (DPA) and PEG [95]. The antigen OVA peptide was encapsulated in the 200-nm polymersomes for anti-tumor vaccine development [95]. As pH dropped from neutral to pH 5.5 (e.g. in endosomal compartments), the polymersomes could be perforated and thus promoted permeability, enabling sustained release of the loaded antigens, while additionally allowing extended access of the antigens to the cytosols promoting CD4+ and CD8+ T cell responses [95,226]. In vivo administration of the OVA-loaded pH-responsive polymersomes induced more than 3-fold increase in the IgG2a/IgG1 ratio compared to the antigen alone, along with secretion of IL-2, IL-12 and IFN-γ indicative of a Th1-skewed response. Using an EG.7-OVA T-lymphoma tumor model in mice, it was demonstrated that the polymersome group remained tumor-free 13 d postchallenge, whereas tumors in the saline group progressed to reach tumor volume of 1250 mm3 [95].
Core-shell polymeric particles
Core-shell polymeric particles are composed of a core-forming polymeric particle that is surrounded by a shell formed of lipids, cell-membranes, or proteins. Such a hierarchical structure synergistically combines the merits of polymer cores (physical stability, antigen-encapsulation space, payload for molecular adjuvants and controlled release properties) with the biomimetic characteristics of bacterial or viral surfaces imparted by the shells, expected to enhance antigen-specific immune responses. Therefore, such core-shell particles have emerged as an attractive platform technology for immune modulation (Table 2). In this section, three different types of core-shell polymeric particles shells will be described as follows.
Lipid-coated polymeric particles
Learning the lesson from the lipid-based delivery vehicles like Doxil® approved by the US FDA for human use [227], there have been considerable efforts to develop lipid coated polymeric particles for vaccine applications. A variety of polymer structures has been coated with lipid components, including predominantly solid-core polymeric particles having sizes ranging from micro-to nanometer [65,80,140,148,228,229], and, to a lesser extent, hollow-core polymeric nanoparticles [96] and polymersomes [230]. It has been demonstrated with liposomes that their outer lipid layer's positive charges promoted their adherence to negatively charged cell membranes via ionic interaction, leading to sustained antigen release (i.e. depot effect) [231]. Therefore, many cationic lipid components have been used to coat polymeric particles, including lecithin [80], 3β-[N-(N′,N′-dimethylaminoethane)-carbamoyl] cholesterol (DC-Chol) [140,228], dimethyldioctadecylammonium (DDA) [148,229], and 1,2-dioleoyl-3-trimethylammonium propane (DOTAP) [65,230]. In addition, cationic lipid shells offer facile functionalization through physical adsorption of antigens, molecular adjuvants, and targeting moieties such as hyaluronic acid [65] or mannose [230] that bind to CD44 or calcium dependent (C-type) lectin receptors on DCs, respectively, to enhance immune responses. Similar to the general methods to prepare polymeric structures, lipid-coated polymeric particles can be synthesized based on emulsion/solvent evaporation methods, where the lipid components are added along with the polymer precursor in an oil phase followed by emulsification and then solvent evaporation.
Lipid-coated PLGA nanoparticles have been developed to elicit potent systemic and gastrointestinal immune responses, potential for vaccination against gastrointestinal infections [228]. To achieve this, the PLGA core was loaded with all-trans retinoic acid (atRA) derived from gut-residing DCs known able to produce signals inducing gut-tropism of lymphocytes [232], while the lipid shell was functionalized with both immunostimulatory adjuvant CpG-ODN (TLR9 agonist) and antigen OVA. In this way, atRA could be slowly released expectedly to incur persistent activation of the atRA signal at low concentrations, thus inducing trafficking to the draining LNs rather than triggering gut tropism at the injection site. As compared to the controls (OVA-atRA-CpG and OVA-Alum-CpG), mice immunized with the sub-200 nm nanoparticles showed increased OVA-specific IgG and IgA titers, as well as CTL responses in the spleen and OVA-specific T cell activation in the gut [228].
Hollow PLGA nanoparticles enveloped with lipid having a diameter of 100 nm have been synthesized in efforts to make virus-mimetic vaccines against Middle East Respiratory Syndrome Corona Virus (MERS-CoV) (Fig. 7 ) [96]. These nanostructures facilitated loading of water-soluble molecular adjuvants cdGMP (STING agonist) and surface-display of receptor binding domain (RBD) protein antigens. PLGA nanoshells are relatively easy to be acid-hydrolyzed compared to the solid counterpart, which would be beneficial for unloading cdGMP in acidic endosomal compartments, hence enabling cdGMP to function against STING localized at an endoplasmic reticulum. Mice immunized with these nanoparticles generated long-lasting humoral and Th1/Th2 balanced immune responses with high proliferation of central memory CD4+ T cells and IFN-γ-secreting CD4+ and CD8+ T cell population, as compared to that induced by the control formulations (free RBD antigen admixed with either free cdGMP or MF59 adjuvant). Antibodies raised by the nanoparticle vaccination were able to neutralize live MERS-CoV. To demonstrate the protective efficacy, mice immunized with cdGMP-loaded nanoparticles displaying RBD antigen were completely protected against the MERS-CoV challenge on day 24 post-infection and exhibited significantly reduced viral titers, as compared to the nanoparticles only containing cdGMP (without the antigen) (Fig. 7) [96]. This approach might also be considered for development of a vaccine against the pandemic causing SARS-CoV-2 [233].
![Viromimetic nanoparticle vaccines against Middle East Respiratory Syndrome Coronavirus (MERS-CoV). (A) The nanoparticles are composed of lipid coated PLGA nanoshells that are loaded with the molecular adjuvant cdGMP at the core and conjugated with receptor binding domain (RBD) of the MERS-CoV spike antigen at the surface (a) and the representative cryo-TEM image of the nanoparticles (b). (B) Immunogenicity and protective efficacy of the nanoparticles after subcutaneous vaccination of human DPP4-transgenic mice on days 0 and 21: Titers of 100% neutralizing serum antibody after four weeks of the last administration (a), viral loads in the lungs of immunized mice following intranasal challenge with MERS-CoV EMC/2012 strain quantified using a Vero E6 cell-based assay (b) and quantitative PCR (c), and the survival curve for the challenged mice (d). Reproduced with permission from Ref. [96]. Copyright (2019) John Wiley and Sons.](/dataresources/secured/content-1765799170241-d106e8c1-f09f-4b76-8335-3dc3d2fa44f7/assets/gr7_lrg.jpg)
Viromimetic nanoparticle vaccines against Middle East Respiratory Syndrome Coronavirus (MERS-CoV). (A) The nanoparticles are composed of lipid coated PLGA nanoshells that are loaded with the molecular adjuvant cdGMP at the core and conjugated with receptor binding domain (RBD) of the MERS-CoV spike antigen at the surface (a) and the representative cryo-TEM image of the nanoparticles (b). (B) Immunogenicity and protective efficacy of the nanoparticles after subcutaneous vaccination of human DPP4-transgenic mice on days 0 and 21: Titers of 100% neutralizing serum antibody after four weeks of the last administration (a), viral loads in the lungs of immunized mice following intranasal challenge with MERS-CoV EMC/2012 strain quantified using a Vero E6 cell-based assay (b) and quantitative PCR (c), and the survival curve for the challenged mice (d). Reproduced with permission from Ref. [96]. Copyright (2019) John Wiley and Sons.
Cell membrane-coated polymeric particles
Inspired by the biointerfacing capability of cells that enables them to perform complex biological functionalities, polymeric nanoparticles (mostly PLGA) have been directly coated with cell membranes – the outermost layer of a cell (e.g. red blood cells (RBC), macrophages, or cancer cells), expectedly to replicate the properties of the source cells [[234], [235], [236]]. Such nanostructures can be spontaneously formed by mixing the membranes with the preformed nanoparticles through extrusion or sonication.
PLGA nanoparticles coated with RBC membranes have been developed as a safe “nanotoxoid” vaccine with a size of approximately 100 nm, taking advantage of the natural ability of RBC membranes to entrap bacterial toxins which become deactivated upon complexation into the membrane's lipid bilayer [154,155]. Multiple virulence enhancing toxins secreted from Staphylococcus aureus cultures, including α-toxin, γ-toxin and Panton–Valentine leukocidin, could be simultaneously retained in the RBC membrane-coated nanoparticles to form multiantigenic nanotoxoids [155]. As compared to the traditional heat-treated toxoid, mice vaccinated with the nanotoxoids promoted more germinal center B cell induction and elicited significantly higher IgG titers, which conferred enhanced protection against subcutaneous and intravenous toxin challenges [155].
Macrophage membranes have also been used to coat PLGA nanoparticles as multivalent nanotoxoid vaccines against Gram-negative bacteria Pseudomonas aeruginosa infections, due to their broad interactions with the bacteria (Fig. 8 ) [138]. The nanotoxoids were synthesized by coating PLGA nanoparticles with macrophages, followed by incubation of the macrophage-coated nanoparticles in bacterial culture supernatant to capture P. aeruginosa secretion proteins (PaS) (Fig. 8A). Subcutaneous immunization of mice with the nanotoxoids resulted in higher antigen-specific IgG titers, when compared to blank solution (10% sucrose) (Fig. 8B). After intratracheal bacteria challenges, the immunized mice generated high anti-PaS IgG titer in the lungs with substantial reduction of bacteria counts within the lungs (Fig. 8B) [138]. Intranasally administered nanotoxoids in mice were also elicited high level of anti-PaS IgG titers in sera, with additional anti-PaS secretory IgA and IgG titers in bronchoalveolar lavage fluid, enabling protection of the immunized mice against intratracheal challenge with P. aeruginosa [138].
![Multiantigenic nanotoxoids against Pseudomonas aeruginosa. (A) Synthesis and use of the nanotoxoids: (i) PLGA nanoparticles were coated with macrophages to enable capture, retention and hence neutralization of P. aeruginosa secretions (PaS) within the macrophage; (ii) subcutaneous or intranasal immunization of these nanotoxoids in mice elicited potent humoral immune responses for antibacterial protection against P. aeruginosa. (B) Immunogenicity and protective efficacy of the nanotoxoids as compared to blank solutions (10% sucrose) after subcutaneous vaccination in mice on days 0, 7, and 14: IgG titers in mice sera on day 21 (a), IgG titers in the lungs of immunized mice following intratracheal challenge with P. aeruginosa on day 35 (b), and bacterial loads in the lungs of immunized mice after the challenge (c). Reproduced with permission from Ref. [138]. Copyright (2019) American Chemical Society.](/dataresources/secured/content-1765799170241-d106e8c1-f09f-4b76-8335-3dc3d2fa44f7/assets/gr8_lrg.jpg)
Multiantigenic nanotoxoids against Pseudomonas aeruginosa. (A) Synthesis and use of the nanotoxoids: (i) PLGA nanoparticles were coated with macrophages to enable capture, retention and hence neutralization of P. aeruginosa secretions (PaS) within the macrophage; (ii) subcutaneous or intranasal immunization of these nanotoxoids in mice elicited potent humoral immune responses for antibacterial protection against P. aeruginosa. (B) Immunogenicity and protective efficacy of the nanotoxoids as compared to blank solutions (10% sucrose) after subcutaneous vaccination in mice on days 0, 7, and 14: IgG titers in mice sera on day 21 (a), IgG titers in the lungs of immunized mice following intratracheal challenge with P. aeruginosa on day 35 (b), and bacterial loads in the lungs of immunized mice after the challenge (c). Reproduced with permission from Ref. [138]. Copyright (2019) American Chemical Society.
Cancer cell membrane-coated PLGA nanoparticles are commonly used as a personalized anticancer vaccine. They are generally composed of a PLGA nanoparticle core encapsulating an immunostimulatory adjuvant while an autologously derived cancer cell membrane that already contains diverse tumor antigens forms the shell [101,102]. In addition, mannose can be conjugated to the cell membrane-coated nanoparticles via a lipid anchoring method to enhance the binding and uptake by DCs in the LNs [102]. Prophylactic administration of B16-OVA melanoma cancer cell membrane-coated, R873 (TLR7 agonist)-loaded, mannose modified PLGA nanoparticles increased the frequency of antigen-specific T cells producing IFN-γ and generating CTL activation, which resulted in a significant delay of tumor occurrence 17 d after challenge with the tumor cells compared to the control groups [102].
Protein-coated polymeric particles
In the past five years, there have been several efforts to develop polymeric particles that display protein antigens with controlled orientation and repetitive structures mimicking the surface features of a virus or a bacterium. In general, such particles can be synthesized based on the self-assembly of the polymers either in vitro in bulk solutions or in vivo in engineered bacteria.
Poly(ε-caprolactone) (PCL) has been used for making sub-200 nm nanoparticles in vitro displaying either dengue virus serotype-2 envelope protein (DV2EP) or Plasmodium falciparum malaria circumsporozoite protein (CSP) [188]. To achieve organized antigen display, PCL was grafted with pyridine, providing a docking site for linking the PCL with the antigens through hydrogen bonding upon nanoparticle formation [237]. The mice trials using these nanoparticle vaccines in both DV2EP and CSP groups demonstrated a significant increase of antigen-specific IgG1 and IgG2a titers as compared to the antigens alone [188].
Similarly, poly(styrene) (PS) nanoparticles displaying hemagglutinin 1 protein (H1) of influenza A virus (H1N1) in bulk have been reported [103,104]. Functionalization of PS with Ni2+-nitrilotriacetic acid facilitated selective deposition of hexahistidine-tagged H1 (His6H1) driven by metal–histidine coordination bonds during the self-assembly. These nanoparticles generated concerted CD4+ and CD8+ T cell responses promoting antigen-specific Th1 and CTL activation. Furthermore, mice vaccinated with His6H1-coated PS nanoparticles elicited robust IgG antibody titer and provided superior protection against lethal challenge with H1N1 [104]. Due to their effective induction of DC maturation without any adjuvant [104], His6H1-coated PS nanoparticles were subsequently applied for cancer immunotherapy by co-displaying His10-tagged ovalbumin peptides (His10OVA) [238]. Mice immunized with PS nanoparticles presenting both H1 and OVA at the surfaces led to higher OVA-specific IFN-γ production and more effective suppression of OVA-expressing-B16-melanoma tumor growth compared to PS nanoparticles that only displayed either H1 or OVA. Although there were no complication from coagulation, allergenic response or pulmonary embolism in mice during the course of immunization using the PS nanoparticles [104], it should be noted that various in vitro and in vivo tests using PS nanoparticles demonstrated cytotoxicity due to the nanoparticles’ chemical reactivity and tendency to aggregate [239], which necessitates more thorough risk assessments when translating PS nanoparticle into particulate vaccines.
The bacterial biopolymer poly(3-hydroxybutyric acid) (PHB) is an emerging material for the synthesis of particulate vaccines [43] enabling rapid design and cost-effective manufacture at scale [42,44,240,241]. Through protein engineering, protein antigens can be fused, at the DNA level, to the dimeric protein synthase (PhaC) [242] that mediates the intracellular polymerization, leading to the in vivo formation of 200–1000 nm spherical PHB particles coated with the fusion proteins (Fig. 9 ) [240]. In addition, these protein-coated PHB particles can be further encapsulated by hydrogels, such as alginate [243], to potentially enhance vaccine performance by enabling oral administration routes and/or by generating antigen depot effects. Escherichia coli (E. coli) is predominantly used as the microbial cell factories producing the particles [244], as it offers high volumetric productivity through high cell-density fermentation technology that had already been well-established for commercial biopharmaceutical production for decades [245]. Recent development of endotoxin-free mutant of E. coli, ClearColi™ BL21(DE3) [246], provides cost reduction as laborious downstream removal of bacterial endotoxin that is immunogenic and cytotoxic is not required. As such, protein-coated PHB particles have to date been developed as a vaccine platform technology for the delivery of infectious-disease associated antigens originated from virus (e.g. hepatitis C virus [150]) and bacteria (e.g. Streptococcus pneumoniae [158,159], Mycobacterium tuberculosis [131], and Neisseria meningtidis [160]) (Fig. 9 and Table 2). Mice immunized with antigen-coated PHB particle vaccines demonstrated robust humoral- and cell-mediated immune responses.
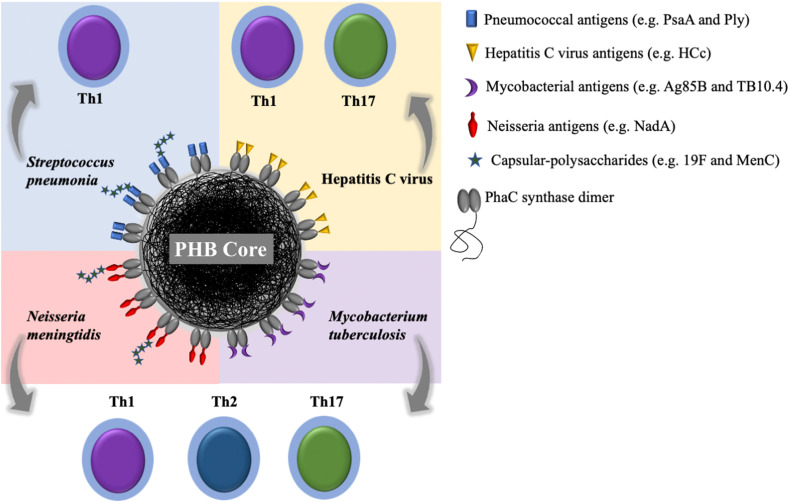
Overview of the particulate vaccine platform technology based on a core-shell protein-coated poly(3-hydroxybutyric acid) (PHB) particle against viral and bacterial pathogens, and their robust immune responses. The particles coated with protein antigens can be formed in vivo through self-assembly in engineered, endotoxin-free E. coli., and are composed of (i) PHB and (ii) dimeric PhaC protein synthase that bridges the PHB at the core (through covalent bonds) with the protein antigens at the shells. Capsular-polysaccharide (CPS) antigens can also be chemically conjugated in vitro on the particle surface, e.g. 19F (CPS from S. pneumoniae serotype 19F) and MenC (CPS from N. meningtidis serogroup C).
Examples of bioproduced PHB particle vaccines include multiantigenic PHB called particle-H28 that have been developed as tuberculosis vaccines (Fig. 10 ) [131]. This was achieved by engineering PhaC protein with the fusion proteins comprised of three Mycobacterium tuberculosis antigens (H28), i.e. Ag85B, TB10.4 and Rv2660c, which was then used to transform ClearColi BL21(DE3) to enable the bioproduction of PHB particles displaying the three antigens (Fig. 10A and B). Subcutaneous immunization of mice using particle-H28 adjuvanted with dioctadecyl ammonium bromide (DDA) micelles induced highly antigen-specific IgG1and IgG2c titers with strong cytokine profiles, indicating Th1-, Th2- and Th17-cell mediated immune responses, as compared to DDA micelles, control particles (containing PhaC proteins without the antigens) and soluble antigens (Fig. 10C) [131]. In an attempt to develop vaccines against Streptococcus pneumoniae, PHB particles displaying pneumolysin (Ply) (a serotype independent conserved protein found on the cytoplasm of S. pneumoniae) were produced in engineered ClearColi BL21(DE3) [160]. Mice immunized subcutaneously with the Ply-PHB particles adjuvanted with alum generated significantly higher Ply-specific IgG, IgG1 and IgG2b titers as well as IL-17A as compared to soluble Ply, PHB particles only (without displaying Ply) and alum only. In the same study, PHB particles were chemically conjugated with capsular polysaccharide serotype 19F (CPS 19F) from S. pneumoniae and formed 19F-PHB particles [160]. Mice subcutaneously immunized with the 19F-PHB particles adjuvanted with alum generated significantly high, specific and persistence IgG titers for at least six months, as compared to 19F conjugated to tetanus toxoid (as positive control), nonconjugated CPS 19F mixed with PHB particles, and alum only. Furthermore, serum antibodies from mice vaccinated with 19F-PHB particles demonstrated opsonophagocytic killing activity against cells of S. pneumoniae serotype 19F. Overall, the vast design space, i.e. the ability to incorporate diverse and multiple antigens, combined with rapid development for large scale manufacture, suggest that the PHB particles vaccine approach would be ideally suited to respond to pandemic threats such as caused by SARS-CoV-2 [233].
![Bacterial polyester particles for the development of tuberculosis vaccines. The particles are composed of hydrophobic poly(3-hydroxybutyric acid) (PHB) displaying PhaC protein linked to the fusion protein antigens Ag85B-TB10.4-Rv2660c (H28) from Mycobacterium tuberculosis, called particle-H28. (A) Representative TEM images of Escherichia coli containing particle-H28 (left) and the purified particle-H28 (right). (B) Schematic overview of particle-H28 displaying multiple protein antigens. (C) Cytokine responses after subcutaneous injection of particle-H28 adjuvanted with dimethyl dioctadecyl ammonium bromide (DDA) micelles in mice as compared to placebo (DDA only), particle (displaying PhaC only), and soluble His6-H28 peptides. Reproduced with permission from Ref. [131]. Copyright (2019) John Wiley and Sons.](/dataresources/secured/content-1765799170241-d106e8c1-f09f-4b76-8335-3dc3d2fa44f7/assets/gr10_lrg.jpg)
Bacterial polyester particles for the development of tuberculosis vaccines. The particles are composed of hydrophobic poly(3-hydroxybutyric acid) (PHB) displaying PhaC protein linked to the fusion protein antigens Ag85B-TB10.4-Rv2660c (H28) from Mycobacterium tuberculosis, called particle-H28. (A) Representative TEM images of Escherichia coli containing particle-H28 (left) and the purified particle-H28 (right). (B) Schematic overview of particle-H28 displaying multiple protein antigens. (C) Cytokine responses after subcutaneous injection of particle-H28 adjuvanted with dimethyl dioctadecyl ammonium bromide (DDA) micelles in mice as compared to placebo (DDA only), particle (displaying PhaC only), and soluble His6-H28 peptides. Reproduced with permission from Ref. [131]. Copyright (2019) John Wiley and Sons.
Prospects of translating polymeric nanoparticle vaccines
A variety of polymeric nanostructures has been synthesized for vaccine development directed towards infectious diseases (Table 2). In general, there are several advantageous factors in polymeric nanostructures that motivate their utilities as an antigen carrier platform (Table 3 ). First, polymeric structures can be synthesized in micro- or nanosize range mimicking bacterial or viral sizes, respectively, and be loaded with diverse vaccine constituents, including antigens and pattern recognition receptor agonist (as molecular adjuvants), in/on a single polymeric nanostructure synchronizing their co-delivery and mimicking the natural process of pathogen recognition by APCs. In addition, polymeric nanostructures, such as biobased PHB nanoparticles (Fig. 9), can serve as plug-and-play platform technologies that enable loading of different antigens in parallel, which are beneficial to provide rapid responses should disease outbreaks emerge. Second, polymeric nanostructures can be easily surface-engineered to enhance their stability and loading capacity, and/or to allow their targeting ability towards APCs for enhanced cellular internalization. Third, by judiciously selecting polymer particle precursors, stimuli-responsive polymeric nanostructures can be synthesized to facilitate controlled release of loaded antigens towards cytosols where cross-presentation of the processed antigens occur. Further, advanced molecular biotechnology allows the development of engineered bacteria for mediating the production of functional polymer particle-core protein-shell in one step in vivo, thus simplifying the manufacturing process and likely able to be scaled up. All these attributes have played important roles in the immunological properties of polymeric nanostructures as demonstrated in preclinical studies against various emerging diseases (Table 2). Some vaccine prototypes were able to elicit mucosal immunity through generation of secretory IgA which would be beneficial to provide immune defense on mucosal surfaces – the first point of entry of SARS-CoV-2.
| Structure | Synthesis methods | Uses | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
Solid particles  | •Homogenization of oil-in-water emulsion (single emulsion) or water-in-oil-in-water emulsion (double emulsion), and followed by solvent evaporation •Ionic complexation between polyelectrolytes and nucleic acid encoding the antigens | •As antigen carriers against many emerging diseases (see Table 2) •PLGA microparticles with adsorbed DNA encoding HIV antigens used as prophylactic vaccines reached Phase I clinical trial •PEI complexed with DNA encoding HIV antigens used as therapeutic vaccines is directed towards Phase III clinical trial •As adjuvant by loading with immunostimulant molecules | •Tunable physicochemical properties (e.g. hydrophobic, size and charge) simply by tuning synthesis parameters (e.g. composition, molecular weight of precursors, energy shearing, etc.) •Wide range of size and aspect ratio •Tunable surface characteristics for antigen loading and targeting moieties •Enable high antigen loading and sustained antigen release | •Acidic degradation of PLGA may pose adverse effects on protein antigen integrity •Pre-loading of antigens into the particles may lead to degradation of the antigens that are sensitive to organic solvents, temperature or shearing forces •Higher levels of both molecular weight and charge density of polycationic are better for transfection efficiency, but may lead to in vivo cytotoxicity | [42,127,251] |
Nanogels | •Physical self-assembly of interactive polymer chains •Emulsion polymerization •Crosslinking of self-assembled polymer aggregates or preformed polymer chains •Template-assisted nanofabrication with PRINT technology | •As antigen carriers against respiratory-based infections caused by viruses (e.g. H1N1) and bacteria (e.g. Streptococcus pneumoniae) •As cancer vaccines, in which cholesteryl-group-bearing pullulan nanogels have already reached clinical trials •As adjuvants by loading with immunostimulant molecules | •Controllable swelling for cargo loading and release depending on chemical structure of polymers, crosslinking density, charge density, or environmental parameters like pH, temperature, ionic strength •Tailorable deformability for efficient tissue penetration by engineering the crosslinking density and/or moiety | •The cargo-loading and stimuli-responsive release are often based on the charged groups incorporated into polymeric network, in which the global charges (especially positive) could induce cytotoxicity •Physically crosslinked nanogels pose stability problems during storage or in vivo delivery | [191,252,253] |
Micelles | Self-assembly of amphiphilic block copolymers (with >50% of block copolymers are hydrophilic block) | •As antigen carriers against bacterial pathogens and cancers •As adjuvant by loading with immunostimulant molecules | •The core can be loaded with a poorly water-soluble cargo, while the shell is for water-soluble ones •Small size for facilitating efficient tissue penetration or lymph node retention •Stimuli-responsive cargo release | •Limited loading capacity •Limited translation of micelle platforms between protein antigen cargoes | [34,254] |
Polymersomes | Self-assembly of amphiphilic block copolymers (with 35% of block copolymers are hydrophilic block) through thin film rehydration or flash nanoprecipitation | •As antigen carriers against cancers, lassa virus, influenza virus, and Mycobacterium tuberculosis | •The core domain allows loading of hydrophilic antigens, while the membrane shells can entrap poorly water-soluble immunostimulants | •Novel polymers are developed to make polymersomes along with organic solvents and/or additives, which require rigorous evaluation on cytotoxicity | [39,218,255,256] |
| •As adjuvants by loading with immunostimulant molecules | •Tunable membrane-shell permeability or thickness for better retention of loaded cargo | •Incorporation of highly hydrophobic copolymers makes the polymer aggregation and hydration challenging | |||
| •Tunable crosslinking and targeting moieties for enhanced stability and targeting ability | •Limited experience for scale up | ||||
| •Stimuli-responsive cargo release | |||||
Lipid coated particles  | Homogenization of oil-in-water emulsion (single emulsion) or water-in-oil-in-water emulsion (double emulsion), and followed by solvent evaporation | •As antigen carriers against cancers and viral infections such as MERS-CoV | The lipid shells facilitate: •Loading of antigens and/or immunostimulant molecules •Affinity of particles towards cells and their cell membrane penetration, which can lead to enhanced cell uptake | •The manufacture involves multiples steps which would pose scalability and reproducibility problems | [96,228,230] |
Cell membrane coated particles | Extrusion or sonication of a mixture of preformed solid polymeric nanoparticles with cell membranes purified from red blood cells, macrophages, or cancer cells | •As antigen carriers against bacterial pathogens •As personalized cancer vaccines | •Enable to display wide repertoire of antigens, replicating the properties of the sourced cells | •Large scale manufacture is limited for personalized vaccine use | [234,235] |
Protein coated particles | •Chemically synthesized particles are based on a layer-by-layer functionalization of preformed solid polymeric nanoparticles •Biologically synthesized particles are based on in vivo self-assembly of PHB driven by polymer synthase protein in engineered bacteria | •Chemically synthesized particles: antigen carriers against cancers, influenza, and malaria •Biologically synthesized particles: antigen carriers against bacterial and viral infections (Fig. 9) | •Controlled orientation and repetitive structure of protein antigen display on chemically synthesized particles •Antigen loading and particle formation of biologically synthesized particles occur in one step in vivo, scalable and reproducible through high-cell-density fermentation, and the degraded PHB is not cytotoxic | •Manufacture of chemically synthesized particles involves multiple steps which would pose problems in large scale production and reproducibility •It is difficult to control size and predict yields of biologically synthesized particles with potentials of difficult-to-remove host-cell-protein impurities | [42,104,188] |
Abbreviation: HIV, human immunodeficiency virus; MERS-CoV, Middle East Respiratory Syndrome Coronavirus; MPLA, monophosphoryl lipid A; PEI, poly(ethyleneimine); PHB, poly(3-hydroxybutyric acid) PLGA, poly(lactic-co-glycolic acid), PRINT, particle replication in nonwetting templates.
Despite the promising results of polymeric nanostructures against many emerging diseases in preclinical studies, only a few of those has reached clinical trials (Table 2). The translation of polymeric nanostructures as vaccine candidates to clinical trials and ultimately to the market face several bottlenecks which are partly related to the disadvantages of the polymeric nanostructures (Table 3). While sophisticated polymeric nanostructures can be made through a layer-by-layer functionalization to enhance antigen delivery, it also adds complexity layers into the manufacturing process, posing great challenges in scaling up and maintaining batch-to-batch reproducibility. In addition, each of those additional manufacturing steps requires quality controls in compliance with Good Manufacturing Practice (GMP) which would extend production timeline and incur additional cost, although the public health benefits would potentially outweigh the technological and logistical investment. Learning the lesson from polymeric nanostructures that have reached clinical trials (Table 2), PEI polyplexes loaded with DNA encoding antigens for HIV are based on the self-assembly between cationic PEI and anionic DNA [144,145], suggesting process simplicity, speed and scalability. Another obstacle for translation is thorough safety evaluation of the polymers, including the released products after biodegradation which may associate with cytotoxicity, accumulation and unexpected nonspecific immune responses. Vaccine efficacy tests are also a challenge due to lack of immune correlates of protection and difficulties in translating the immunological results from preclinical to clinical studies caused by inter-species differences. For example, intramuscular immunization of human volunteers during Phase I clinical trial with DNA encoding HIV antigens adsorbed on PLGA microparticles were shown minimal neutralizing breadth against the heterologous HIV, despite the previous preclinical trial in rhesus macaques that demonstrated the opposite [135]. Lastly, several vaccine formulations especially those containing temperature-sensitive materials such as recombinant protein antigens require cold chain for stability, which make their global distribution, storage and administration logistically challenging. Freeze-drying the nanoparticle vaccine formulation in the presence of cryoprotectant (e.g. sucrose) could be used to enhance their stability [247], as long as the physicochemical and immunological properties of the nanoparticles are not compromised and the lyophilized products can be easily reconstituted by healthcare professionals on site. Other technologies have also been developed, including single-dose polymeric implant prepared via melt-processing [248], thin film based vaccines [249], and microneedle-based patches [250]. For future development, these technologies could be integrated with polymeric nanoparticle vaccines to reduce reliance on the cold chains.
Conclusions
Recent advances in design and assembly of polymeric nanostructures have generated tremendous diversity of novel polymeric nanoparticle designed toward particulate vaccines. It is clear that physicochemical properties of the polymeric nanoparticles, including size, shape, charge, and hydrophobicity, play crucial roles in modulating the delivery and trafficking into lymph nodes, the activation of CD4+ and CD8+ T cells, and the subsequent induction of humoral and/or cellular immune responses. Emerging methods have been developed to synthesize different polymeric platforms, including particles, micelles, nanogels, polymersomes, and core-shell particles, with different physicochemical properties that mimic viral or bacterial pathogen characteristics. In addition, antigens and/or immunostimulatory agents can be encapsulated into or displayed onto these nanostructured polymers through physical, chemical and/or synthetic biology approaches, which pave ways to novel formulation designs that promise controlled immune modulation. In particular, pre-clinical trials of these polymeric nanoparticles demonstrated improved efficacy against infectious diseases over equivalent dose of soluble subunit vaccines, aligned with cross-presentation of antigens and cellular immunity which are important for infection with intracellular pathogens and cancer. The possibility to design antigen-bearing polymer nanoparticles for induction of strong and specific immune responses needed to establish long-lasting protective immunity provides competitive advantages over other vaccine approaches. Extensive preclinical safety and efficacy studies are paving the way for polymeric nanoparticles to be tested as particulate vaccines in human clinical trial. It is also important to consider large-scale manufacturing capability of the designed polymeric nanostructures. Simple particle design with minimal fabrication steps may be of preference to ensure reproducibility of the particles and their rapid production in industrial settings, as long as the vaccine safety and efficacy are not compromised. Polymer particle-based vaccine approaches hold the promise to serve as rapid response platform technologies to timely contain emerging pandemic threats such as e.g. the pandemic causing SARS-CoV-2 [233].
Author contributions
DW outlined the manuscript and designed the figures. DW, SJ, ZJG, BE, SZ, and BHAR participated in researching and discussing literature and writing parts of the manuscripts. All authors read and approved the final version.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1
2
3
4
5
6
7
8
11
12
13
14
15
16
17
18
19
20
21
22
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
49
50
51
52
53
54
55
56
57
58
59
60
62
63
64
65
66
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
243
244
245
246
247
248
249
250
251
252
253
254
255
256
List of abbreviations
| APC | Antigen presenting cell |
| BCG | Bacillus Calmette–Guèrin |
| cdGMP | Cyclic diguanylate monophosphate (CDN) |
| CDN | Cyclic dinucleotide |
| CTL | Cytotoxic T-cell lymphocyte |
| CpG-ODN | Oligodeoxynucleotides with unmethylated CpG motifs |
| DC | Dendritic cell |
| DC-Chol | 3β-[N-(N′,N′-Dimethylaminoethane)-carbamoyl]cholesterol (DC-Chol) |
| DDA | Dimethyldioctadecylammonium |
| dLNs | Draining lymph nodes |
| DOTAP | 1,2-dioleoyl-3-trimethylammoniumpropane |
| US FDA | The Food and Drug Administration |
| IFN-γ | Interferon gamma |
| IL | Interleukin |
| MERS-CoV | Middle East Respiratory Syndrome Corona Virus |
| MHC-I | Major histocompatibility complex class I |
| MHC-II | Major histocompatibility complex class II |
| MPLA | Monophosphoryl lipid A |
| NLR | NOD-like receptor |
| NOD | Nucleotide-binding oligomerization domain |
| OVA | Ovalbumin |
| PAN | Poly(anhydride) |
| PCL | Poly(ε-caprolactone) |
| PDCP | Poly(dicholorophosphazene) |
| PEG | Poly(ethylene glycol) |
| PEI | Poly(ethylene imine) |
| PGA | Poly(glycolic acid) |
| PHB | Poly(3-hydroxybutyric acid) |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactic-co-glycolic acid) |
| Poly(I:C) | Polyinosinic:polycytidylic acid |
| PPS | Poly(propylene sulfide) |
| PRR | Pattern-recognition receptor |
| PS | Poly(styrene) |
| RBC | Red blood cell |
| RIG-I | Retinoic acid-inducible gene I-like receptors |
| RLR | RIG-I-like receptors |
| Th1 | T helper type 1 |
| Th2 | T helper type 2 |
| TLR | Toll-like receptor |
| TMC | Trimethyl chitosan |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| SLR | 5′-triphosphorylated stem-loop RNA |
| STING | Stimulator of interferon genes |
Acknowledgments
B.H.A. Rehm acknowledges financial support from the award of the