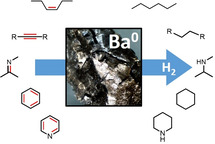
 Metallic Barium: A Versatile and Efficient Hydrogenation Catalyst
Metallic Barium: A Versatile and Efficient Hydrogenation Catalyst



- Altmetric
Ba metal was activated by evaporation and cocondensation with heptane. This black powder is a highly active hydrogenation catalyst for the reduction of a variety of unactivated (non‐conjugated) mono‐, di‐ and tri‐substituted alkenes, tetraphenylethylene, benzene, a number of polycyclic aromatic hydrocarbons, aldimines, ketimines and various pyridines. The performance of metallic Ba in hydrogenation catalysis tops that of the hitherto most active molecular group 2 metal catalysts. Depending on the substrate, two different catalytic cycles are proposed. A: a classical metal hydride cycle and B: the Ba metal cycle. The latter is proposed for substrates that are easily reduced by Ba0, that is, conjugated alkenes, alkynes, annulated rings, imines and pyridines. In addition, a mechanism in which Ba0 and BaH2 are both essential is discussed. DFT calculations on benzene hydrogenation with a simple model system (Ba/BaH2) confirm that the presence of metallic Ba has an accelerating effect.
Just a pinch of Ba metal: Efficient hydrogenation of a wide range of substrates is achieved with metallic barium previously activated by metal vapor synthesis. A mechanism in which Ba0 and BaH2 are both essential is discussed.
Introduction
Two major milestones stand at the cradle of heterogeneous metal catalysis. [1] Döbereiner observed in 1823 that hydrogen spontaneously burns in air upon contact with finely divided platinum. In 1912 Sabatier was awarded the Nobel prize in chemistry for the development of nickel catalysed alkene hydrogenation. The scientific basis for both discoveries is bond activation and cleavage at the surface of a metal.
It is hardly known that, following these groundbreaking studies on the catalytic activity of the late transition metals, catalytic ethylene hydrogenation with the much less noble alkaline earth (Ae) metal Ca has been described as early as 1925. [2] Schmidt observed that the larger group 2 metal Ba is an even more reactive hydrogenation catalyst. [3] In his extensive review on heterogeneous catalytic reactions, discrimination between two groups of metal catalysts is made. The first are the more noble transition metals with high ionization potentials that form solid solutions of hydrogen. The second class is represented by electropositive metals that form saline hydrides. It was proposed that the catalytic activity of the latter metals is strongly related to metal hydride formation. [3] Indeed, later studies by Wright and Weller show that also CaH2 and BaH2 are active in ethylene hydrogenation but only after prior thermal treatment under high vacuum, advocating that the presence of the metallic state is of importance. [4]
Although these early reports are limited only to ethylene hydrogenation, they become topical again in light of the recent interest in alkene hydrogenation with heavier group 2 metal catalysts.[ 5 , 6 , 7 , 8 , 9 ] We reported alkene hydrogenation using simple Ae metal amides like Ae[N(SiMe3)2]2 (Ae=Ca, Sr, Ba) [5b] with activities and scope sharply increasing from Ca to Ba. Contrary to expectation, these metal amides react with hydrogen to give HN(SiMe3)2 and larger metal hydride clusters of which the surface is decorated with amide ligands (Scheme 1). [9] These capping ligands solubilize the Ae hydride clusters but also prevent further aggregation to insoluble (AeH2)n salts. It was found that increasing the size of the amide ligand from N(SiMe3)2 to N(SiiPr3)2 led to a considerable boost in catalytic activity and broadened the substrate scope from alkenes to arenes. [5d] This has been rationalized by the fact that larger capping ligands like N(SiiPr3)2 form smaller, more active, metal hydride clusters. Although one would expect that the catalyst activity may be increased further using even bulkier ligands, we now report that ligand‐free metallic Ba is a very simple but highly active hydrogenation catalyst.

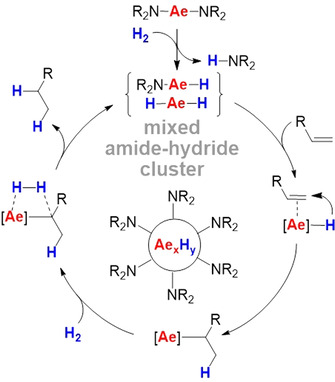
Simplified catalytic cycle for alkene hydrogenation with Ae metal amide (pre)catalysts.
Results and Discussion
In order to shed light on the historically documented catalytic activity of Ba metal and BaH2,[ 3 , 4 ] we first tested commercially available BaH2 (white powder, 60 mesh) in hydrogenation catalysis. Under various conditions (120–150 °C, 20–50 bar H2), no conversion of benzene and cyclohexene was found. Also thermal vacuum treatment of BaH2 (200 °C, 10−2 bar, 17 h) did not result in an active catalyst. Similarly, small pieces of Ba metal freshly cut under N2 are not catalytically active for alkene hydrogenation and it was found that commercially available Ba metal is essentially inert to H2 gas. This stands in strong contrast with one classical application of Ba in radio or television tubes as a getter, that is, a metal that reacts with hydrogen or other gases in order to maintain the excellent tube vacuum. [10] In this role, a barium mirror is condensed on the inside of the tube as a highly activated thin metal film. With this application in mind, we activated bulk Ba metal by evaporation and cocondensation with heptane under high vacuum, using home‐build metal‐evaporation equipment as described previously. [11] This gave after solvent removal an oxide‐free black Ba metal powder that is a highly potent catalyst for alkene, alkyne, imine and arene hydrogenation (Table 1 and Table 2).

|
Entry |
Substrate |
mol % |
H2 [bar] |
T [°C] |
t [h] |
Product(s) |
Conv. [%] |
|---|---|---|---|---|---|---|---|
|
1 |
|
5 |
12 |
25 |
3 [3] |
|
99 [99] |
|
2 |
5 |
12 |
80 |
0.5 [3] |
99 [99] | ||
|
3 |
2.5 |
12 |
100 |
1 [3] |
99 [99] | ||
|
4[a] |
|
5 |
20 |
120 |
6 [1] |
|
99 [99] |
|
5 |
|
10 |
20 |
150 |
24 [24] |
|
99 [81] |
|
6[a] |
|
10 |
20 |
150 |
24 [24] |
|
99 [73] |
|
7 |
|
5 |
12 |
100 |
3 [0.5] |
|
99 [99] |
|
8 |
|
5 |
12 |
120 |
1 [7] |
|
99 [99] |
|
9 |
|
10 |
20 |
120 |
24 [22] |
|
99 [99] |
|
10 |
|
5 |
12 |
120 |
2.5 [3] |
|
99 [99] |
|
11[a] |
|
5 |
12 |
120 |
6 [1] |
|
99 [99] |
|
12 |
|
10 |
20 |
120 |
24 [24] |
|
99 [99] |
|
13 |
|
5 |
12 |
120 |
3.5 |
|
99 |
|
14 |
|
10 |
20 |
120 |
24 |
|
99 |
|
15[a] |
|
5 |
20 |
120 |
8 |
|
99 |



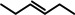

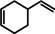
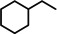




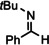
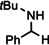
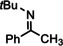
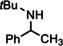
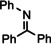
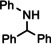
[a]=reactions performed in C6D6.
|
Entry |
Substrate |
mol % |
H2 [bar] |
T [°C] |
t [h] |
Product(s) |
Conv. [%] |
|---|---|---|---|---|---|---|---|
|
1[a] |
|
10 |
50 |
150 |
6 days [3 days] |
|
99 [18][b] |
|
2 |
|
10 |
20 |
120 |
3.5 [2] |
|
99 [99] |
|
3 |
|
5 |
20 |
120 |
12 [10] |
|
99 [99] |
|
4 |
|
5 |
12 |
120 |
24 [2.5] |
|
<5 % [94/2] |
|
5 |
|
10 |
50 |
150 |
24 [48] |
|
31/69[c] [49/51] |
|
6 |
|
5 |
20 |
120 |
25 [24] |
|
86/13 [84/15] |
|
7 |
|
10 |
50 |
120 |
24 |
|
85[c]/12[d] |
|
8 |
|
10 |
50 |
150 |
48 [24] |
|
2/34/ 43/21 [94/6/ 0/0]
|
|
9 |
|
10 |
50 |
150 |
24 [24] |
|
50 [0] |
|
10 |
|
10 |
20 |
135 |
24 |
|
99 |
|
11 |
|
10 |
50 |
150 |
2.5 days |
|
99 |
|
12 |
|
10 |
50 |
150 |
24 |
|
99[c] |
|
13[e] |
|
10 |
50 |
150 |
3.7 days |
|
99 |
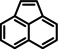
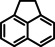
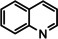
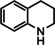
[a] neat; [b] Conditions: 2.5 mol % Ba[N(SiiPr3)2]2, 50 Bar H2, 140 °C; [c] mixture of cis‐ and trans‐isomers; [d]+ 3 % octahydro‐dimethyl‐anthracene; [e]=1 M in C6D12.
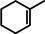
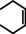
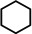
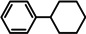
As a first test case we chose the reduction of cyclohexene, a challenging substrate [12] that previously only could be converted by the hitherto most reactive group 2 metal catalyst with a superbulky amide ligand: Ba[N(SiiPr3)2]2. [5d] Noticeably, with a catalyst loading of 5 mol % (H2 12 bar, 80 °C) cyclohexene was fully reduced within 0.5 hour (Table 1, entry 1). It is even more remarkable that at room temperature cyclohexene was fully hydrogenated within three hours (entry 2) and the catalyst concentration could easily be lowered to 2.5 mol % (entry 3). Activities generally increase with increasing pressure and temperature (Figures S80,81). Table 1 demonstrates that ligand‐free Ba metal is at least as effective in hydrogenation catalysis than Ba[N(SiiPr3)2]2; values for the latter are shown between squared brackets. Attempts to activate bulk Ba metal by dissolution in liquid ammonia and subsequent solvent evaporation gave a Ba0 powder that is essentially non‐active in cyclohexene hydrogenation (Table S5), implying that catalyst synthesis by the metal vapor method is crucial.
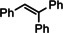
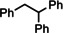
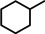
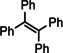
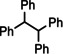
Encouraged by these preliminary results, hydrogenation of tri‐substituted alkenes was probed. While the activated (conjugated) alkene Ph2C=C(H)Ph could be fully converted at 120 °C (entry 4), the unactivated alkene 1‐Me‐cyclohexene needed a somewhat higher temperature of 150 °C (entry 5). For the first time, also the tetra‐substituted alkene, Ph2C=CPh2, could be quantitatively reduced with a group 2 metal catalyst (entry 6).
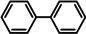
Further comparison (entries 7–10) shows that the catalytic performance of Ba metal is at least at par with that of Ba[N(SiiPr3)2]2, the hitherto most active group 2 metal hydrogenation catalyst. Noticeable, is the unproblematic full conversion of non‐cyclic internal alkenes like cis‐ and trans‐3‐hexene (entries 8,9). Also di‐substituted alkynes were fully hydrogenated at rates similar to those reported for Ba[N(SiiPr3)2]2 (entries 11,12).
For further comparison of Ba metal and Ba amide catalysts, also imine hydrogenation has been investigated (entry 13). The activity of Ba metal is comparable to that of Ba[N(SiMe3)2]2. [13] Interestingly, while the latter catalyst could only reduce aldimines, Ba metal fully converted more challenging ketimines, further extending the applicability of group 2 metal catalysis (entry 14,15).
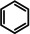
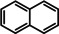
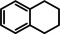
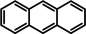
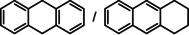
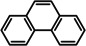
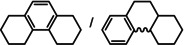
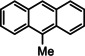
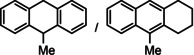
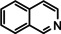
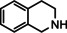
As a last showcase for the full scope of metallic Ba in hydrogenation catalysis, the reduction of aromatic rings was demonstrated (Table 2). Under forced conditions (150 °C, 50 bar), benzene was quantitatively converted to cyclohexane (entry 1). Although a prolonged reaction time of six days is needed, it should be noted that with Ba[N(SiiPr3)2]2 after three days only 18 % conversion could be reached. [5d] Conjugated rings in naphthalene or biphenyl reacted smoothly (entries 2,3) but anthracene gave essentially no conversion (entry 4), also not under forced conditions (150 °C, 50 bar). Since Ba amide catalysts gave fast reduction, failure of Ba metal to catalyse anthracene reduction may be related to the formation of insoluble, polymeric, {[Ba2+][C14H10 2−]}n salts. Indeed, hydrogenation of phenanthrene or more soluble alkylated anthracenes was successful (entries 5–7). While Ba[N(SiiPr3)2]2 was not able to reduce pyrene or acenaphthylene, catalytic quantities of Ba metal reduced up to two out of four of the pyrene rings (entry 8) while also acenaphthylene could be reduced (entry 9).
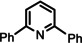
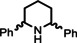
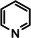
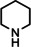
One disadvantage of the bulky Ba amide catalyst Ba[N(SiiPr3)2]2 is its inability to hydrogenate substrates that contain heteroatoms like O or N. This was attributed to substrate‐metal coordination, blocking potential coordinations sites for H2 activation. [5d] Metallic Ba, however, is able to catalyze the hydrogenation of (iso)quinoline and pyridines (entries 10–13). Despite the somewhat longer reaction times, even pyridine itself could be fully and cleanly converted to piperidine without formation of a bipyridine by‐product.
It is clear from these data that Ba metal is a highly effective, universal, hydrogenation catalyst. In contrast, it is difficult to fully understand the underlying mechanism(s). Other than in homogeneous catalysis, in heterogenous catalysis intermediates are not easily detected and characterized. Moreover, there is a growing body of opinion that a clear distinction between homogeneous or heterogeneous catalysis is often problematic. [14]
Given the low ionization potential of Ba, the logical first step is the conversion of Ba0 with H2 to BaH2. This reaction has been reported to start at 120 °C [15] but most recent literature mention an onset point of 80 °C. [16] This would not explain the rather facile cyclohexene hydrogenation at room temperature (Table 1, entry 2). It is, however, also known that oxide‐free thin films of Ba absorb H2 already at room temperature which is the basis of its application as a H2 getter. [17] Reacting black Ba powder, activated by cocondensation, overnight in a reactor with 20 bar H2, either at 20 °C or 120 °C, led in both cases to a color change to grey. It is well‐known that, although barium hydride is referred to as BaH2, often this composition is never attained and generally substoichiometric compounds are formed. [18] Therefore, we presume the product to be a Ba0/BaH2 mixture. Indeed, reaction with CH3OD led to formation of H‐D, which proves the presence of hydride. On the other hand, reaction with benzophenone gave a color change to intense deep purple and addition of pivaldehyde led to pinacolate coupling, both providing evidence for metallic Ba; Scheme 2 a (Figures S67–70).
![a) Formation of highly active Ba0 and reaction with H2 to a catalyst for alkene hydrogenation. Cycle A: metal hydride mechanism. Cycle B: metal mechanism. b) Proposed dual site catalysis:
[4a]
the alkene is activated by Ba0 and attacked by Ba hydride.](/dataresources/secured/content-1765943831275-634c947e-f685-4cab-b0ec-fa7478ffe109/assets/ANIE-60-4252-g003.jpg)
a) Formation of highly active Ba0 and reaction with H2 to a catalyst for alkene hydrogenation. Cycle A: metal hydride mechanism. Cycle B: metal mechanism. b) Proposed dual site catalysis: [4a] the alkene is activated by Ba0 and attacked by Ba hydride.
Based on an insoluble Ba0/BaH2 catalyst, two different catalytic cycles could be envisioned (Scheme 2 a): (A) the classical hydride cycle in which alkene insertion is followed by hydrogenolysis, or (B) the Ba metal cycle in which Ba first reacts with the alkene to a metallacycle after which the product is formed by hydrogenolysis.
The hydride cycle A is proposed for isolated (unactivated) alkenes, for example, 1‐hexene. This cycle might operate on the metal surface but, as previously described for Pd metal catalyzed reactions, [14c] during catalysis also smaller Ba hydride clusters, solubilized by organic groups, may go in solution. After catalytic hydrogenation generally grey turbid suspensions were obtained. Separation of the mother liquor by filtration and evaporation of all volatiles (solvent, educt and product) did not lead to visible residues and the residue can be reused as a catalyst, which suggests a heterogenous mechanism. This is supported by mercury poisoning: addition of liquid Hg to the Ba metal catalyst killed all activity (Ba amalgam reacts with H2 at 1400 °C). [18] Addition of Hg after reaction of Ba with H2, however, keeps the catalyst active. We propose therefore a heterogenous Ba0 catalyst with Ba hydride functions at its surface.
The Ba metal cycle B could be operative for easily reducible substrates like conjugated systems (e.g. Ph2C=CPh2), extended arenes, alkynes, imines or pyridines. This is exemplified by the well‐known oxidative addition of Ae metals to dienes [19] which especially for the heavier Ae metals (Ca, Sr, Ba) already proceeds at room temperature. [20] Since also imines [21] and extended π‐systems (like anthracene) [22] react with the heavier Ae metals to dianionic ions, cycle B may also in these cases be operative. Note that BaH2 is formed in the last step of cycle B. To reenter the cycle as Ba0, hydrogen must be released. Although reductive elimination (BaH2 → Ba0 + H2) starts at around 330 °C, [23] similar as for MgH2 decreasing particle size will lower the onset temperature. [24] The early work from Weller et al. shows that BaH2 can already be partially converted to Ba0 above 100 °C. [4a] Indicative for this step is the fact that substrates for which the metal mechanism is expected often need the somewhat higher temperature of 150 °C. A strong argument for metal cycle B is the fact that activated Ba0 catalyzes hydrogenation of substrates that are fully inert to Ba amide/hydride catalysts (e.g. Table 1. entries 13–15, Table 2. entries 9–13). It is, however, also possible that in some cases both cycles operate in which case regeneration of Ba0 is no longer a requirement.
The feasibility of the Ba metal mechanism B was validated by stoichiometric conversions. Reaction of Ph2C=NPh with activated Ba0 metal gave a deep red solution that is typical for the Ph2CNPh2− dianion. [21] The resulting azametallacyclopropane complex crystallized as the dimer [(Ph2CNPh)Ba⋅(THF)3]2 which could be described as two C‐N‐Ba metallacycles connected by bridging nitrogen atoms (Figure 1). The doubly charged Ph2CNPh2− anion is highly reactive and in contact with H2 the expected amine Ph2C(H)N(H)Ph was cleanly formed already at room temperature (Figures S75,76), justifying this route.
![The centrosymmetric crystal structure of [(Ph2CNPh)Ba⋅(THF)3]2; H atoms omitted for clarity. Selected bond distances [Å] and angles [°]: Ba–C1 2.953(2), Ba–N1 2.711(2), Ba–N1′ 2.773(2), Ba–O 2.755(2)–2.821(2); C1‐Ba1‐N1 29.3(7).](/dataresources/secured/content-1765943831275-634c947e-f685-4cab-b0ec-fa7478ffe109/assets/ANIE-60-4252-g001.jpg)
The centrosymmetric crystal structure of [(Ph2CNPh)Ba⋅(THF)3]2; H atoms omitted for clarity. Selected bond distances [Å] and angles [°]: Ba–C1 2.953(2), Ba–N1 2.711(2), Ba–N1′ 2.773(2), Ba–O 2.755(2)–2.821(2); C1‐Ba1‐N1 29.3(7).
As a third possibility, one could discuss a mechanism that is a hybrid between cycles A and B (Scheme 2 b): sorption of an alkene at a Ba0 surface activates the C=C bond for nucleophilic attack by a neighbouring Ba‐H functionality. Adsorption of ethylene on a Ca metal surface was already described as early as 1923 [2] and also has been established for metallic Ba. [25] Mentionable is the recent recognition that d‐orbitals play a role in the chemistry of the heavier Ae metals Ca, Sr and Ba. In metallic form they mimic transition metals and low temperature matrix‐isolation of Ae(CO)8, Ae(N2)8 and Ae(C6H6)3 complexes has been reported. [26] Similarity of Ca and Ba with the transition metals was noticed already by Wright and Weller in the 1950’s. Their comprehensive investigations support a catalyst consisting of Ba0 and BaH2 which in the presence of H2 are in a temperature dependent equilibrium. [4] Thermal treatment of BaH2 under high vacuum gave in some cases a catalyst that hydrogenates ethylene already at −78 °C. [4b] They stress the point that both, metal and metal hydride, need to be present and “the catalytic effect occurs at an interface between free metal and metal hydride”. [4a] These hot spots are called “dual sites” and, already at this very early stage, they explain the high activity of Ca and Ba catalysts with “some overlap of s‐, p‐ and d‐bands”. [4a]
It is difficult to verify heterogeneous catalytic pathways. The possibility for a Ba0/BaH2 pathway needs detailed investigations and advanced calculational studies at surfaces. Although gas phase calculations cannot model complicated heterogeneous systems, they do provide insights in basic steps at an atomic level. Here we present preliminary DFT calculations on very simple model systems. These indicate the activating influence of Ba0 in benzene hydrogenation with BaH2 (Scheme 3). The normal route (in black) shows benzene hydrogenation with BaH2. After formation of a benzene⋅⋅⋅BaH2 complex (I1), an activation energy of 8.2 kcal mol−1 is needed to reach the transition state (TS1). The Meisenheimer anion formed during this process is bound to Ba (I2). Hydrogenolysis in ortho‐position (TS2‐o) is slightly preferred over attack in the para‐position (TS2‐p). Note that reduction of the first C=C bond in benzene is endothermic. The exothermic hydrogenation of the two remaining C=C bonds, however, make benzene hydrogenation overall exothermic. [5d]
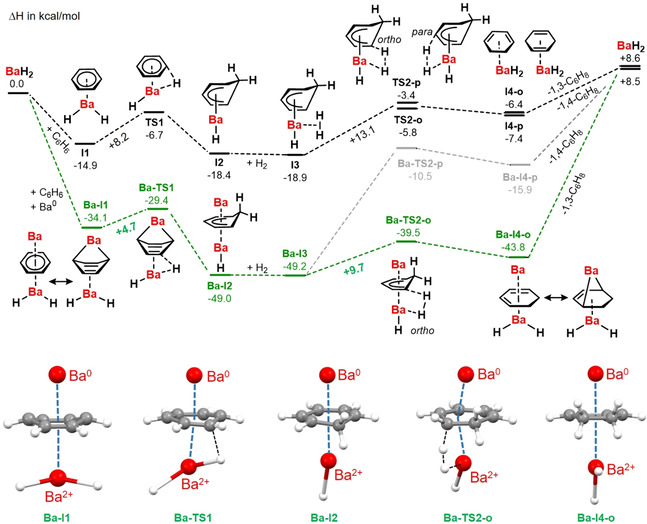
Energy profiles (ΔH in kcal mol−1) for the hydrogenation of benzene (B3PW91/def2TZVPP + GD3BJ). Black: pathway for catalyst BaH2. Green: pathway for catalyst Ba0/BaH2. Some intermediates for the Ba0 assisted route are shown at the bottom.
The same mechanism was calculated in presence of a Ba0 atom (in green). Due to its high reducing power, an interesting starting complex is formed (Ba‐I1) in which the benzene ring is not flat but slightly puckered to a boat form. Different C−C distances in the ring (1.455 Å/1.520 Å) reveal charge transfer from Ba to benzene and the system could be described as having partial Ba2+(1,3‐cyclohexadiene2−) character. Similar distortions have been observed in Ae(C6H6)3 complexes. [26c] NPA charges (Figure S88) confirm a substantial charge transfer from the Ba atom (+1.48) to the ring (−1.50) while the BaH2 unit is close to neutral (Ba +1.53, H −0.76). This reactivity could be compared to the formation of Mg‐anthracene, [19b] the difference being that benzene is much harder to reduce. This process is supported by BaH2 coordination which acts as a Lewis acid that stabilizes the anti‐aromatic 1,3‐cyclohexadiene2− anion. [27] The activation barrier for hydrogenation of 4.7 kcal mol−1 is nearly halved compared to that for the Ba0‐free route (8.2 kcal mol−1). Partial loss of aromaticity in the benzene ring assists the hydrogenation reaction. Also the activation energy for subsequent hydrogenolysis is lowered by additional interaction with Ba0 (+9.7 vs. +13.1 kcal mol−1). Note that this effect is only noticeable for hydrogenation in ortho‐position. Hydrogenolysis in para‐position leads to a drastic increase of the barrier. The preference for the ortho‐position can be explained by the favorable interaction of Ba0 with conjugated dienes (Ba‐I4‐o) compared to isolated alkenes (Ba‐I4‐p). Complex Ba‐I4‐o should rather be described as the product of 1,4‐addition of Ba0 to a diene.[ 19 , 20 ] The positive charge of +1.55 on Ba and negative charge of −1.57 on the ring confirm this view. Although these models are rudimentary, they reinforce the thesis that Ba0 can have an accelerating effect on catalytic hydrogenation.
Conclusion
Metallic barium is shown to be an all‐round catalyst for effective hydrogenation of challenging alkenes, alkynes, arenes and imines. In strong contrast with our previous approach to increase the activity of Ba amide catalysts by increasing ligand size, use of metallic Ba as a versatile, highly active, early main group metal hydrogenation catalyst not only extends the scope of substrates but is also remarkably simple. Although the metal needs to be activated, the current method represents a welcoming short‐cut in respect to the syntheses of superbulky Ba amide catalysts. Precise understanding of the fundamental mechanisms is at this stage difficult but the idea that the combination of Ba metal and Ba hydride dual sites is key to its activity has been demonstrated by DFT calculations on a simplified model system. This important aspect will be comprehensively investigated and exploited further in future research.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We kindly acknowledge the Deutsche Forschungsgemeinschaft (DFG) for funding of this project (HA 3218/9‐1). Open access funding enabled and organized by Projekt DEAL.
References
1
1a
1b
1c
2
3
4
4a
4b
4c
5
5a
5a
5b
5b
5c
5c
5d
5d
6
6a
6a
6b
6b
6c
6c
7
7
8
8a
8a
9
9a
9a
9b
9b
10
10a
10b
11
11a
11b
12
13
14
14a
14c
15
16
17
18
19
19a
19b
19b
19c
19d
20
21
22
22a
22b
23
24
24a
24b
24b
25
26
26a
26b
26c
26c
27
27