
Contributed by Nieng Yan, November 17, 2020 (sent for review August 24, 2020; reviewed by Xiaoguang Lei, Jingshi Shen, and Jian Zhang)
Author contributions: J.H., Y.Y., N.Z., X.J., N.K., N.Y., and H.Y. designed research; J.H., Y.Y., N.Z., D.P., Q.T., S.Z., S.L., X.Y., N.W., Y.X., T.Z., Z.L., T.S.-K., and X.J. performed research; J.H., Y.Y., N.Z., D.P., Q.T., S.Z., S.L., X.Y., N.W., Y.X., T.Z., Z.L., T.S.-K., X.J., N.K., N.Y., and H.Y. analyzed data; and J.H., Y.Y., N.Z., X.J., N.K., N.Y., and H.Y. wrote the paper.
Reviewers: X.L., Peking University; J.S., University of Colorado Boulder; and J.Z., Shanghai Jiao Tong University.
1J.H., Y.Y., and N.Z. contributed equally to this work.
3Present address: Department of Molecular Biology, Princeton University, Princeton, NJ 08544.
There is an urgent need for alternative antimalarials with the emergence of artemisinin-resistant malaria parasites. Blocking sugar uptake in Plasmodium falciparum by selectively inhibiting the hexose transporter P. falciparum hexose transporter 1 (PfHT1) kills the blood-stage parasites without affecting the host cells, making PfHT1 a promising therapeutic target. Here, we report the development of a series of small-molecule inhibitors that simultaneously target the orthosteric and the allosteric binding sites of PfHT1. These inhibitors all exhibit selective potency on the P. falciparum strains over human cell lines. Our findings establish the basis for the rational design of next-generation antimalarial drugs.
Artemisinin-resistant malaria parasites have emerged and have been spreading, posing a significant public health challenge. Antimalarial drugs with novel mechanisms of action are therefore urgently needed. In this report, we exploit a “selective starvation” strategy by inhibiting Plasmodium falciparum hexose transporter 1 (PfHT1), the sole hexose transporter in P. falciparum, over human glucose transporter 1 (hGLUT1), providing an alternative approach to fight against multidrug-resistant malaria parasites. The crystal structure of hGLUT3, which shares 80% sequence similarity with hGLUT1, was resolved in complex with C3361, a moderate PfHT1-specific inhibitor, at 2.3-Å resolution. Structural comparison between the present hGLUT3-C3361 and our previously reported PfHT1-C3361 confirmed the unique inhibitor binding-induced pocket in PfHT1. We then designed small molecules to simultaneously block the orthosteric and allosteric pockets of PfHT1. Through extensive structure–activity relationship studies, the TH-PF series was identified to selectively inhibit PfHT1 over hGLUT1 and potent against multiple strains of the blood-stage P. falciparum. Our findings shed light on the next-generation chemotherapeutics with a paradigm-shifting structure-based design strategy to simultaneously target the orthosteric and allosteric sites of a transporter.
Plasmodium falciparum is the deadliest species of Plasmodium, responsible for around 50% of human malaria cases and nearly all malarial death (1). Despite intensive malaria-eradication efforts to control the spread of this disease, malaria prevalence remains alarmingly high, with 228 million cases and a fatality tally of 405,000 in 2018 alone (2). The situation has become even more daunting as resistance to the first-line antimalarial agents has emerged and is rapidly spreading. For instance, artemisinin resistance, primarily mediated by P. falciparum Kelch13 (PF3D7_1343700) propeller domain mutations (3, 4), severely compromises the campaign of antimalarial chemotherapy (5678–9). Novel antimalarial agents overcoming the drug resistance are therefore urgently needed (10).
The blood-stage malaria parasites depend on a constant glucose supply as their primary source of energy (11). P. falciparum hexose transporter 1 (PfHT1; PF3D7_0204700) (12) is transcribed from a single-copy gene with no close paralogue (13) and has been genetically validated as essential for the survival of the blood-stage parasite (14). A possible approach to kill the parasite is to “starve it out” by the chemical intervention of the parasite hexose transporter (13, 15). The feasibility of this approach would depend on the successful development of selective PfHT1 inhibitors that do not affect the activities of human hexose transporter orthologs (e.g., human glucose transporter 1 [hGLUT1]).
Previously, Compound 3361 (C3361) (15), a glucose analog, has been reported to moderately inhibit PfHT1 and suppress the growth of blood-stage parasites in vitro (16). Nonetheless, the modest potency and selectivity of C3361 had limited its further development. Structural determination of PfHT1 and human glucose transporters provides an unprecedented opportunity for rational design of PfHT1-specific inhibitors (171819–20). While hGLUT1 is the primary glucose transporter in erythrocyte, its structure was determined only in the inward-open state (17). Fortunately, the neuronal glucose transporter hGLUT3, which shares over 80% sequence similarity with hGLUT1, was captured in both outward-open and outward-occluded conformations (18). A reliable homology model of outward-facing hGLUT1 could thus be generated based on the structure of hGLUT3.
Comparing the structures of PfHT1 (19, 20) and hGLUT1, we identified an additional pocket adjacent to the substrate-binding site. Coadministration of allosteric and orthosteric drugs is generally applied to tackle drug resistance when these two pockets were spatially separated (21). However, this discovery led to a hypothesis that simultaneously targeting the orthosteric and allosteric sites by tethering a pharmacophore to the carbohydrate core might render selective inhibitors for PfHT1. Based on this hypothesis, we designed a class of small molecules containing a sugar moiety and an allosteric pocket-occupying motif connected by a flexible linker. Among them, TH-PF01, TH-PF02, and TH-PF03 have exhibited selective biophysical and antiplasmodial activities with moderate cytotoxicity. Furthermore, in silico computational simulations also confirmed their binding mode, lending further support to the dual-inhibitor design. Taken together, our studies validated an antimalaria development strategy that simultaneously targets the orthosteric and allosteric sites of PfHT1.
Recently, we reported the structures of PfHT1 in complex with d-glucose and C3361 at resolutions of 2.6 and 3.7 Å, respectively (20). Structural comparison between PfHT1 and hGLUT1 shows that residues around their glucose-binding site are nearly identical (Fig. 1 and SI Appendix, Fig. S1). Interestingly, we discovered an additional pocket adjacent to the substrate-binding site, linked by a narrow channel that is highly hydrophobic in PfHT1 but more hydrophilic in hGLUT1 (Fig. 1). This proposed allosteric site was also independently confirmed by a computational method (SI Appendix, Fig. S1E) (22). Based on this observation, we hypothesized that extended carbohydrate derivatives might render selective inhibitors for PfHT1 by occupying the allosteric site.

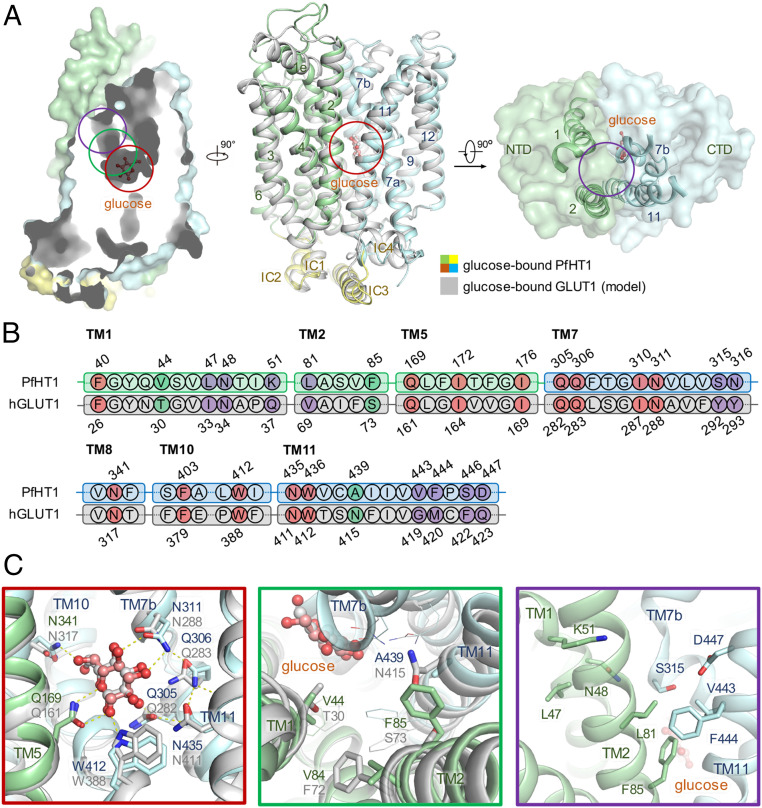
Structural comparison between PfHT1 and hGLUT1 reveals potential druggable site for PfHT1-specific inhibitors. (A) Superimposition of structures between occluded glucose–PfHT1 complex (domain colored, PDB ID code 6M20) and a model of the outward-occluded glucose–hGLUT1 complex (gray). The protein structures are shown in cartoon representation. The amino-terminal (NTD), carboxyl-terminal (CTD), and intracellular helical (ICH) domains of PfHT1 are colored in pale green, pale cyan, and yellow, respectively. (B) Sequence alignment of PfHT1 (green and cyan) and hGLUT1 (gray) highlights the portion that engages with glucose. The residues involved in the glucose-binding site, the allosteric pocket, and the connecting channel are colored in red, purple, and green, respectively. Residue numbers for PfHT1 and hGLUT1 are shown above and below the alignment, respectively. (C) Close-up views of the glucose-binding site, connecting channel, and the extended pocket are presented below, with red, green, and purple boxes, respectively.
To confirm whether the allosteric pocket can be utilized to improve the selectivity, we set up to resolve structures of human glucose transporters in the presence of C3361. Despite extensive trials, we were unable to crystalize hGLUT1 bound to C3361. It may be due to that the preferred inward-facing conformation of hGLUT1 when purified in detergents is not compatible with C3361 binding. We therefore focused on hGLUT3 and determined its crystal structure in complex with C3361 to 2.30-Å resolution (Fig. 2 and SI Appendix, Fig. S2 and Table S1).
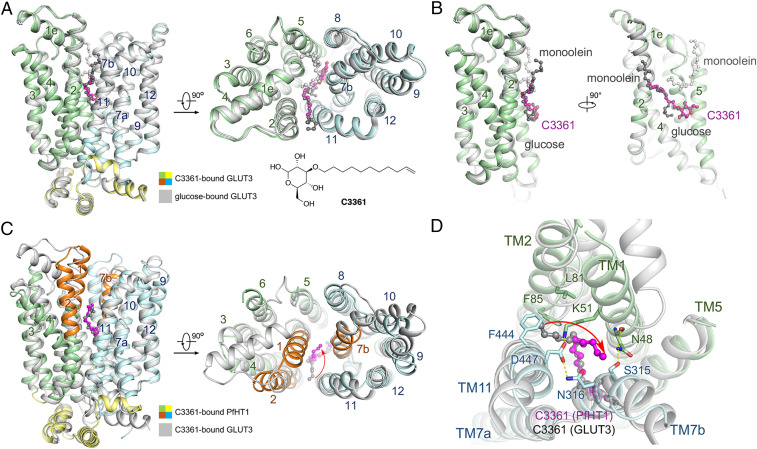
Crystal structure of hGLUT3 bound to C3361 in an outward-occluded conformation. (A) Superimposed structures of glucose–hGLUT3 complex (gray, PDB ID code 4ZW9) and C3361–hGLUT3 complex (domains colored) presented in both side and intracellular views of overall structures. The protein structures are shown in cartoon representation, and the ligands are shown in ball-and-stick representation. The amino-terminal, carboxyl-terminal, and intracellular helical domains of C3361-bound hGLUT3 are colored in pale green, pale cyan, and yellow, respectively. (B) The coordination of the sugar moiety of C3361 is nearly identical to that of d-glucose, and the tail of the C3361 occupied the position where a monoolein molecule located in the glucose-bound hGLUT3 structure. (C) Superimposed structures of C3361–hGLUT3 complex (gray) and C3361–PfHT1 complex (domains colored, PDB ID code 6M2L) presented in both side and intracellular views of overall structures. (D) C3361 demonstrated distinct binding modes when complexed with hGLUT3 or PfHT1; in particular, its tails pointed toward different directions.
The overall structure of C3361-bound hGLUT3 adopts a similar conformation with glucose-bound form (18) (Fig. 2A). The sugar moiety of C3361 is coordinated nearly identical to that of d-glucose by conserved residues in hGLUT3. The conformations of the aliphatic tail of C3361 are different in hGLUT3 and in PfHT1. In hGLUT3, the tail of C3361 occupies the pocket, which accommodates monoolein, a lipid used in lipidic cubic-phase crystallization, in the hGLUT3–glucose complex (Fig. 2B). The tail of C3361 points to the interface between transmembrane helix TM2 and TM11 (Fig. 2C). In the occluded structure of C3361-bound PfHT1 (20), the tail projects into the central cavity (Fig. 2D). These structural differences suggest that PfHT1 possesses unique intradomain flexibility that may be exploited for designing selective inhibitors that target the allosteric site of PfHT1 without inhibiting hGLUTs.
Inspired by the structural insights, we designed a series of substituted carbohydrate derivatives that consist of a sugar moiety, a tail group occupying the allosteric pocket, and an aliphatic linker (SI Appendix, Table S2). All these compounds were successfully prepared using a concise synthetic route (SI Appendix) and tested by a previously established proteoliposome-based counterflow assay against PfHT1 (SI Appendix, Fig. S3). In parallel, the blood-stage parasite growth inhibitory activities and mammalian cytotoxicity were also evaluated (SI Appendix, Table S2).
Firstly, we examined different tail groups with various sizes to fit the allosteric pocket. Among them, the bicyclic rings, such as the naphthyl (2b) (20), quinolinyl (2h) (20), and biphenyl (2d) rings, enhanced the potency, whereas smaller (2a, 2f, 2g) or larger (2q, 2s) rings lowered the potency. Different positions of the linkers have been explored, suggesting that a particular orientation is desired, presumably to fit well with the connecting channel (2b vs. 2c, 2d vs. 2e, 2h vs. 2p). The introduction of nitrogen into the naphthyl ring significantly enhanced the inhibitory potency (2h vs. 2b), suggesting potential polar interactions within the allosteric pocket. Such polar interaction was further supported by the fact that the inhibitory effect is sensitive to the positions of the nitrogen atom (2i, 2j, 2k, 2l, 2m, 2n). We also optimized the skeleton structure of 2h to further explore the pharmacophore. We found that relatively small size of the substituent group was desired; nonetheless, neither electron-withdrawing (3a, 3b, 3c, 3f) nor electron-donating (3e, 3i) groups showed impact on their potency.
Next, a C9 polymethylene linker has been shown to be optimal (1g); both shorter (1f) and longer chains (1h) decreased the activity significantly. The linkers with either ester (1a, 1b, 1d) or amido (1c) functionality lost their inhibitory activity almost completely, which might provide information on the structural constraints of the connecting channel. Installment of the tail group via amidogen ether bond (1e) rather than oxygen ether bond (2h) also decreased the potency. These structure–activity relationship (SAR) results are in good agreement with the hydrophobic channel linking the orthosteric and allosteric pockets of PfHT1 (Fig. 1C).
Finally, we explored the different substitution sites on the carbohydrate core. Various derivatives of glucose, in which the substituents were introduced (5a, 5b, 5c), were successfully prepared. The O-1 or O-4 substituted derivatives showed almost no activity (5a, 5c). By contrast, the O-2 and O-3 derivatives showed improved in vitro potency, demonstrating the necessity of the appropriate orientation (5b, 2h).
From the extensive SAR studies, three glucose derivatives, designated 3a (TH-PF01), 1g (TH-PF02), and 5b (TH-PF03), stood out as the lead compounds (Fig. 3). The half-maximal inhibitory concentration (IC50) values of the glucose transport activity for TH-PF01, TH-PF02, and TH-PF03 were determined as 0.615 ± 0.046, 0.329 ± 0.028, and 1.22 ± 0.09 μΜ for PfHT1, respectively, and 111 ± 17, 92.3 ± 7.3, and 97.3 ± 5.9 μΜ for hGLUT1, respectively. In a similar trend, the half-maximal effective concentration (EC50) values of TH-PF01, TH-PF02, and TH-PF03 for the parasite growth inhibition assay were shown to be 0.371 ± 0.026, 0.308 ± 0.004, and 0.165 ± 0.003 μΜ against the 3D7 strain, respectively, and 0.349 ± 0.003, 0.298 ± 0.021, and 0.141 ± 0.002 μΜ against the Dd2 strain of P. falciparum, respectively. Furthermore, these lead compounds showed a reasonable therapeutic window indicated by their high 50% cytotoxic concentration (CC50)/EC50 ratio values ranging from 36.1 to 107.2 (Fig. 3C). It was worth noting that all these rationally designed compounds showed equipotency to both the drug-sensitive strain (P. falciparum 3D7) and the multidrug-resistant strain (P. falciparum Dd2), starkly in contrast to quinine that only showed activity against P. falciparum 3D7 (Fig. 3A).
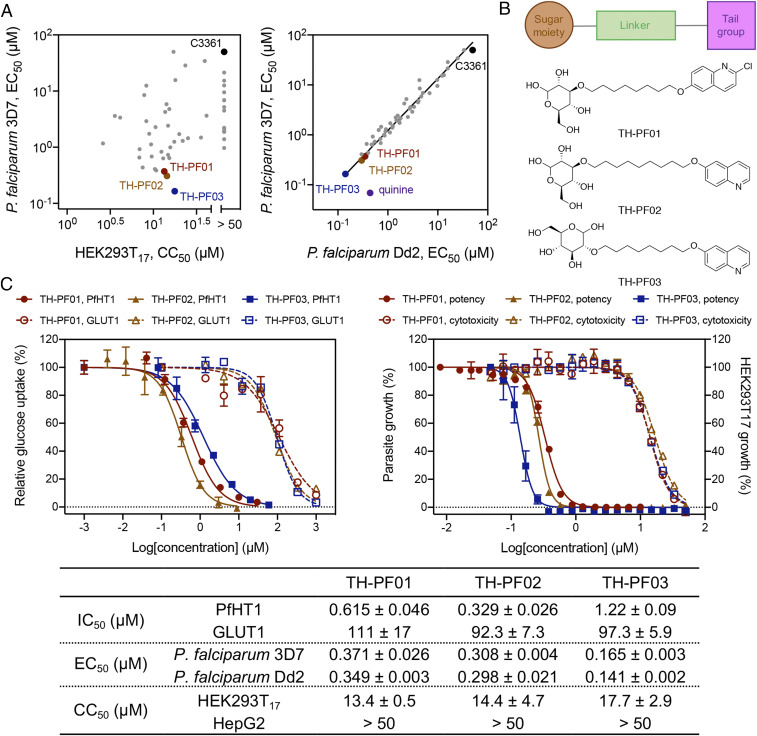
Rational design of selective inhibitors targeting PfHT1. (A) Potency against P. falciparum (3D7 and Dd2 strains) and cytotoxicity in HEK293T17. PfHT1 inhibitors showed equal potency to quinine-resistant strain, Dd2, as well as 3D7. All EC50 and CC50 values are an average of two or three biological replicates. The entire dataset is in SI Appendix, Table S2. C3361 is shown as a reference compound reported in ref. 20. (B) The generic chemical structures of TH-PF01, TH-PF02, and TH-PF03 containing a sugar moiety, a substituted heteroaromatic tail, and a flexible linker. The predicted ClogP values calculated by ChemDraw for each compound were 4.01, 3.79, and 2.06. (C) TH-PF01, TH-PF02, and TH-PF03 demonstrated robust parasite growth inhibitory activities, selectivity, and cytotoxicity. The IC50 values were determined by proteoliposome-based counterflow assay. The blood-stage P. falciparum (3D7 and Dd2) and mammalian cells (HEK293T17 and HepG2) were incubated with the compounds for 72 h, and the cell growth was quantified with SYBR Green I and Cell Titer Glo correspondingly. The data are representative of three bioreplicates and shown as average ± SD of three technical replicates.
To further elucidate the binding mode of TH-PF inhibitors against PfHT1, we next employed in silico molecular docking simulation. The results confirmed that the sugar moiety of TH-PF01 could fit into the orthosteric glucose-binding pocket of PfHT1. On the other hand, the quinoline fragment was suggested to occupy the allosteric site, forming a hydrogen bond network between TH-PF01, Lys51, and Asp447 of the protein (Fig. 4A). To validate these specific interactions observed, we performed an in silico alanine scan of residues within the TH-PF01–binding site. Reduced association energy predicted with mutants harboring K51A, Q169A, Q305A, and N341A agreed with the rational design that emphasizes the importance of the polar interactions (Fig. 4B). To further test this hypothesis, several PfHT1 recombinant mutants that contained a single point mutation (Q169A, Q305A, N341A, K51A, F85S, F85Y, V44T) or a double mutation (K51A/D447A) were experimentally prepared and measured using the counterflow assay with 1 μM TH-PF01 or isovolumetric DMSO as control. Compared with the wild type, TH-PF01 showed reduced potency to all these mutants, further confirming the computational simulation results. More specifically, Q169A, Q305A, and N341A demonstrated the interaction between the PfHT1 and the sugar moiety of TH-PF01. Additionally, both K51A and K51A/D447A showed the additional hydrogen network formed between PfHT1 and the quinoline tail group. Finally, mutations of the residues within the connecting channel to their corresponding ones in hGLUT1(F85S and V44T) or merely enhancing the polarity (F85Y) decreased the inhibitory potency, indicating the hydrophobicity of the lead compounds was indeed critical to their selectivity (Fig. 4C). Taken together, molecular simulation combined with the experimental mutagenesis strongly supported that TH-PF compounds recognize both the orthosteric and allosteric sites connected via the narrow channel as predicted (Fig. 4D).
![Biophysical characterizations of TH-PF01 binding to PfHT1. (A) The identical semitransparent cut-open view of the protein surface is shown for PfHT1 with TH-PF01 docked into the protein. Hydrogen bonds are shown as yellow dashed lines. The amino-terminal, carboxyl-terminal, and intracellular helical domains of PfHT1 are colored in pale green, pale cyan, and yellow, respectively. (B) In silico alanine scanning results. Positive values indicate that the alanine substitution interacts less favorably with TH-PF01 than the native residue. WT: wild type. (C) Key residues involved in TH-PF01 recognition were tested by protein mutagenesis. (D, Left) The curves represent the best fit of data to the competitive inhibition equation, v = Vmax[S]/((1 + [I]/Ki)Km + [S]), where Ki is the apparent inhibition constant of TH-PF01. (D, Right) Lineweaver–Burk plot of experimental kinetic data for inhibition of PfHT1 by TH-PF01, confirming a competitive inhibition mode. All experiments have been repeated three times, and the data are shown as mean ± SD.](/dataresources/secured/content-1765840672029-b7a27c9c-7a96-4112-851a-16cb224ef610/assets/pnas.2017749118fig04.jpg)
Biophysical characterizations of TH-PF01 binding to PfHT1. (A) The identical semitransparent cut-open view of the protein surface is shown for PfHT1 with TH-PF01 docked into the protein. Hydrogen bonds are shown as yellow dashed lines. The amino-terminal, carboxyl-terminal, and intracellular helical domains of PfHT1 are colored in pale green, pale cyan, and yellow, respectively. (B) In silico alanine scanning results. Positive values indicate that the alanine substitution interacts less favorably with TH-PF01 than the native residue. WT: wild type. (C) Key residues involved in TH-PF01 recognition were tested by protein mutagenesis. (D, Left) The curves represent the best fit of data to the competitive inhibition equation, v = Vmax[S]/((1 + [I]/Ki)Km + [S]), where Ki is the apparent inhibition constant of TH-PF01. (D, Right) Lineweaver–Burk plot of experimental kinetic data for inhibition of PfHT1 by TH-PF01, confirming a competitive inhibition mode. All experiments have been repeated three times, and the data are shown as mean ± SD.
We further examined whether the disruption of the PfHT1 activity can explain the growth inhibition of the TH-PF compounds. We reasoned that if the primary antiplasmodial mechanism of the TH-PF compounds was via inhibition of PfHT1, then IC50 values for the glucose transport activity of PfHT1 should correlate with EC50 values obtained in parasite growth inhibition assays. Indeed, a reasonable correlation between the two parameters was observed using 12 analogs from the TH-PF series covering a wide range of activities (Fig. 5A). We also confirmed that the EC50 values of TH-PF01 and TH-PF03 improved in the culture media with lower glucose concentration, while other antimalarial drugs quinine, mefloquine, and dihydroartemisinin showed no effects by glucose concentration (Fig. 5B). These results indicate that TH-PF01 and TH-PF03 are competing with glucose for the same substrate-binding site of PfHT1, confirming the on-target effects of the TH-PF series.
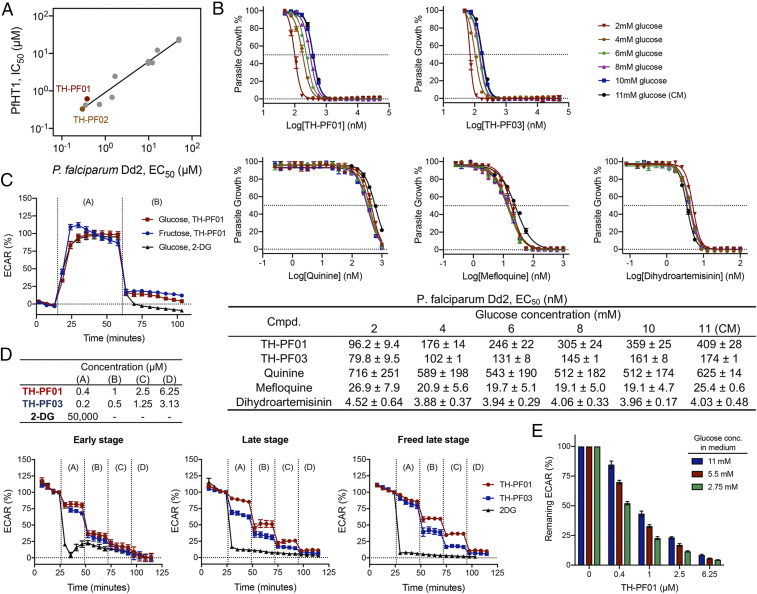
TH-PF01 derivatives selectively target PfHT1. (A) PfHT1 inhibitors showed a reasonable correlation between blood-stage parasite growth inhibition (EC50) and biochemical inhibition (IC50) of PfHT1 glucose transport activity. (B) Glucose concentration in culture media offsets EC50 values of TH-PF01 and TH-PF03 in a dose–response manner but not common antimalarial drugs quinine, mefloquine, and dihydroartemisinin. The blood-stage P. falciparum Dd2 was exposed to the test compounds in assay media containing glucose at different concentrations for 48 h starting at the ring stage, and the parasite growth was determined by SYBR Green I. The data represent four (TH-PF01) or two (TH-PF03, quinine, mefloquine, and dihydroartemisinin) independent experiments and are shown as average ± SD of two technical replicates. CM: culture medium. (C) The inhibition of glycolytic activity by TH-PF01 was observed by Seahorse extracellular flux analyzer. Dd2 schizont-stage parasites in RBCs were seeded in medium without glucose and exposed to glucose (11 mM as final concentration) or fructose (40 mM as final concentration) at 15 min (the first vertical dotted line), resulting in robust increases of ECAR. The addition of TH-PF01 (20 µM as final concentration) or 2-deoxy-d-glucose (2-DG) (50 mM as the final concentration) at 61 min (the second vertical line) lowered the ECAR, indicating glycolytic activity was inhibited. Data were normalized with ECAR values before and after the glucose additions as 0 and 100%, respectively. All data were average values pooled from two independent experiments with three technical replicates. Error bars represent SEM. (D) Extracellular flux analysis showed that TH-PF01 and TH-PF03 inhibit glycolytic activity in a dose-dependent manner in the early (rings) and late stages (trophozoites/schizonts) of the parasites in RBCs as well as freed late stages from RBCs. The Dd2 parasites were seeded in the assay medium containing glucose (11 mM), and TH-PF01 or TH-PF03 was added four times in sequence at the final concentrations of 0.4, 1, 2.5, and 6.25 or 0.2, 0.5, 1.25, and 3.13 µM. Glycolysis inhibitor, 2-DG, was added at 50 mM once. ECAR values were normalized with the values before the first compound addition as 100% and the values of background as 0%. All data were average values pooled from two independent experiments with two or three technical replicates. Error bars represent SEM. (E) The glucose concentration in the assay media and the potency of TH-PF01 against the glycolysis activity show a negative correlation. PfDd2 schizont-stage parasites in RBCs were seeded in an assay medium supplemented with glucose at 11, 5.5, or 2.75 mM, and TH-PF01 was sequentially added four times with the final concentrations of 0.4, 1, 2.5, and 6.25 µM. ECAR values after compound addition were normalized with the value before the first compound addition as 100% and the value of background as 0%. All data were average values pooled from two independent experiments with two technical replicates. Error bars represent SEM.
We further assessed whether the TH-PF series disrupts the glycolysis activity of the blood-stage P. falciparum. Seahorse extracellular flux analyzer has been used to simultaneously monitor glycolysis and mitochondrial respiration in live cells through extracellular acidification rate (ECAR) and oxygen consumption rate, respectively (23). Using purified infected red blood cells (RBCs), we observed robust initiation of glycolysis in late-stage parasites after the addition of glucose or fructose (Fig. 5C). This increased ECAR was abolished by the addition of TH-PF01 (20 μM) and 2-deoxy-d-glucose (2-DG, 50 mM), a glycolysis inhibitor, clearly demonstrating TH-PF01's inhibitory activity of glycolysis. It should be noted that 2-deoxyglucose had no effect on the increased ECAR at 50 μM. Furthermore, we confirmed that TH-PF01 and TH-PF03 reduced the ECAR at EC50 values and that the ECAR was decreased in a dose-dependent manner. This glycolysis inhibition was also observed with early-stage parasites in red blood cells and late-stage parasites extracted from red blood cells (Fig. 5D). Lastly, we measured ECAR reduction by TH-PF01 in media containing three different glucose concentrations (Fig. 5E). Similar to the EC50 shift (Fig. 5B), ECAR reduction was negatively correlated with glucose concentration. All of these findings provide strong evidence that the TH-PF compounds disrupt the glycolytic activity of the blood-stage parasites.
We further examined the glucose dependency of blood-stage P. falciparum (Dd2). First, the blood-stage parasites were incubated in Roswell Park Memorial Institute (RPMI) media with different sugars at various concentrations for 72 h, and parasite growth was quantified by a DNA dye (Fig. 6A). The blood-stage parasites grew only in the presence of glucose or fructose. Since the physiological concentrations in human blood are 3.9 to 6.9 mM for glucose (24) and 0.005 to 0.317 mM for fructose (25), glucose seems to be the sole energy source for the blood-stage P. falciparum, validating the rationale of targeting PfHT1 for antimalarial chemotherapy. We also examined how glucose consumption level changes over the blood-stage development of P. falciparum (Dd2) by monitoring the glucose consumption rate (Fig. 6B) and glycolysis activity (ECAR) (Fig. 6C). The blood-stage P. falciparum has a 2-d life cycle comprising merozoite invasion, proliferation from ring stage to trophozoite and then multicellular schizont, and egress from red blood cells (26). Both glucose consumption rate and glycolysis activity increase as the blood-stage progresses, and the schizont-stage parasites consume the majority of glucose in the culture media as an energy source (Fig. 6 B and C). Next, we investigated the substage-specific activity of the TH-PF series using lactate dehydrogenase-based assay (27). First, the ring-stage parasites (P. falciparum 3D7) were treated with TH-PF01 or dihydroartemisinin (control) for 24, 36, 48, and 72 h, and we found that the EC50 values were very similar to the previously determined 72-h growth inhibition assay (Fig. 6 D, i and E, i). Next, we treated the early ring, late ring, trophozoite, or schizont stage with TH-PF01 for 12 h. The parasites were subsequently washed with growth media and further incubated for an additional 36 h without the compound. TH-PF01 was less potent against the early blood-stage parasites (early and late ring stages) than the late blood-stage parasites (trophozoite and schizont stages). The early-stage parasites (ring stage) are less sensitive to TH-PF01, likely because they are less dependent on glucose consumption than the late-stage parasites (trophozoite and schizont) (Fig. 6 D, ii and E, ii). On the contrary, dihydroartemisinin was found to be less potent against the late stages. Lastly, we treated parasites with the compounds for 24, 36, 48, and 72 h from the early ring stage and then incubated them without the compounds for an additional 36 h (Fig. 6 D, iii and E, iii). The obtained EC50 values also showed that the ring-stage parasites were less sensitive to the PfHT1 inhibitor than late-stage parasites (24-h treatment), but longer than 36 h of treatment showed similar potency as 72-h assay. Light microscopic observations of the compound-treated parasites suggested that exposure to TH-PF01 induced the ring-stage parasites to arrest development, but the arrested parasites restart growth after removal of TH-PF01 (Fig. 6F and SI Appendix, Fig. S4).
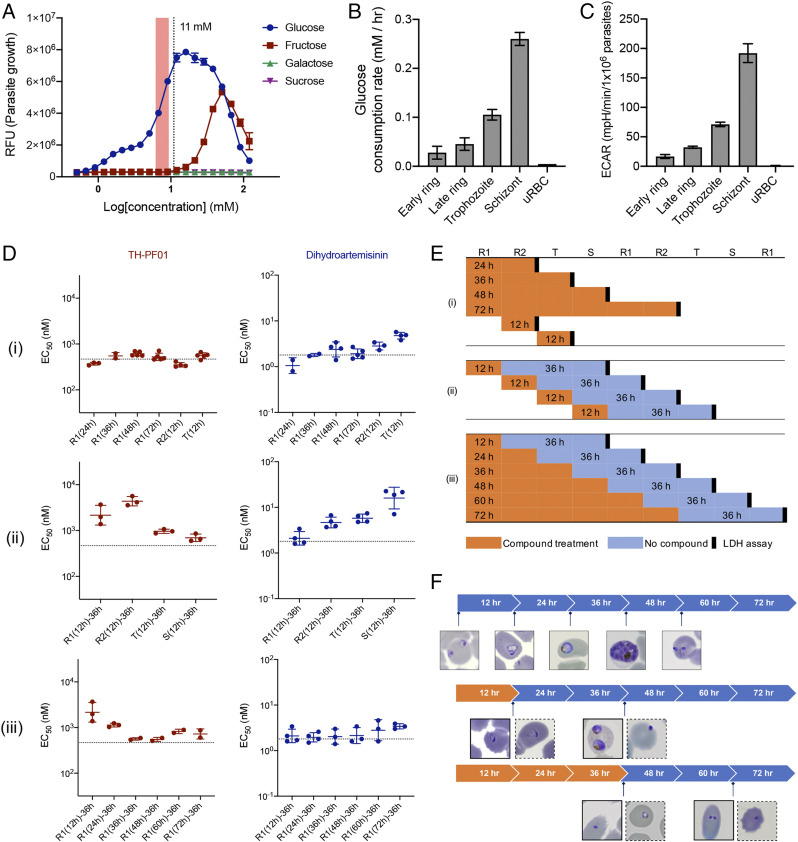
TH-PF01 suppressed parasite growth at different blood substages. (A) Glucose is essential for the survival of the blood-stage parasite. The P. falciparum (Dd2) grows only in the presence of glucose (peaking at 16 mM) or fructose (peaking at 50 mM). The physiological glucose concentration range in human blood was highlighted (3.9 to 6.9 mM). The RPMI media used for parasite culture contain 11 mM glucose (indicated by the vertical dotted line) and no fructose. The data represent two independent experiments and are shown as an average of three technical replicates. RFU: relative fluorescence units. Error bars represent SD. (B) The increased glucose consumption during the parasite developmental cycle was observed by monitoring glucose concentration in the culture medium (RPMI containing 11 mM glucose with 3% parasitemia and 1% hematocrit). The glucose concentration of two culture flasks containing tightly synchronized parasites (P. falciparum Dd2) and a flask containing uninfected RBC (uRBC) was monitored every 4 h, and parasite substages were assessed by microscopic observations. The data of parasite culture show the average of two flasks. The data of uRBC were calculated as the change of glucose concentration over 48 h. Error bars represent SD. (C) Basal ECAR shows that the schizont stage conducts the highest glycolysis among the substages. Early ring-, late ring-, trophozoite-, and schizont-stage parasites (PfDd2) in RBCs were seeded in an assay medium containing glucose (11 mM), and the basal ECAR was determined as the difference before and after 2-deoxy-d-glucose (2-DG, 50 mM as a final concentration) addition. Data were normalized with seeding density and parasitemia. All data were average values pooled from two to five independent experiments with three technical replicates. Error bars represent SEM. (D) TH-PF01 appeared as equipotent to all substages when the survival was assessed immediately after compound treatment; however, it required longer incubation time against ring-stage than late-stage parasites to show the same potency when incubated for an additional 36 h after washed. EC50 values of TH-PF01 and dihydroartemisinin against substages were determined by lactate dehydrogenase (LDH) assay in the time course experiments depicted in E, i, ii, and iii. The horizontal dashed lines indicate the EC50 value determined by a 72-h SYBR assay. The data show an average EC50 with SD of two to five bioreplicates. (E) A schematic representation of the substage assay. Tightly synchronized parasites (Pf3D7) were exposed to TH-PF01 or dihydroartemisinin for various periods at the substages indicated. (F) Representative images of compound-treated parasites (SI Appendix, Fig. S5). Solid outline, TH-PF01 treated parasites; dotted outline, dihydroartemisinin-treated parasites.
With the continuous emergence and spread of drug resistance, current antimalaria chemotherapies are facing serious limitations (3). Innovative drug discovery strategies, novel targets, and therapeutic agents for malaria treatment are an urgent need. Given that proliferation of the malaria parasites depends on d-glucose, we have conceptualized a “selective starvation” strategy. Decreasing the uptake of d-glucose via PfHT1, the sole hexose importer in P. falciparum could be a potential venue to kill drug-resistant malaria parasites. A comparison of the crystal structures of PfHT1 (20) and hGLUT1 led to the discovery of an allosteric site that further prompted us to design and develop selective PfHT1 inhibitors over its human orthologs. The TH-PF series demonstrated a strong correlation between blood-stage growth inhibition and the biophysical inhibition of glucose transporting activity. Moreover, the inhibitors quickly shut down the glycolysis of the blood-stage parasites and demonstrated even higher potency when glucose concentration in the growth media was lowered, confirming that PfHT1 is not only the molecular target of the TH-PF series but also, the sole sugar transporter at least in the blood stage. Approximately 15 to 20% of malaria patients suffer from life-threatening hypoglycemia and other complications, including irreversible brain damage and neurological sequelae (28). Thus, inhibiting PfHT1 cannot only starve and kill the blood-stage parasites but also, might quickly relieve hypoglycemia symptoms. Lastly, these PfHT1 inhibitors may validate an approach for structure-based drug design. This is an example of dual inhibition of the orthosteric and allosteric sites for a transmembrane protein. Our findings serve as proof of the concept that PfHT1 is a druggable target for next-generation antimalarials, laying a foundation for future therapeutic development.
Protein expression and purification methods of hGLUT1 (N45T), hGLUT3 (N43T), and PfHT1 were described previously (17, 18, 20). In brief, PfHT1 and mutants were expressed in Sf9 cells and extracted in lysis buffer (25 mM 2-(N-morpholino)ethanesulfonic acid [MES], pH 6.0, 150 mM NaCl) plus 2% (wt/vol) n-dodecyl-β-D-maltopyranoside (DDM). After loading with protein extraction, the nickel-nitrilotriacetic acid (Ni-NTA) resin was washed with lysis buffer plus 0.02% (wt/vol) DDM and 30 mM imidazole. Protein was eluted with wash buffer supplemented with 270 mM imidazole and applied to size-exclusion chromatography in lysis buffer plus 0.02% (wt/vol) DDM.
Proteoliposome preparation and counterflow assay were performed as previously described (20).
To determine the inhibition mechanism of inhibitors, the nonradiolabeled glucose concentration in the external reaction solution was from 0.5 to 4 mM, and the initial velocities were measured at 15 s. The data were fitted to the competitive inhibition equation, v = Vmax[S]/((1 + [I]/Ki)Km + [S]), in Prism 8. All experiments were performed three times and expressed as mean ± SD.
Lipidic cubic-phase (LCP) crystallization was performed as previously described (18, 20). In brief, hGLUT3 protein purified in 0.06% (wt/vol) Cymal-6 was concentrated to ∼40 mg/mL and incubated with 50 mM C3361 for 1 h. Seventy-five nanoliters protein mixed with monoolein was loaded into well with 1 μL precipitant solution containing 0.1 M 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (Hepes), pH 7.0, 0.1 M NH4Cl, and 40% polyethylene glycol 400 (PEG400). Crystals grew at 20 °C and reached full size in 1 wk.
The X-ray diffraction data were collected at the BL32XU beamline of SPring-8, Japan. Due to the small size of LCP crystals, a 10- × 15-μm microfocus beam with 1.0-Å wavelength was applied for data collection. Wedges of 10° were collected for every single crystal with a 0.1° oscillation angle through the EIGER X 9M detector. Hundreds of datasets were screened and automatically collected by ZOO (29), followed by first-round automatic data processing through KAMO (30). Datasets with good diffraction and low R-merge factor were manually picked out and merged through XDS (31). Further processing was carried out using the CCP4 suite (32). The phase was solved by molecular replacement with a search model of hGLUT3 (Protein Data Bank [PDB] ID code 4ZW9) through PHASER (33). The structural model was adjusted through COOT (34) and refined by PHENIX (35). SI Appendix, Table S1 summarizes the statistics for data collection and structure refinement.
AlloSite server (http://mdl.shsmu.edu.cn/AST/) was used to predict potential allosteric sites on the basis of the structure of PfHT1 (PDB ID code 6M2L). The outward-occluded models of hGLUT1 were built and refined based on the crystal structure of glucose-bound hGLUT3 (PDB ID code 4ZW9) as a template within Modeller-9.19 (36), and the best model was chosen by PROCHECK (37). Molecules (glucose and TH-PF01) were drawn in two-dimensional sketcher in Schrödinger suite 2018–1 (38), and three-dimensional structures were processed by default setting using the Ligprep program (39). The protein structures were processed by default setting using the Protein Preparation Wizard. Molecules were docked against PfHT1 (PDB ID code 6M2L) or hGLUT1 model using the extraprecision docking (Glide XP) method within the Glide program.
The binding pocket was defined by identifying residues in direct contact with TH-PF01, including F40, V44, L47, N48, K51, L81, F85, Q169, I172, T173, I176, Q305, Q306, I310, N311, V312, S315, N316, N341, F403, W412, N435, W436, A439, V443, and S446. To validate the role of these residues in the inhibitor binding, each pocket residue was mutated to alanine in silico using PyRosetta (40, 41). Then, the relative binding free energy change (ΔΔG) of each mutant over the wild type was calculated using the Prime-molecular mechanics/generalized Born surface area (MM/GBSA) method, keeping the ligand and other residues fixed.
Compounds were prepared by a concise synthetic route. SI Appendix, Supplementary Information Text has details.
The blood stage of P. falciparum (Dd2 and 3D7) was cultured following standard methods (20, 42). The parasite cultures (regularly synchronized by 5% sorbitol [Sigma-Aldrich] treatment) were maintained at 37 °C in an atmosphere of 5% O2, 5% CO2, and 90% N2.
In vitro potency of PfHT1 inhibitors was determined by the parasite growth assay as previously described (20). Briefly, synchronized ring-stage parasites were cultured in the presence of the test compounds in culture medium (50 µL) at 1.0% parasitemia and 0.8% hematocrit in the regular culture condition. Dihydroartemisinin (5 µM) was used as killing control. After 72-h incubation, SYBR Green I fluorescent dye (Invitrogen) in lysis buffer (20 mM Tris⋅HCl [Sangon Biotech], 5 mM ethylene diamine tetraacetic acid (EDTA) [Thermo Scientific], 0.16% [wt/vol] Saponin [Sangon Biotech], 1.6% [vol/vol] Triton X-100 [Sangon Biotech]) was added, and fluorescence intensity was measured by Envision (PerkinElmer). EC50 values were calculated using Prism Software version 8 (GraphPad). The reported EC50 values were the results of two technical and at least two biological replicates.
Glucose (Sigma-Aldrich) was dissolved in glucose-free RPMI 1640 media (Thermo Fisher Scientific) to prepare glucose-containing culture media of the concentrations of 2, 4, 6, 8, 10, and 11 mM; adjusted to 7.4 pH; and sterile filtered. The compound susceptibility against Dd2 parasites was measured in the same way described above, but the incubation period was extended to 48 h (20).
A tightly synchronized 3D7 culture was obtained by a combination of sorbitol synchronization and heparin treatment. After sorbitol synchronization, the obtained ring-stage parasites were cultured with the presence of heparin sodium salt from Porcine Intestinal Mucosa (230 μg/mL; Sigma-Aldrich) until the majority of parasites developed to the late schizont stage. Then, heparin was removed from culture to allow merozoites to invade erythrocytes. After 6 h, the culture was sorbitol synchronized, resulting in early ring-stage parasites (∼0 to 6 hours post infection, hpi), and resuspended in culture medium at 1.0% parasitemia and 0.8% hematocrit. The test compounds were prepared in the same way described above, and the obtained tightly synchronized culture was exposed to the tested compounds in the schedule scheme in Fig. 6. After incubation with test compounds, the culture was freeze thawed, and drug susceptibility was determined by lactate dehydrogenase (LDH) assay (43).
HEK293T17 and HepG2 were purchased from ATCC and then seeded in suitable plates filled with 89% DMEM (Gibco) supplemented with 10% inactivated fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Gibco) maintained in an atmosphere at 37 °C with 5% CO2.
Cytotoxicity was determined by cell viability assay as previously described (20). The test compounds dissolved in dimethyl sulfoxide (DMSO) were transferred into 384-well white, solid-bottom plates (Corning) by D300e Digital Dispenser (Tecan). Puromycin was used (5 µM) as killing control. HEK293T17 and HepG2 cells were seeded into the assay plates at ∼2,000 and 2,500 cells per well, respectively, and incubated for 72 h at 37 °C. The cell viability was measured using Cell-Titer Glo (Promega) according to the manufacturers' instructions, and luminescence signals were measured by Envision (PerkinElmer) with US LUM 384 setting. CC50 values were calculated using a nonlinear regression curve fit in Prism Software version 8 (GraphPad). The reported CC50 values were the results of two technical and at least two biological replicates.
All assays were conducted according to the manufacturer’s manual with some modifications. A sensor cartridge was hydrated overnight in XF Calibrant Solution at 37 °C. Two assay media were employed for the analysis of parasites: RPMI medium 1640 (Thermo Fisher Scientific; containing glucose 11 mM) and Seahorse XF RPMI Medium (Agilent Technologies). Injection solutions containing test compounds were prepared in assay medium at 10× of final concentration and loaded in the reagent delivery chambers of the sensor (20, 22, 24.5, and 27 μL for the first, second, third, and fourth injections, respectively). Late-stage Dd2 parasites in RBCs were magnetically purified from 5% sorbitol-synchronized cultures using MACS LD columns (Miltenyi Biotec) and seeded at 0.8 million RBCs per well in a Seahorse miniplate, which was precoated with Cell-Tak cell and tissue adhesive (Corning). Ring-stage Dd2 parasites in RBCs were obtained at roughly 20% parasitemia by culturing magnetic-activated cell sorting (MACS)-purified schizonts with a small amount of fresh blood overnight and seeded at 0.8 million RBCs per well. Freed schizonts were prepared by saponin lysis (23) and seeded at 2.5 million RBCs per well. After centrifugation at 500 rpm for 5 min with slow acceleration and no braking, assay medium was added to all wells (180 μL as final volume), and the miniplate was loaded into the flux analyzer to start measurements (mix time: 30 s; wait time: 1 min and 30 s; measure time: 3 min). In an assay plate, two wells were used for background correction.
For analysis of HEK293T17 cells, Seahorse XF DMEM (Agilent Technologies) supplemented with glucose (25 mM), pyruvate (1 mM), and glutamine (4 mM) was used. HEK293T17 cells were preseeded at 60,000 cells per well in culture medium (80 μL) in a Seahorse miniplate precoated with Cell-Tak and adhesive in the same way described above. After culturing overnight, the medium was exchanged to the assay medium (180 μL per well), and the cells were placed in the non-CO2 incubator at 37 °C for 1 h. The assay was conducted in the same way described above.
This work was supported by National Natural Science Foundation of China Grants 21825702, 31630017, and 81861138009. H.Y. acknowledges Beijing Outstanding Young Scientist Program Grant BJJWZYJ-H01201910003013. We thank the Beijing Municipal Government, the Bill & Melinda Gates Foundation and Global Health Drug Discovery Institute (GHDDI) for support. We also thank Kunio Hirata at Super Photon ring-8 (SPring-8, Japan) BL32XU beamline for the help of crystal screening and data collection and the X-ray Crystallography Platform of the Tsinghua University Technology Center for Protein Research for the crystallization facility.
The coordinates and structure factors for hGLUT3 bound to C3361 have been deposited in the PDB (ID code 7CRZ). All additional study data are included in the article and SI Appendix.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
18
19
20
21
22
23
24.
25
27
28
29
30
32
34
35
36
37
38.
39
40
41
42
 Orthosteric–allosteric dual inhibitors of PfHT1 as selective antimalarial agents
Orthosteric–allosteric dual inhibitors of PfHT1 as selective antimalarial agents
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

