 Time-Resolved
Small-Angle X-ray Scattering
Studies during Aqueous Emulsion Polymerization
Time-Resolved
Small-Angle X-ray Scattering
Studies during Aqueous Emulsion Polymerization


- Altmetric
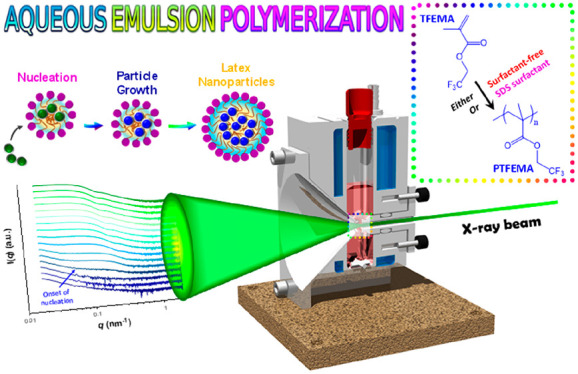
The persulfate-initiated aqueous emulsion polymerization of 2,2,2-trifluoroethyl methacrylate (TFEMA) is studied by time-resolved small-angle X-ray scattering (SAXS) at 60 °C using a stirrable reaction cell. TFEMA was preferred to styrene because it offers much greater X-ray scattering contrast relative to water, which is essential for sufficient temporal resolution. The evolution in particle size is monitored by both in situ SAXS and ex situ DLS in the absence or presence of an anionic surfactant (sodium dodecyl sulfate, SDS). Post-mortem SAXS studies confirmed the formation of well-defined spherical latexes, with volume-average diameters of 353 ± 9 nm and 68 ± 4 nm being obtained for the surfactant-free and SDS formulations, respectively. 1H NMR spectroscopy studies of the equivalent laboratory-scale formulations indicated TFEMA conversions of 99% within 80 min and 93% within 60 min for the surfactant-free and SDS formulations, respectively. Comparable polymerization kinetics are observed for the in situ SAXS experiments and the laboratory-scale syntheses, with nucleation occurring after approximately 6 min in each case. After nucleation, scattering patterns are fitted using a hard sphere scattering model to determine the evolution in particle growth for both formulations. Moreover, in situ SAXS enables identification of the three main intervals (I, II, and III) that are observed during aqueous emulsion polymerization in the presence of surfactant. These intervals are consistent with those indicated by solution conductivity and optical microscopy studies. Significant differences between the surfactant-free and SDS formulations are observed, providing useful insights into the mechanism of emulsion polymerization.
Introduction
Aqueous emulsion polymerization is an environmentally friendly process that is widely used on an industrial scale to polymerize many water-immiscible vinyl monomers, including styrene, methacrylates, acrylates, vinyl acetate, vinyl chloride, etc.1,2 Such heterophase polymerizations account for approximately 25% of synthetic polymers produced globally,3 with tens of millions of tons of copolymer latexes being prepared each year.3,4 Importantly, microcompartmentalization enables the efficient production of high molecular weight polymer chains in convenient low-viscosity form while offering good control over heat dissipation.2,5−9 The resulting latex particles are used for many applications, including architectural paints, anticorrosion coatings, adhesives, varnishes, cement and concrete additives, rheology modifiers; they can also serve as the mobile phase for immunodiagnostic assays.10
Aqueous emulsion polymerizations are inherently heterogeneous in nature. Thus, such formulations usually require vigorous stirring to generate micrometer-sized monomer droplets. Such droplets act as reservoirs and provide a sufficiently high interfacial area to ensure efficient mass transport of the water-immiscible monomer to the growing particles during polymerization. Various in situ techniques have been utilized to monitor the kinetics of aqueous emulsion polymerization, including 1H NMR spectroscopy combined with a flow cell,11 Raman spectroscopy,12 and near-IR spectroscopy.13,14 However, such studies do not enable the evolution in particle morphology to be assessed, hence they can provide only rather limited insights regarding the complex mechanism of emulsion polymerization.2,15,16
The kinetics of emulsion polymerization has been extensively studied.6,17−19 The generally accepted mechanism comprises three distinct regions, which are denoted as Intervals I, II, and III (see Figure 1).1,6−9,16,20−22 A typical batch emulsion polymerization formulation comprises a vinyl monomer of relatively low water solubility (e.g., styrene), water, surfactant, and a water-soluble initiator. Prior to polymerization, the hydrophobic monomer mainly resides in the monomer droplets, with a relatively small fraction solubilized within surfactant micelles and a further (minor) fraction dissolved within the aqueous continuous phase. Free radicals derived from the water-soluble initiator polymerize monomer dissolved in the aqueous phase to form oligomeric radicals. At some critical chain length, these oligomeric radicals become sufficiently hydrophobic to enter the monomer-swollen micelles, which vastly exceeds the monomer droplets in terms of both number density and overall interfacial area.6,7 Further monomer then diffuses from the monomer droplets into these nascent particles and new polymer chains are initiated within the monomer-swollen particles, which continue to grow in size. To maintain colloidal stability, the remaining surfactant micelles undergo dissociation to supply additional surfactant and hence ensure monolayer coverage of the surface of the growing polymer particles. Furthermore, the surfactant molecules that act as an emulsifier desorb from the (shrinking) monomer droplets to coat these particles. Once there are no remaining surfactant micelles, particle nucleation (i.e., Interval I) is complete, see Figure 1a. Thereafter, the number of latex particles remains relatively constant. Polymerization continues primarily within monomer-swollen particles with monomer droplets serving as reservoirs to supply the growing particles with further monomer (and surfactant). This particle growth stage (Interval II, Figure 1b) is complete when there are no remaining monomer droplets. This leads to so-called “monomer-starved” conditions and the polymerization proceeds at a slower rate until all the monomer is consumed (Interval III, Figure 1c).
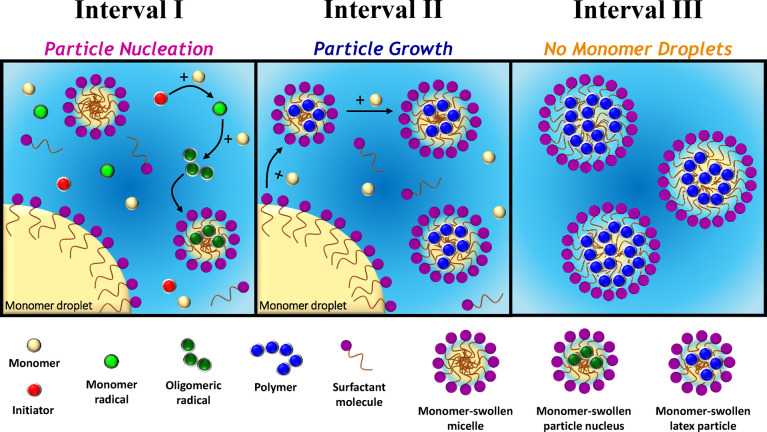
However, aqueous emulsion polymerization can also occur under surfactant-free conditions.23−29 In this case, thermal decomposition of an ionic initiator (e.g., persulfate) generates charged water-soluble radicals that react with dissolved monomer within the aqueous phase. This generates a growing polymer radical with a terminal anionic sulfate group that becomes insoluble at some critical chain length to form a primary particle. These primary particles are colloidally unstable and thus undergo aggregation. The ensuing increase in surface charge density produces colloidally stable mature particle nuclei into which monomer can diffuse from the droplet reservoirs and/or the aqueous phase. Latexes prepared under surfactant-free conditions tend to be significantly larger than those prepared in the presence of surfactant, which has been attributed to a coagulative nucleation mechanism.30
Small-angle X-ray scattering (SAXS) is ideally suited for characterizing systems exhibiting multiple colloidal length scales.31 Furthermore, using a synchrotron X-ray source ensures superb temporal resolution for in situ studies, thus providing unique insights into various phenomena, including particle formation and growth,32 kinetics33 and self-assembly.34,35 Recently, we have demonstrated that time-resolved SAXS is a powerful technique for studying the evolution in block copolymer morphology that occurs during polymerization-induced self-assembly (PISA).36−38 Herein we utilize our recently reported stirrable reaction cell36 to conduct in situ SAXS studies during the free persulfate-initiated aqueous emulsion polymerization of 2,2,2-trifluoroethyl methacrylate (TFEMA) at 60 °C. More specifically, we examined two formulations: one was conducted in the presence of an anionic surfactant (sodium dodecyl sulfate, SDS) while the other was performed under surfactant-free conditions,23 (see Figure 2).

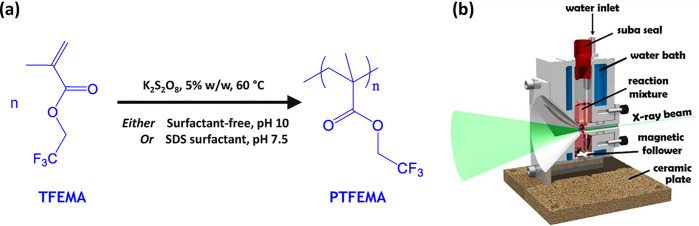
(a) Representation of the synthesis of PTFEMA latex particles formed via aqueous emulsion polymerization of 2,2,2-trifluoroethyl methacrylate (TFEMA) using an anionic free radical initiator (potassium persulfate, KPS) at 60 °C either in the presence of an anionic surfactant (SDS) or under surfactant-free conditions targeting 5.0% w/w solids. (b) Schematic cross-section of the stirrable reaction cell used for time-resolved small-angle X-ray scattering (SAXS) studies of such formulations. The volume of the reaction solution within this cell is approximately 2.0 mL, which is sufficient to enable post-mortem analysis using 1H NMR spectroscopy, TEM, and dynamic light scattering.
Results and Discussion
Preliminary Experiments
From an academic perspective, the most widely studied aqueous emulsion polymerization formulation involves the homopolymerization of styrene.39−42 Although the glass transition temperature of polystyrene is too high for paints and coatings applications, polystyrene latexes are widely used as calibration standards for particle size analysis43 and for visual agglutination immunodiagnostic assays.44 Given these considerations, our original aim was to conduct in situ SAXS studies of the aqueous emulsion polymerization of styrene. Unfortunately, this prototypical formulation offers very poor X-ray contrast between the polystyrene particles and the aqueous continuous phase owing to the remarkably similar scattering length densities (SLD, or ξ) of water (ξwater = 9.42 × 1010 cm–2) and polystyrene (ξPolystyrene = 9.41 × 1010 cm–2).45 Indeed, there is almost no difference between the scattering curve recorded at 40% styrene conversion during the surfactant-free aqueous emulsion polymerization of styrene and that for pure water (see Figure S1 in the Supporting Information, SI). With the benefit of hindsight, these observations are not surprising: X-ray contrast correlates quite closely with density, and the solid-state density of polystyrene (1.05 g cm–3) is simply too close to that of water (1.00 g cm–3). In view of this problem, we sought an alternative vinyl polymer with a significantly higher density (and hence SLD) than that of water. Previously, we had prepared sterically stabilized diblock copolymer nanoparticles via RAFT aqueous emulsion polymerization of TFEMA.46 This monomer has a density of 1.18 g cm–3 and the corresponding PTFEMA homopolymer has a solid-state density of 1.47 g cm–3. Therefore, we elected to use TFEMA monomer instead of styrene because it provides much stronger contrast relative to water during in situ SAXS studies (ξPTFEMA = 12.76 × 1010 cm–2). TFEMA has an aqueous solubility of approximately 2.9 g dm–3 at 25 °C, which is approximately an order of magnitude higher than that of styrene (0.31 g dm–3 at 25 °C). Nevertheless, the aqueous solubility of TFEMA is sufficiently low to ensure a genuine aqueous emulsion polymerization formulation. Furthermore, given that the Tg of PTFEMA homopolymer is around 55 °C, the nanoparticles retain their original morphology during TEM analysis.
In principle, particle growth during emulsion polymerization can be monitored by analyzing aliquots periodically extracted from the reaction mixture using techniques such as DLS. However, for formulations involving an ionic surfactant such as SDS, significant changes in solution conductivity also occur during polymerization. This is because the solution conductivity depends mainly on the concentration of free surfactant dissolved in the aqueous continuous phase (Figure 1). Initially, most of this surfactant is either present in the form of micelles or is adsorbed at the surface of the monomer droplets. At the end of the polymerization, the majority of the surfactant is adsorbed at the surface of the final latex particles. At intermediate monomer conversions, the solution conductivity—which can be readily monitored in situ—depends on the relative populations of monomer droplets, micelles, free (dissolved) surfactant, and growing latex particles. Thus, the evolution in solution conductivity during aqueous emulsion polymerization can provide valuable information on the polymerization kinetics,47 particle nucleation,5 and the underlying mechanism of emulsion polymerization.48,49Figure 3 shows in situ solution conductivity data recorded during the laboratory-scale synthesis of PTFEMA latex particles in the presence of SDS surfactant under the conditions shown in Figure 2. Once particle nucleation has occurred, free surfactant molecules adsorb onto the growing nascent particles to confer anionic surface charge and hence colloidal stability. This depletion of free surfactant leads to a gradual reduction in conductivity and the period between 0 and 11 min corresponds to Interval I (Figure 1a).49 At the end of Interval I, there is no more molecularly dissolved surfactant in the aqueous continuous phase. Thereafter, there is an increase in conductivity, which indicates the onset of Interval II (Figure 1b).49 This occurs because, as the polymerization proceeds and monomer is consumed, surfactant molecules desorb from the surface of the shrinking monomer droplets, which leads to higher solution conductivities for the aqueous continuous phase. A local maximum in conductivity is observed after approximately 28 min. This period corresponds to the onset of Interval III. At this point, essentially all the monomer droplets have been consumed, so the remaining monomer is mainly located within the growing PTFEMA latex particles (because TFEMA monomer is a good solvent for PTFEMA homopolymer). Any free surfactant molecules remaining within the aqueous phase adsorb onto the latex particles during the latter stages of their growth, which accounts for the gradual reduction in solution conductivity observed over the following 32 min. After 60 min, the conductivity remains constant, suggesting that the TFEMA polymerization is complete.
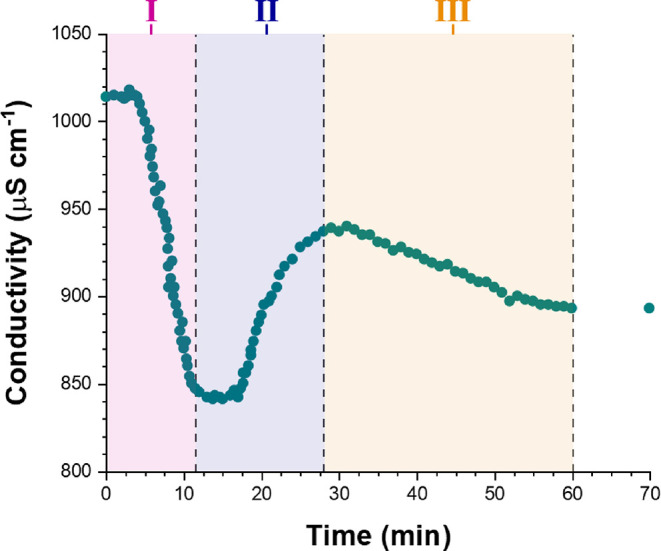
Solution conductivity measurements recorded in situ during the aqueous emulsion polymerization of TFEMA in the presence of SDS surfactant at 60 °C targeting 5.0% w/w solids. The highlighted three time intervals (I, II, and III) are known to occur when such polymerizations are performed in the presence of a surfactant above its CMC (Figure 1).
The kinetics of TFEMA polymerization and the corresponding evolution in particle size were simultaneously monitored during a laboratory-scale synthesis (conducted under the conditions shown in Figure 2) by periodically withdrawing aliquots from the heterogeneous reaction mixture for analysis. The polymerization was quenched by immediately immersing each aliquot into an ice bath with concomitant exposure of the reaction mixture to air. Instantaneous TFEMA conversions were determined via 1H NMR spectroscopy by diluting 80 μL of each extracted aliquot in 500 μL CDCl3, with anhydrous MgSO4 being used to remove residual water. The evolution in particle size was monitored by dynamic light scattering (DLS) studies of 0.20% w/w aqueous dispersions obtained by diluting 40 μL of each extracted aliquot using deionized water (960 μL). Volume-average size distributions were calculated from the intensity-average size distributions obtained by DLS using the Mie theory that is embedded within the instrument manufacturer’s software, see SI for further details. Accordingly, Figure 4 shows the conversion vs time curves and the evolution in volume-average particle diameter over time during the aqueous emulsion polymerization of TFEMA obtained either under surfactant-free conditions or in the presence of SDS surfactant. Furthermore, the time intervals corresponding to those determined by in situ conductivity measurements for the SDS formulation (Figure 3) are also included in Figure 4b. For the surfactant-free formulation, no further increase in TFEMA conversion is observed after 80 min (Figure 4a). At this time point, 1H NMR spectroscopy indicated a final monomer conversion of 98%, while DLS studies reported a volume-average particle diameter of 485 nm (DLS polydispersity = 0.02). TEM studies confirm the formation of relatively uniform spherical latex particles with a number-average diameter of 464 nm, see Figure 5c. A slightly faster rate of polymerization occurs in the presence of SDS, with no further increase in conversion being observed after 60 min (see Figure 4b). This agrees rather well with the overall time scale required for this polymerization indicated by the solution conductivity measurements (Figure 3). Furthermore, this is consistent with DLS studies, which indicates that the volume- average particle diameter remained constant at 91 nm (DLS polydispersity = 0.01) on the same time scale. TEM studies confirm the formation of well-defined spherical latex particles (see Figure 5d), with a number-average diameter of approximately 89 nm. The DLS data for both formulations shown in Figure 4 is also included in Table S1. For free radical-initiated aqueous emulsion polymerization formulations reported in the literature, smaller latex particles and faster rates of polymerization are typically observed in the presence of surfactant.26,42 Under surfactant-free conditions, latex surface charge is solely conferred by sulfate groups (derived from the persulfate initiator) located on the polymer chain-ends.50−52 In contrast, the PTFEMA particles produced in the presence of surfactant acquire anionic surface charge from both these sulfate end-groups and also the adsorption of SDS at the latex surface.1,2,9 This accounts for the substantial difference in particle size for these two formulations indicated by TEM and DLS studies. Furthermore, microcompartmentalization of the growing polymer radicals within surfactant micelles suppresses their termination, which leads to a faster overall rate of polymerization and a significantly higher molecular weight than the equivalent solution polymerization.1 This is supported by the molecular weight distributions obtained for each formulation using gel permeation chromatography (see Figure S2).
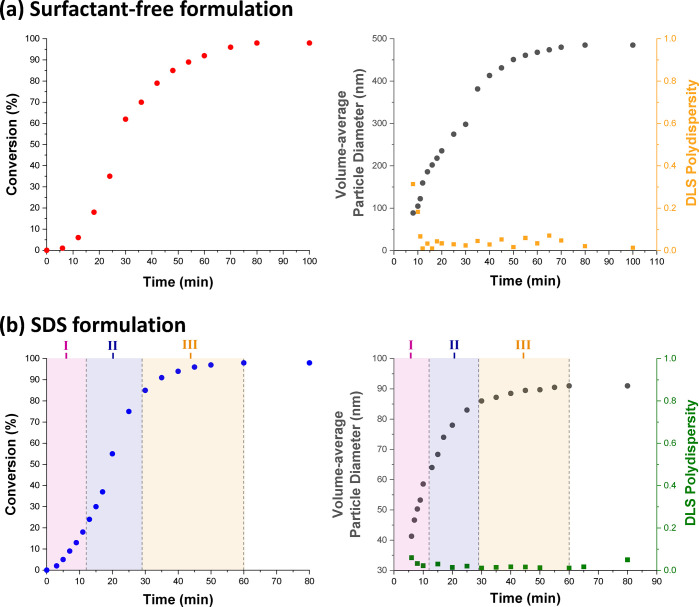
(a) Conversion vs time curve obtained by 1H NMR spectroscopy and evolution in volume-average particle diameter and polydispersity determined by DLS for the laboratory-scale surfactant-free aqueous emulsion polymerization of TFEMA at 60 °C targeting 5.0% w/w solids. (b) Equivalent data for the laboratory-scale aqueous emulsion polymerization of TFEMA at 60 °C targeting 5.0% w/w solids in the presence of SDS surfactant (2.0 mol % based on TFEMA), where the three regions corresponding to Intervals I, II, and III indicated by solution conductivity measurements (Figure 3) are also shown.

TEM images recorded for PTFEMA latex particles prepared during the aqueous emulsion polymerization of TFEMA at 60 °C in the absence or presence of SDS surfactant. Images a–d correspond to PTFEMA nanoparticles formed during the laboratory-scale synthesis. Images e and f correspond to post-mortem analysis of PTFEMA nanoparticles formed during the in situ SAXS synthesis using the stirrable reaction cell.
The three distinct time intervals observed during emulsion polymerization (see Figure 1) and identified by solution conductivity measurements (see Figure 3) are consistent with the NMR-derived kinetic data obtained for the SDS formulation shown in Figure 4b. According to Figure 3, Interval I lies between 0 and 11 min. During this time period, 1H NMR analysis indicates a discernible increase in the rate of polymerization, suggesting that particle nucleation occurs within this time frame. Furthermore, Interval I typically exists up to 10–20% monomer conversion2 and, according to the data presented in Figure 4b, the end of Interval I corresponds to approximately 20% TFEMA conversion. Inspecting Figure 3, Interval II is complete after around 28 min, which is consistent with the period of rapid polymerization observed by 1H NMR spectroscopy over this time period. According to the literature, the typical monomer conversion at the end of Interval II is around 60%.2 However, for the specific formulation studied herein, the TFEMA conversion at the end of Interval II is approximately 84%. After this time point, 1H NMR indicates a reduction in the rate of reaction until the TFEMA polymerization is more or less complete after 60 min. This matches the concomitant reduction in solution conductivity—and hence the beginning of Interval III—observed in Figure 3. Furthermore, DLS studies indicate a significant reduction in the rate of particle growth over the second half of the polymerization.
In situ SAXS Studies during TFEMA Polymerization
In the literature, in situ experiments have been conducted during aqueous emulsion polymerization using Raman12 or FT–IR spectroscopy.13,14 However, such techniques can only monitor the instantaneous monomer concentration; they provide no particle size information. SAXS is a well-established structural characterization technique in colloid science.31,53−56 It has been used to characterize the particle size distributions of various polymer latexes prepared via free radical-initiated aqueous emulsion polymerization.57−61 However, as far as we are aware, there have been no in situ studies for such heterogeneous formulations. One likely reason for this surprising omission is the requirement for efficient stirring during aqueous emulsion polymerization, which is simply not feasible using the capillary cells that are normally used for SAXS measurements. However, we have recently reported in situ SAXS studies during the reversible addition–fragmentation chain transfer (RAFT) aqueous emulsion polymerization of 2-methoxyethyl methacrylate using a stirrable reaction cell (Figure 2b).36 In essence, this cell comprises a capillary positioned above a 2.0 mL chamber (equipped with a magnetic flea), which is encased within an aluminum block that enables efficient heat transfer via a circulating water jacket. The reaction mixture can be stirred sufficiently vigorously to create the micrometer-sized monomer droplets that are required for aqueous emulsion polymerization to proceed. One important feature of this stirrable reaction cell is that its sample volume is sufficient to enable post-mortem characterization of the final latex particles using multiple techniques. Herein, we use this new experimental setup to perform time-resolved SAXS studies during the aqueous emulsion polymerization of TFEMA. The in situ polymerization formulations were conducted on a smaller scale than the equivalent laboratory-scale reactions reported in Figures 3 and 4. A synchrotron source is essential for such in situ SAXS experiments: it ensures sufficient temporal resolution to monitor the relatively fast kinetics of the TFEMA polymerization (Figure 4). This enables many high-quality scattering patterns to be recorded on a relatively short time scale, thus providing information regarding both nucleation and subsequent particle growth.
Particle Nucleation
Figure 6 shows the X-ray scattering intensity, I(q), plotted against the scattering vector, q, for selected SAXS patterns recorded in situ during the aqueous emulsion polymerization of TFEMA at 60 °C in the presence or absence of surfactant, with 5.0% w/w solids being targeted in each case. This relatively low monomer concentration was chosen to minimize interparticle interactions, which nevertheless still required the introduction of a structure factor (see later). Figure 6 also includes selected scattering patterns (and representative data fits) recorded at specific time points for both formulations. For scattering patterns that are not scaled by an arbitrary factor and also residual fits to final scattering patterns, see Figures S8 and S9 in the SI. In the low q regime, I(q) is proportional to the volume of a scattering object; this relationship can be used to identify the onset of particle nucleation. Thus, I(q) was plotted against time during the early stages of the polymerization at arbitrary q values of 0.02 nm–1 and 0.1 nm–1 for the surfactant-free and SDS formulations respectively, see Figure 7. [N.B. It was necessary to select a suitably low q value for the surfactant-free formulation to avoid the multiple fringes within the associated scattering patterns.] The pronounced upturns in I(q) highlighted by the blue arrows in Figure 7 indicate the point at which particle nucleation occurs (with the corresponding scattering pattern also highlighted in Figure 6). Nucleation is observed after approximately 8 and 6 min for the surfactant-free and SDS formulations, respectively. Identifying the onset of nucleation for either formulation during the equivalent laboratory-scale syntheses (see Figure 4) is somewhat problematic owing to the difficulty in sampling such heterogeneous reaction mixtures during the early stages of polymerization. However, DLS studies revealed a pronounced upturn in the scattered light intensity (derived count rate) after 6 min for the SDS formulation and 10 min for the surfactant-free formulation respectively, see Figure 7. In principle, this should correspond to nucleation. These approximate nucleation times agree well with those observed by measuring I(q) during the equivalent in situ SAXS syntheses. Nucleation appears to occur on a shorter time scale for the surfactant-free formulation during the in situ SAXS experiments. In principle, this could constitute evidence for an X-ray beam-induced rate enhancement. Indeed, our prior in situ SAXS studies conducted during PISA syntheses in mineral oil revealed a significant rate enhancement that was attributed to an additional radical flux generated by the high-energy X-ray beam.36,38 However, TEM analysis conducted on aliquots extracted from the laboratory-scale syntheses confirmed the formation of nascent nuclei within the short time scales indicated by the in situ SAXS experiments, see Figure 5a,b. Therefore, nucleation seems to occur on similar time scales in both cases. The spherical nuclei observed by TEM after 6 min (SDS formulation) and 8 min (surfactant-free formulation) have number-average particle diameters of 39 and 62 nm, respectively. DLS studies of the same aliquots indicated volume-average particle diameters of 41 and 88 nm for the corresponding SDS and surfactant-free formulations, respectively. As expected, DLS studies of aliquots extracted prior to nucleation exhibited significantly low scattered light intensity after serial dilution, suggesting no particle formation.
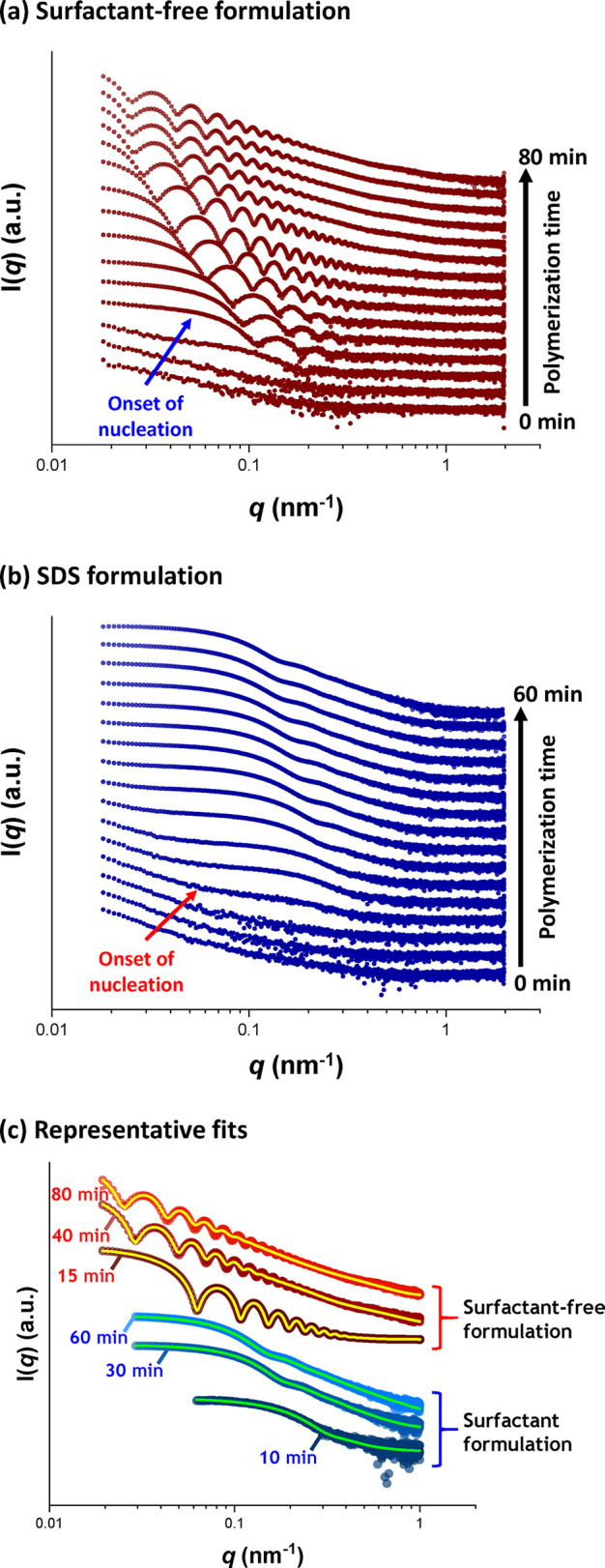
SAXS patterns recorded in situ during the aqueous emulsion polymerization of TFEMA at 60 °C targeting 5% w/w solids (a) under surfactant-free conditions and (b) in the presence of SDS surfactant. The onset of particle nucleation is indicated by the arrow. (c) Representative fits for scattering patterns recorded at specific time points for both formulations with data fits represented by either yellow (surfactant-free formulation) or green (SDS formulation) lines, respectively. All scattering patterns are scaled by an arbitrary factor to avoid overlap and improve clarity.
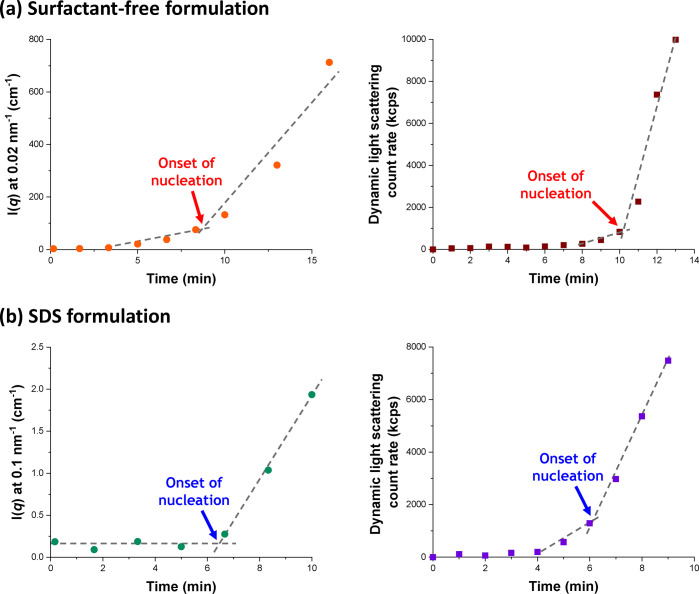
Evolution in I(q) recorded at arbitrary q values during the in situ synthesis and light scattering count rate determined by DLS studies of the equivalent laboratory-based aqueous emulsion polymerization of TFEMA at 60 °C when targeting 5% w/w solids for (a) a surfactant-free formulation and (b) in the presence of SDS surfactant. The onset of particle nucleation is indicated in each case.
The aqueous emulsion polymerization of TFEMA was judged to be complete when no discernible difference was observed between consecutive scattering patterns. This occurred after reaction times of 80 and 60 min for the surfactant-free and SDS formulations, respectively (see Figure S3). These time scales are equivalent to those observed for the equivalent laboratory-scale syntheses (see Figure 4). It is also noteworthy that the surfactant-free formulation produced SAXS patterns with multiple fringes (see Figure 6a), whereas the SDS formulation led to relatively featureless scattering patterns (see Figure 6b). This indicates that the surfactant-free formulation produces PTFEMA particles with a significantly narrower size distribution.62 Post-mortem 1H NMR analysis of quenched reaction mixtures retrieved from the stirrable reaction cell indicated final TFEMA conversions of 99% and 93% for the surfactant-free and SDS formulations, respectively. The corresponding intensity-average particle diameters indicated by DLS studies were 444 nm (DLS polydispersity = 0.114) and 113 nm (DLS polydispersity = 0.076). These data are consistent with the equivalent laboratory-scale syntheses. Post-mortem TEM analysis confirmed the formation of PTFEMA latex particles with a well-defined spherical morphology in each case, with final number-average particle diameters of 328 and 75 nm being estimated for the surfactant-free and SDS formulations respectively, see Figure 5e,f.
Particle Growth
The surface character of the two PTFEMA latexes was assessed by aqueous electrophoresis. In both cases, highly negative zeta potentials were obtained over the entire pH range investigated, see Figure S4. Given the strongly anionic character of the latex particles, the scattering patterns obtained after particle nucleation were fitted using a well-known scattering model for spheres63 by incorporating a hard-sphere structure factor (solved with the Percus–Yevick closure relation64) to account for interparticle interactions.
Figure 8 shows the evolution in volume-average diameter for the growing latex particles during the TFEMA polymerization as determined by in situ SAXS studies using the stirrable reaction cell. For the surfactant-free formulation, two distinct regimes are observed after nucleation (see Figure 8a). First, there is a period of linear growth up to 27 min. Thereafter, there is a brief increase in the rate of particle growth, which then slows down until the TFEMA polymerization is more or less complete, producing colloidally stable latex particles with a volume-average particle diameter of 353 ± 9 nm after 80 min. Bearing in mind the effect of polydispersity, this final particle size is reasonably consistent with the volume-average particle diameter of 444 nm determined by post-mortem DLS analysis. Furthermore, the evolution in particle diameter determined by in situ SAXS (Figure 8a) is similar to that indicated by DLS studies of the equivalent laboratory-scale synthesis (Figure 4a). For example, mean diameters of the nascent particles observed after 8 min are 88 and 94 nm for the SAXS and DLS data, respectively. The rate of particle growth begins to slow down after approximately 40 min for both the SAXS and equivalent laboratory-scale syntheses. At this time point, the mean particle diameters are 312 and 413 nm, respectively. During the early stages of the TFEMA polymerization (8–27 min), the rate of particle growth is constant. Both in situ SAXS and DLS studies suggest a brief increase in the rate of growth after approximately 27 min. In principle, this feature should correspond to the end of Interval II (Figure 1b). Since there are no remaining monomer droplets, polymerization proceeds under monomer-starved conditions, which explains the slower rate of particle growth observed after 30 min. The solution conductivity was also monitored in situ during the aqueous emulsion polymerization of TFEMA using the surfactant-free formulation, see Figure S5. In this case, there is no measurable solution conductivity for the first 28 min of the TFEMA polymerization, at which point the instantaneous monomer conversion is approximately 60%, see Figure 4a. Subsequently, there is a dramatic increase in solution conductivity in the 29–31 min interval. Interestingly, this time point corresponds to a discernible inflection point during the evolution in particle size indicated by DLS studies during the equivalent laboratory-scale synthesis, see Figure 4a. Moreover, optical microscopy studies confirm essentially full consumption of the monomer droplets after approximately 30 min, see Figure S7. Thus, it seems likely that this time point corresponds to the Interval II/Interval III boundary as shown in Figure 8a. Thereafter, the rate of increase in solution conductivity is reduced, with a maximum solution conductivity of 640 μS cm–1 being observed after 55 min followed by a gradual reduction in conductivity to a limiting value of 590 μS cm–1 after 75 min. According to the NMR kinetic data in Figure 4a, this latter time point corresponds to the end of the polymerization. In summary, solution conductivity measurements undertaken during the surfactant-free aqueous emulsion polymerization of TFEMA may provide some useful information during the latter stages of the reaction, but it appears that this technique cannot be used to pinpoint the Interval I/Interval II boundary for such formulations.
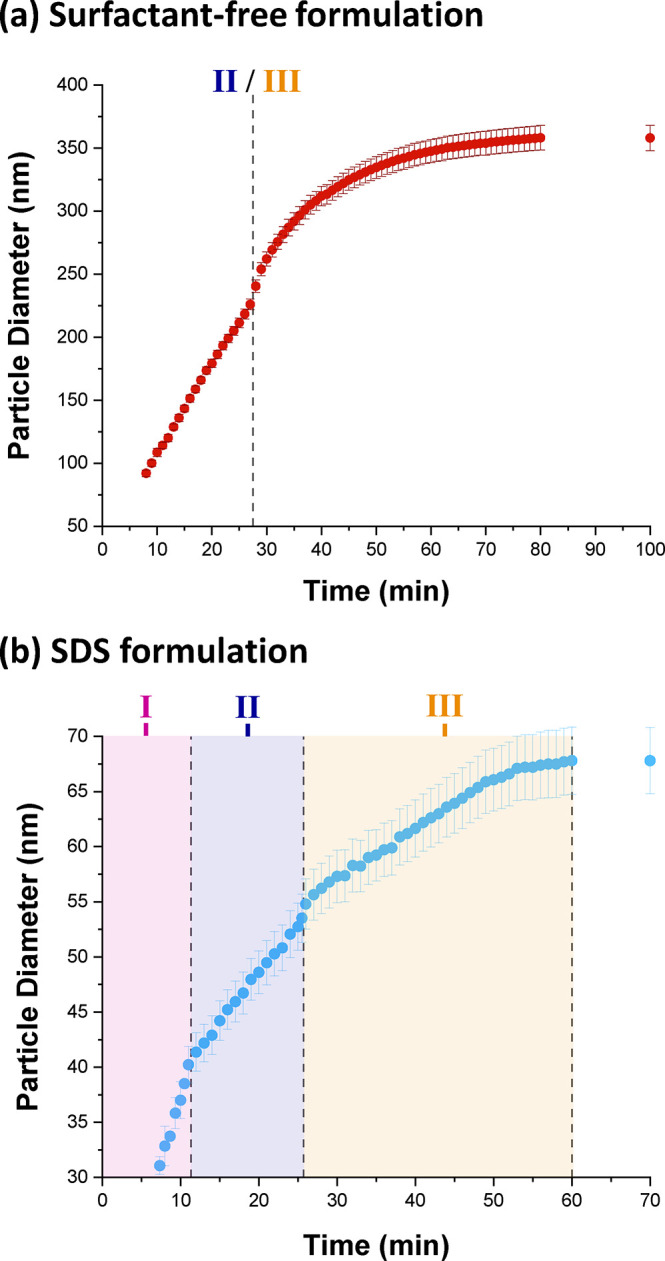
Evolution of the PTFEMA latex particle diameter determined by in situ SAXS studies conducted during the aqueous emulsion polymerization of TFEMA at 60 °C targeting 5.0% w/w solids for (a) a surfactant-free formulation and (b) an SDS formulation. The three characteristic time intervals (I, II, and III) identified by solution conductivity measurements for the SDS formulation (Figure 3) are shown for comparison. The Interval II/III boundary for the surfactant-free formulation is also shown.
Figure 8b shows the evolution in particle diameter during the in situ polymerization of TFEMA conducted in the presence of SDS surfactant. There are two regimes of linear particle growth during the first 25 min. Thereafter, there is a brief but discernible increase in the rate of particle growth, similar to that observed for the surfactant-free formulation. However, this feature is not observed by DLS for the equivalent laboratory-scale SDS synthesis (Figure 4a). Subsequently, the rate of particle growth remains constant until the TFEMA polymerization is essentially complete, producing spherical latex particles with a final volume-average particle diameter of 68 ± 4 nm. This final particle size is smaller than the volume-average particle diameter of 113 nm (polydispersity = 0.076) determined by post-mortem DLS analysis of the quenched reaction mixture. The points of inflection observed in Figure 8b occur at strikingly similar time scales to the Interval I/II and II/III boundaries indicated by in situ conductivity measurements (Figure 3). For example, the latter technique indicates that the II/III boundary occurs at around 28 min, whereas the in situ SAXS data suggests approximately 26 min. Furthermore, optical microscopy confirms that no monomer droplets are present after 30 min, see Figure S6. Such discrepancies are small and are most likely within experimental error, especially if the differing experimental set-ups (reaction volumes, heating rates etc.) are taken into account. Accordingly, the three main Intervals for the aqueous emulsion polymerization of TFEMA in the presence of SDS are assigned on Figure 8b and the likely Interval II/III boundary for the corresponding surfactant-free formulation is indicated in Figure 8a.
Conclusions
The persulfate-initiated aqueous emulsion polymerization of TFEMA at 60 °C leads to the formation of well-defined spherical latex particles when performed either under surfactant-free conditions or in the presence of SDS surfactant. This semifluorinated vinyl monomer was preferred to styrene because it ensures much stronger X-ray contrast for the corresponding latex particles relative to water. Nucleation and subsequent particle growth has been monitored in situ for both formulations utilizing a stirrable reaction cell to perform time-resolved SAXS studies. This cell has a reaction solution volume of approximately 2.0 mL, which is sufficient to allow post-mortem analysis of the final latex particles by 1H NMR spectroscopy, DLS, and TEM.
For both formulations, the rate of polymerization appears to be unaffected when subjected to synchrotron X-ray irradiation. This is in marked contrast to our prior in situ SAXS study of the synthesis of diblock copolymer nanoparticles via RAFT dispersion polymerization in mineral oil, whereby the enhanced rate of polymerization was attributed to an additional radical flux generated by the high-energy X-ray beam.36,38 Time-resolved SAXS measurements indicate that nucleation occurs after 8 min in the absence of surfactant and after 6 min in the presence of SDS surfactant, respectively. Following the nucleation event, nascent spherical nanoparticles are observed by TEM and are also detected by DLS. X-ray scattering patterns could be fitted using a simple sphere model, which enabled the evolution in particle diameter to be elucidated for both formulations. The PTFEMA latex particles prepared under surfactant-free conditions are significantly larger and also have a narrower particle size distribution, as judged by the multiple fringes observed for the corresponding patterns. Moreover, a faster rate of particle growth is observed for both formulations at intermediate monomer conversion, suggesting a transition during the polymerization. Similar behavior is also indicated by DLS analysis of the equivalent laboratory-scale synthesis of PTFEMA under surfactant-free conditions. The subsequent reduction in the rate of particle growth most likely corresponds to the disappearance of monomer droplets and hence the transition from Interval II to Interval III. Indeed, optical microscopy studies of laboratory-scale syntheses confirm that monomer droplet depletion occurs on this time scale. Furthermore, the boundaries between these three time intervals can be identified from in situ SAXS measurements for the SDS formulation and are comparable with those indicated by solution conductivity data. This observation may be important for formulations involving non-ionic surfactants, which are not amenable to solution conductivity measurements. SAXS analysis indicates final volume-average particle diameters of 353 ± 9 nm (TFEMA conversion = 99%) and 68 ± 4 nm (TFEMA conversion = 93%) for the surfactant-free and SDS formulations, respectively. These values are consistent with those obtained by 1H NMR and DLS analyses of the equivalent laboratory-scale syntheses, which confirms that the stirrable reaction cell provides sufficiently efficient stirring for representative experiments. In summary, this time-resolved SAXS study has enhanced our understanding of the mechanism of aqueous emulsion polymerization, which suggests that further studies with other vinyl monomers are warranted.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c11183.
Full experimental details for PTFEMA particles; further characterization data including aqueous electrophoresis, additional SAXS patterns, optical microscopy images of monomer droplets recorded at various TFEMA conversions, and GPC analysis of PTFEMA chains; and details and examples of the supporting analysis, along with the hard sphere scattering model used to analyze the PTFEMA latexes (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
Notes
The authors declare no competing financial interest.
Acknowledgments
S.P.A. acknowledges an EPSRC Established Career Particle Technology Fellowship grant (EP/R003009) and also postdoctoral support from The Leverhulme Trust (RPG-2016-330) for A.C. Diamond Light Source is acknowledged for granting synchrotron SAXS beam-time at I22 (proposal number SM21776). Finally, Prof. P. D. Topham (Aston, University, U.K.) is thanked for his design of the stirrable reaction cell used for these SAXS studies.