
Edited by Tullio Pozzan, University of Padova, Padova, Italy, and approved December 7, 2020 (received for review August 5, 2020)
Author contributions: R.K. designed research; Z.Z., K.O., J.O., T.H., M.A., and R.K. performed research; M.I. and K.F.T. contributed new reagents/analytic tools; Z.Z., Y.I., and R.K. analyzed data; and Z.Z. and R.K. wrote the paper.
Astrocytes have been suggested as important modulators of memory. However, intracellular signaling that regulates astrocytic function in memory modulation remains to be clarified. Here, we generated a transgenic mouse line, Mlc1-bPAC, in which astrocytes increase their intracellular cAMP levels upon blue light stimulation. Using this method, we have uncovered that light-induced increase in astrocytic cAMP is sufficient to induce synaptic plasticity and modulate memory. Furthermore, we have shown that an increase in astrocytic cAMP activates the astrocyte–neuron lactate shuttle, which is essential for synaptic plasticity and memory modulation. These findings indicate the importance of astrocytic cAMP in synaptic plasticity and memory and provide a method for investigating astrocyte functions in vivo in the future.
Astrocytes play a key role in brain homeostasis and functions such as memory. Specifically, astrocytes express multiple receptors that transduce signals via the second messenger cAMP. However, the involvement of astrocytic cAMP in animal behavior and the underlying glial–neuronal interactions remains largely unknown. Here, we show that an increase in astrocytic cAMP is sufficient to induce synaptic plasticity and modulate memory. We developed a method to increase astrocytic cAMP levels in vivo using photoactivated adenylyl cyclase and found that increased cAMP in hippocampal astrocytes at different time points facilitated memory formation but interrupted memory retention via NMDA receptor–dependent plasticity. Furthermore, we found that the cAMP-induced modulation of memory was mediated by the astrocyte–neuron lactate shuttle. Thus, our study unveils a role of astrocytic cAMP in brain function by providing a tool to modulate astrocytic cAMP in vivo.
Astrocytes are abundant glial cells in the brain and play essential roles in brain homeostasis and functions (1). For example, manipulation of astrocytic intracellular signaling, such as the activation of Ca2+ signals, has been shown to modulate numerous domains of animal behaviors, including attention, fear, decision making, and memory (234–5).
Stable long-term memory is formed through initial memory acquisition followed by memory consolidation, during which synaptic plasticity occurs in memory-related neural circuits. The neurons undergoing synaptic plasticity during memory formation are considered to be responsible for the encoding of memory. After memory formation, memory is maintained through a process known as memory retention, which is believed to involve the preservation of changes in neural circuits (6). Then, memory can be retrieved through the reactivation of related neural circuits (7).
Previous reports have shown that astrocytic gliotransmitters play an important role in memory (3, 89–10). For example, astrocyte-derived lactate is essential for synaptic plasticity and the formation of long-term memory (8, 9). D-serine, another astrocytic gliotransmitter, has also been shown to be necessary for contextual memory formation (10). In addition, astrocytic release of glutamate and adenosine triphosphate (ATP) is reported to modulate fear memory (3). However, the astrocytic intracellular signaling that governs the release of these gliotransmitters is not fully understood.
Among intracellular signaling molecules, cyclic adenosine monophosphate (cAMP) is one that has attracted the attention of researchers. cAMP is constantly produced in astrocytes in vitro, as evidenced by the immediate decrease in cAMP levels following adenylyl cyclase blockade (11). Astrocytic cAMP pathways have also been confirmed in vivo via gene expression analyses (12). In addition, in vivo cAMP imaging has shown that the cAMP levels in cortical astrocytes increase in response to repeated sensory stimulation (13). In vitro studies have shown that cAMP signals regulate basic astrocyte functions, including energy metabolism, gap junction formation, and glutamate recycling in vitro (141516–17). Importantly, an increase in astrocytic cAMP in acute brain slices has been shown to induce lactate release, which is essential in synaptic plasticity and memory (8, 14). However, it has not been determined whether and how astrocytic cAMP modulates memory, and the underlying astrocyte–neuron interaction remains unknown due to a lack of methods to increase cAMP levels specifically in astrocytes.
Here, we developed a double-transgenic mouse line to manipulate cAMP signals specifically in astrocytes both in vitro and in vivo with high temporal resolution. Using this method, we aimed to investigate the role of astrocytic cAMP in the modulation of synaptic plasticity and memory. Specifically, we examined the impact of cAMP increases in the hippocampus during memory formation and retention, both of which affect memory performance. Our results show that increasing astrocytic cAMP is sufficient to facilitate memory formation and impair memory retention. Furthermore, we elucidated the mechanisms underlying the behavioral changes induced by astrocytic cAMP increases. We found that astrocytic cAMP activates lactate release, which subsequently activates NMDAR receptors and induces synaptic plasticity.
By manipulating astrocytic cAMP, our study has provided solid in vivo evidence for astrocytic cAMP–dependent memory modulation via lactate, filling a previous gap in the literature, and has clearly highlighted a potential role of astrocytic cAMP in controlling memory formation and retention.
To increase the intracellular cAMP levels in mouse astrocytes with high temporal resolution in vivo, we adopted an optogenetic approach using photoactivated adenylyl cyclase (PAC). PAC is known to be activated by blue light to convert ATP to cAMP and immediately returns to its inactivated form in the dark (18) (Fig. 1A). We utilized photoactivated cyclase from Beggiatoa sp. (bPAC), which is found in the bacterium Beggiatoa (19), because of its small size and low enzyme activity in dark conditions. To generate a mouse line that expressed bPAC specifically in astrocytes, we used the bigenic tTA-tetO system (20). First, we generated a mouse line expressing bPAC downstream of the Tet operator. We used knockin-mediated enhanced gene expression by improved tetracycline-controlled gene induction (KENGE-tet) (21) to enable sufficient expression of the target genes for generation of the tetO-GFP-2A-bPAC mice (Fig. 1B). Then, the tetO-GFP-2A-bPAC mice were crossed with Mlc1-tTA mice, which were previously used to achieve astrocyte-specific expression of targeted genes (22, 23), to generate an Mlc1-tTA::tetO-GFP-2A-bPAC (Mlc1-bPAC) mouse line (Fig. 1B). We observed GFP expression in astrocytes that were immunopositive for GFAP and S100β throughout the brain in postnatal (P7; SI Appendix, Fig. S1) and adult (P30, Fig. 1 B and C and SI Appendix, Fig. S2 A, B, and D) Mlcl-bPAC mice. Specifically, we found that ∼95% of the GFAP-positive cells and 90% of the S100β-positive cells expressed GFP (SI Appendix, Fig. S2 B–E). We also confirmed that neither the neuronal marker NeuN nor the microglial marker Iba1 was expressed by GFP-positive cells (208 cortical cells and 346 hippocampal cells from four mice; SI Appendix, Fig. S3), indicating astrocyte-specific expression of GFP in Mlc1-bPAC mice. Finally, we confirmed that GFP expression was abolished in the Mlc1-bPAC mice by administrating doxycycline, which is known to turn off tTA-tetO binding (20) (SI Appendix, Fig. S4).

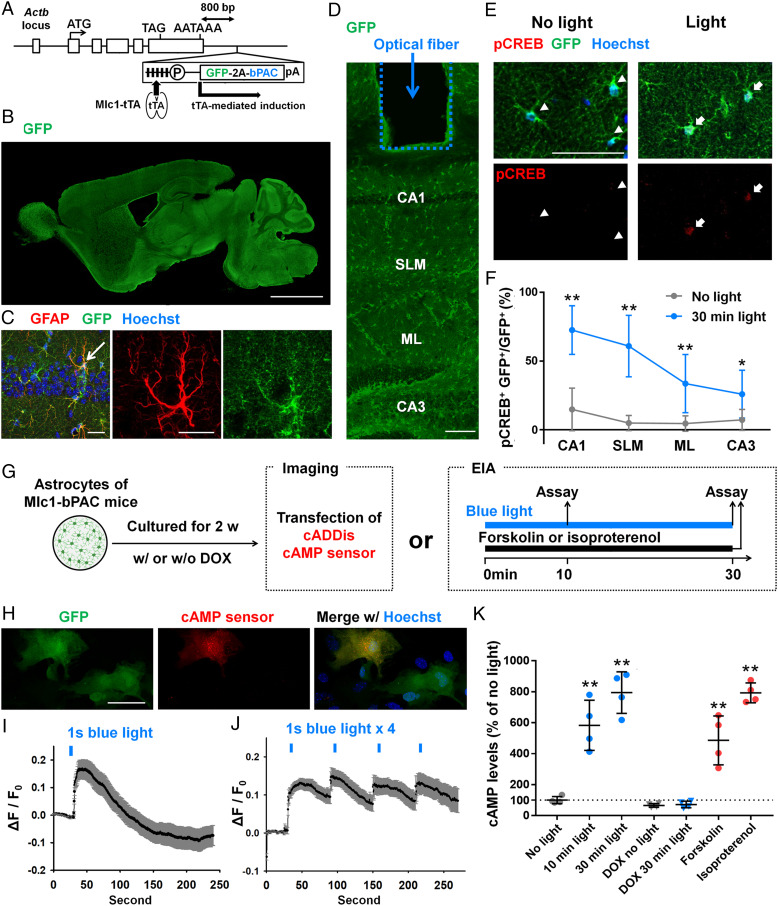
Astrocyte-specific regulation of cAMP using optogenetics. (A) Strategies to generate Mlc1-tTA::tetO-GFP-2A-bPAC mice. Astrocyte-specific GFP-2A-bPAC expression was induced by Mlc1-tTA. (B) A representative image of a sagittal brain section prepared from P30 Mlc1-bPAC mice. GFP expression was detected throughout the brain. (Scale bar, 2 mm.) (C) A representative image of the hippocampal CA1 region immunostained for GFP and GFAP (red). An arrowed astrocyte is shown in the right column. (Scale bar, 20 μm.) (D) A representative image of the trace of an optical fiber. (Scale bar, 100 μm.) (E) Images of hippocampal astrocytes immunostained with GFP and pCREB. Arrowheads indicate astrocytes in the no-light group, and arrows indicate astrocytes in the light group. (Scale bar, 100 μm.) (F) The pCREB-positive rate in GFP-positive astrocytes. n = 6 mice for each group (801 cells were analyzed in the no-light group, and 802 cells were analyzed in the 30 min light group). **P < 0.01 versus the Mlc1-bPAC + no-light group by Student’s t test. SLM = stratum lacunosum-moleculare; ML = molecular layer. (G) Schematics of the experimental paradigm. Astrocytes were cultured from P1 Mlc1-bPAC mouse cortices for 2 wk with or without 1 μM doxycycline before treatment and subsequently processed for cAMP imaging or enzyme immunoassay (EIA). (H) A representative image of cultured astrocytes derived from Mlc1-bPAC mice that also express the cAMP sensor (red). (Scale bar, 100 μm.) (I and J) Change in red fluorescent protein fluorescence of the cAMP sensor in response to single (I) or multiple (J) blue light stimulation. n = 19 cells for I. n = 49 cells for J. Data are presented as the mean ± SEM (K) cAMP levels measured by EIAs after blue light or reagent stimulation. n = 4 cultures for each group. **P < 0.01 versus the no-light group by Tukey’s test after one-way ANOVA. Data are presented as the mean ± SD (except I and J).
Next, we examined the function of bPAC in the primary culture of astrocytes prepared from Mlc1-bPAC mice (Fig. 1G). We first examined the dynamics of the intracellular cAMP levels in response to blue light stimulation (Fig. 1G). We transfected cultured astrocytes with a genetically encoded red fluorescent cAMP sensor, cADDis, to monitor the cAMP changes (24, 25) (Fig. 1 G and H). Stimulation with blue light for 1 s immediately increased the fluorescence of the cAMP sensor (ΔF/F0), with its peak at ∼10 s after stimulation, and the fluorescence returned to baseline in minutes (Fig. 1I). When a train of blue light stimulation (1 s/min) was applied, the intracellular cAMP levels increased rapidly after each stimulus and were sustained for minutes (Fig. 1J). These results showed that bPAC rapidly produces cAMP in response to blue light stimulation.
Next, we performed enzyme immunoassays (EIAs) to further confirm the bPAC-mediated increase in intracellular cAMP levels in cultured astrocytes (Fig. 1G). We found that 10 min and 30 min of blue light stimulation (pulsed, 1 s/5 s) elevated the intracellular cAMP levels to the same extent as pharmacological induction of cAMP with isoproterenol or forskolin but not in cultures that were pretreated with doxycycline (Fig. 1K).
Finally, we examined whether cAMP is also elevated by blue light in vivo. Optical fibers were implanted in the hippocampal CA1 region to deliver blue light to awake mice (Fig. 1D). We investigated the phosphorylation of cAMP response element binding protein (CREB) as an indicator of the intracellular cAMP increase (26) and found that 30 min of blue light stimulation (pulsed, 1 s/5 s) increased the number of phosphorylated CREB (pCREB)-positive astrocytes (Fig. 1 E and F). The proportion of pCREB-positive astrocytes was higher in the region near the optical fiber (i.e., CA1), and the farther from the optical fiber, the smaller the pCREB positive ratio became (Fig. 1F), which indicated that the increase in astrocytic pCREB was light stimulation dependent. We also attempted nonintrusive light stimulation using light-emitting diodes (LEDs) by attaching LED lamps to one side of the skulls of anesthetized Mlc1-bPAC mice. In both the cortex and hippocampus, light stimulation induced higher pCREB levels in GFP-positive astrocytes than in GFP-negative astrocytes or cells on the contralateral side (SI Appendix, Fig. S5).
We then used Mlc1-bPAC mice to investigate whether and how astrocytic cAMP increases can modulate memory formation and memory retention. We subjected the Mlc1-bPAC mice to a hippocampus-dependent spatial memory test—specifically, the object–location memory test—in which mice learn the location of two identical objects during training and are tasked with distinguishing the relocated object from the unmoved object during the test (27, 28) (Fig. 2A). To examine the influence of astrocytic cAMP increases on memory during different memory stages, we devised different behavioral paradigms in which blue light stimulation was given to the bilateral hippocampus specifically during either memory formation (Fig. 2B) or memory retention (Fig. 2 D and F). First, we applied blue light stimulation (pulsed, 1 s/5 s) using optical fibers to the Mlc1-bPAC mice around the training session to examine the role of astrocytic cAMP in memory formation: 10 min of blue light stimulation was given immediately before, during, or immediately after training, and the memory test was carried out on day 5 (Fig. 2B). The Mlc1-bPAC mice in no light group could not distinguish the relocated object from the other object, on the other hand, the discrimination score was significantly higher when blue light stimulation was conducted during or immediately after training than before training (Fig. 2C). These results indicate that the increase in astrocytic cAMP during and immediately after training contributed to memory formation.
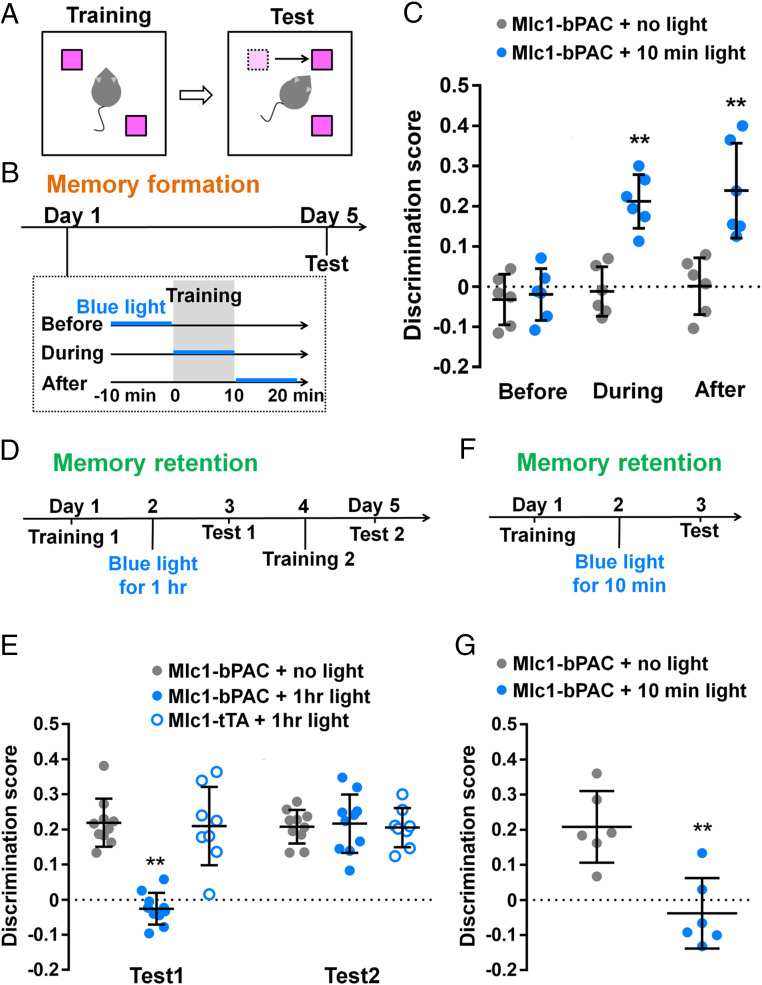
The astrocytic cAMP increase during memory formation or retention differentially modulates memory. (A and B) Schematic diagram of the object–location memory test (A) and its time course: blue light stimulation was given during memory formation (B). (C) Discrimination score of the relocated object. n = 6 mice for each group. **P < 0.01 versus the Mlc1-bPAC + no-light group in each situation by Student’s t test. Discrimination score = (ta-tb)/(ta+tb), where ta is the time the mice spent exploring the relocated object and tb is the time the mice spent exploring the other object. (D) Time course of the object–location memory test: 1 h of blue light stimulation was given during memory retention. (E) Discrimination score of the relocated object. n = 8 to 10 mice for each group. **P < 0.01 versus the Mlc1-bPAC + no-light group and the Mlc1-tTA + 1 h light group by Tukey’s test after ANOVA. (F) Time course of the object–location memory test: a 10-min blue light stimulation was given during memory retention. (G) Discrimination score of the relocated object. n = 6 mice for each group. **P < 0.01 versus the Mlc1-bPAC + no-light group by Student’s t test. Data are presented as the mean ± SD. Circles indicate individual data points of each mouse.
To further examine the effect of the increase in astrocytic cAMP on memory formation, we also performed a Morris water maze test in which mice were trained to learn the location of a hidden platform (SI Appendix, Fig. S6A). The Mlc1-bPAC mice were stimulated with blue light (pulsed, 1 s/5 s) for 20 min/d during training. Blue light stimulation did not affect the latency to reach the platform during training (SI Appendix, Fig. S6B), but the increased preference for the target area was detected only in the light-stimulated group during the probe tests on day 14 and day 28 (SI Appendix, Fig. S6 C and D). These results suggest that the astrocytic cAMP increase during memory formation enhances memory strength.
Next, we stimulated bPAC to increase astrocytic cAMP during memory retention to examine its impact on existing memory. After habituation, the mice were trained on day 1 and subjected to blue light stimulation (1 s/5 s) to the bilateral hippocampus for an hour on day 2 (Fig. 2D). Test 1 on day 3 revealed that the Mlc1-bPAC mice without light stimulation spent more time exploring the relocated object, suggesting that memory was formed and sustained, while the light-stimulated Mlc1-bPAC mice could not distinguish between the two objects (Fig. 2E). Because light stimulation of the bPAC-negative Mlc1-tTA mice did not impair memory performance and all groups of mice exhibited increased discrimination scores in test 2 after the second training without light stimulation, we assume that the impaired memory performance was reversible and not caused by brain damage or blue light itself (Fig. 2E). We also found that shorter blue light stimulation (10 min, Fig. 2F) was sufficient to impair memory performance (Fig. 2G). These results suggested that the increase in astrocytic cAMP in the hippocampus impairs the retention of existing memory.
Taken together, the results of memory tests have shown that astrocytic cAMP increase around memory formation improved memory performance, while cAMP increase during memory retention impaired memory performance. This suggests that astrocytic cAMP increase during different memory stages modulates memory differently. Additionally, we examined locomotor activity and anxiety-like behavior in the open field test and found no significant difference between the light-stimulated and no-light groups (SI Appendix, Fig. S7).
To elucidate the mechanisms involved in astrocytic cAMP–induced memory alteration, we examined the impact of the astrocytic cAMP increase on the activity of hippocampal neurons. Because neuronal activity and long-term plasticity are accompanied by the expression of c-Fos, a well-recognized immediate early gene (29, 30), awake Mlc1-bPAC and Mlc1-tTA mice were stimulated with 10 min or 30 min of blue light (pulsed, 1 s/5 s) and immunostained for c-Fos as a marker of neuronal activity and, especially, plasticity (29, 30). In contrast to the sparse c-Fos expression in the no-light group, a large portion of the CA1 pyramidal neurons were positive for c-Fos in the 30-min light group (Fig. 3A). We found that both 30 min and 10 min of light stimulation significantly increased the density of the c-Fos–positive CA1 pyramidal neurons, and 30 min of light stimulation induced c-Fos expression more substantially (Fig. 3B). Light stimulation–induced increases in c-Fos expression were also observed in CA3 and the dentate gyrus (SI Appendix, Fig. S8 A and B). However, light stimulation of the Mlc1-tTA mice did not induce c-Fos expression, which indicates that neuronal c-Fos expression was caused by an astrocytic cAMP increase (Fig. 3B and SI Appendix, Fig. S8 A and B). The expression of Arc, another immediate early gene used as a marker for synaptic plasticity, was also induced by blue light stimulation in the hippocampus (SI Appendix, Fig. S9).
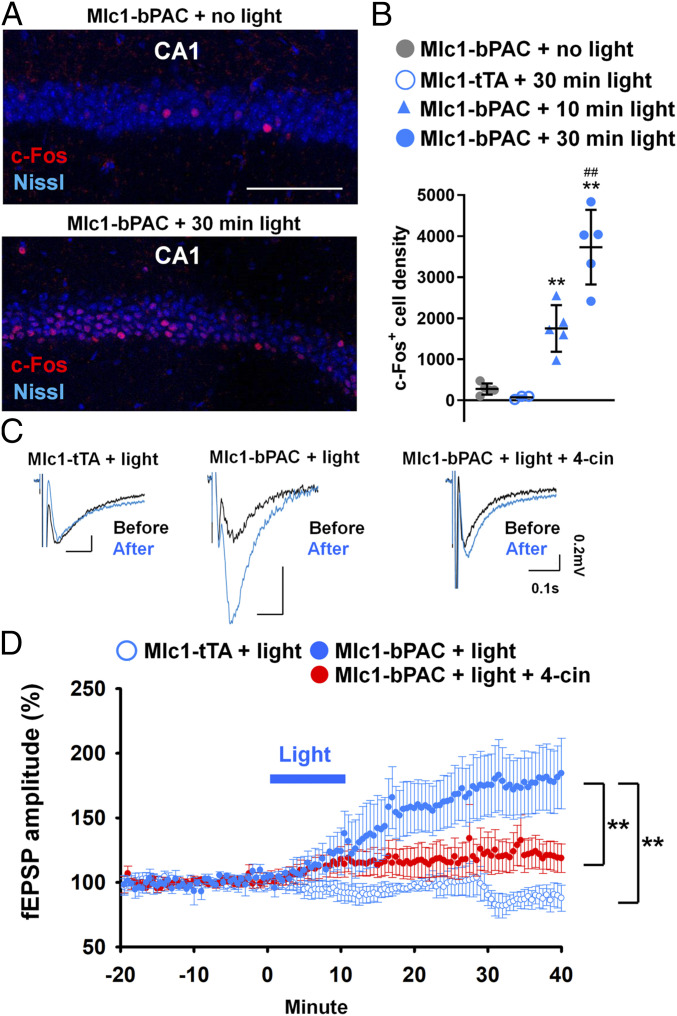
An astrocytic cAMP increase is sufficient to induce synaptic plasticity. (A) Representative images of the hippocampal CA1 region with c-Fos immunostaining (red) and Nissl staining (blue). (Scale bar, 100 μm.) (B) Density of the c-Fos–positive neurons in CA1. n = 3 mice for the Mlc1-tTA + 30 min light group; n = 5 for every other group. Three slices were analyzed for each mouse. **P < 0.01 versus the Mlc1-bPAC + no-light group by Tukey’s test after one-way ANOVA. ##P < 0.01 versus the Mlc1-bPAC + 10 min light group by Tukey’s test after one-way ANOVA. Data are presented as the mean ± SD (C) fEPSP recordings from the CA1 stratum radium under electrical stimulation of SCs. Representative fEPSP traces before (black) and after (blue) blue light stimulation. (D) Time course of changes in the fEPSP amplitude before and after blue light stimulation under SC stimulation. n = 5 slices from 4 mice for each group. **P < 0.01 between the indicated groups by Tukey’s test after two-way ANOVA. Data are presented as the mean ± SEM.
Then, we recorded field excitatory postsynaptic potentials (fEPSPs) in CA1, which were induced by the electrical stimulation of Schaffer collaterals (SCs) in hippocampal slices prepared from Mlc1-bPAC or Mlc1-tTA mice with or without blue light stimulation. In the Mlc1-bPAC + light group, the amplitudes of the fEPSPs gradually increased during the 10-min light stimulation and were maintained between 150 and 200% above the baseline at 30 min after the end of light stimulation. In contrast, the fEPSPs in the Mlc1-tTA group exhibited no significant change (Fig. 3 C and D). Thus, these results suggested that the astrocytic cAMP increase induced long-term potentiation (LTP) in the hippocampus.
We have shown that blue light-induced cAMP astrocytes are sufficient to induce LTP. However, whether astrocytic cAMP–induced memory modulation is caused by synaptic plasticity is unknown. Since long-term synaptic plasticity is dependent on the activation of NMDA receptors (31), we treated Mlc1-bPAC mice with the NMDA receptor blocker MK801 (0.1 mg/kg or 1 mg/kg, intraperitoneal injection [i.p.]) 30 min before blue light stimulation (pulsed, 1 s/5 s) to examine whether the blockade of synaptic plasticity will inhibit astrocytic cAMP–induced memory modulation. We found that MK801 treatment reduced light stimulation–induced c-Fos expression in CA1 (Fig. 4 A and B) as well as CA3 and the dentate gyrus (SI Appendix, Fig. S8 C and D). This result indicates that the astrocytic cAMP increase activates NMDA receptors and subsequently induces synaptic plasticity. We then examined whether astrocytic cAMP–induced memory changes can be reversed by MK801 treatment.
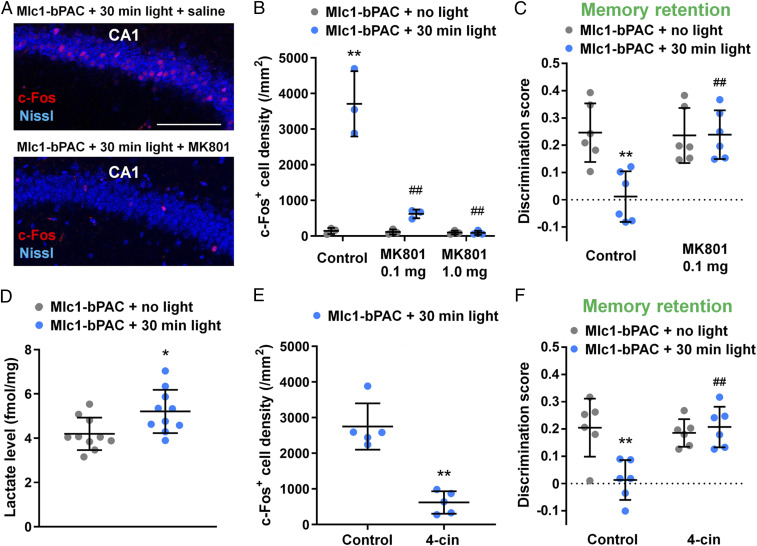
Astrocytic cAMP induces synaptic plasticity and memory changes via lactate shuttle and NMDA receptor activation. (A) Representative images of hippocampal CA1 regions with c-Fos immunostaining (red) and Nissl staining (blue). (Scale bar, 100 μm.) (B) Density of the c-Fos–positive neurons in CA1. n = 3 mice for each group. Three slices were analyzed for each mouse. **P < 0.01 versus the Mlc1-bPAC + no light + saline group; ##P < 0.01 versus the Mlc1-bPAC + light + saline group by Tukey’s test after one-way ANOVA. (C) Discrimination score of the relocated object under NMDA receptor blockade with MK801. n = 6 mice for each group. **P < 0.01 versus the Mlc1-bPAC + no light + saline group; ##P < 0.01 versus the Mlc1-bPAC + light + saline group by Tukey’s test after one-way ANOVA. (D) Lactate levels in the hippocampus determined by lactate assays. n = 10 hippocampi from 5 mice. *P < 0.05 versus the Mlc1-bPAC + no-light group by Student’s t test. (E) Density of c-Fos–positive neurons in CA1. n = 5 mice for each group. Three slices were analyzed for each mouse. **P < 0.01 versus the Mlc1-bPAC + light + vehicle group by Student’s t test. (F) Discrimination score of the relocated object under MCT blockade with 4-cin. n = 6 mice for each group. **P < 0.01 versus the Mlc1-bPAC + no light + vehicle group; ##P < 0.01 versus the Mlc1-bPAC + light + vehicle group by Tukey’s test after one-way ANOVA. Data are presented as the mean ± SD. Circles indicate individual data points of each mouse or hippocampus.
It has been determined that memory formation requires neuronal activity–dependent NMDA receptor activation and subsequent synaptic plasticity, both of which are completely blocked by MK801 treatment (32). Therefore, when MK801 is administered in the experimental paradigm of memory formation, it is not possible to distinguish whether this compound affects changes that depend on neural activity or on light stimulation of astrocytes. Thus, in the present study, we decided to test the NMDAR and its modulation by lactate only in the memory retention experiments. The application of blue light stimulation 24 h after training impaired memory performance in the control group, but the effect of blue light stimulation was blocked by MK801 treatment (Fig. 4C). These results suggest that astrocytic cAMP–induced activation of NMDA receptors impaired memory retention.
To further elucidate the mechanisms underlying the plasticity and memory changes, we examined the astrocyte–neuron lactate shuttle (ANLS) (33) because transportation of lactate from astrocytes to neurons is essential for long-term plasticity and memory (8, 9, 30). Furthermore, astrocytic cAMP has been shown to trigger ANLS in vitro (7). First, we confirmed that the light-stimulated hippocampi from the Mlc1-bPAC mice contained increased levels of lactate (Fig. 4D). Since lactate is transported from astrocytes to neurons via monocarboxylate transporters (MCTs), we examined the effects of an MCT blocker, α-cyano-4-hydroxycinnamic acid (4-cin), on synaptic plasticity and found that 4-cin (100 μM) partially blocked the astrocytic cAMP–induced potentiation of fEPSPs (Fig. 3 C and D). We also injected 4-cin intrahippocampally into awake mice immediately before light stimulation, which blocked both the light-induced increase in c-Fos expression in the CA1 pyramidal neurons (Fig. 4E) and the impaired memory retention in the object–location memory task (Fig. 4F). Thus, these results indicate that the increase in astrocytic cAMP induces synaptic plasticity and modifies memory through ANLS.
Collectively, we have shown that the increase in astrocytic cAMP via optogenetic activation is sufficient to induce synaptic plasticity in surrounding neurons. Astrocyte-induced plasticity, in turn, strengthens the formation of new memory but impairs the retention of existing memory, which results in either improved or impaired memory performance.
An increasing number of studies have highlighted the role of astrocytes in brain function, including memory. However, the role of astrocytic cAMP in memory modulation is still unclear. Therefore, we developed a method to activate the cAMP pathway specifically in astrocytes in vivo to examine whether and how astrocytic cAMP may modulate synaptic plasticity and memory.
First, we have shown that our newly developed double-transgenic mouse line, Mlc1-bPAC, is suited for optogenetic enhancement of cAMP signals specifically in astrocytes both in vitro and in vivo (Fig. 1). Astrocyte-specific regulation of cAMP is indispensable to study astrocytic cAMP in both in vivo and in vitro models including multiple cell types. This approach will benefit studies focusing on the interaction between astrocytes and other cell types as well as the role of astrocytes in behaviors. The use of Mlc1-bPAC mice also provides high temporal resolution, comparable to that of other optogenetic approaches. Differences in the temporal patterns of cAMP increases in neurons have been shown to elicit different bioprocesses in neurons, including synapse elimination, axon guidance, and neurite outgrowth (3435–36). Similarly, the differences in the temporal patterns of the cAMP increase in astrocytes are considered to convey different information to regulate astrocyte functions (37). Thus, our method can provide insights into the impacts of a timing-dependent increase in intracellular cAMP in astrocytes on brain functions.
The bigenic KENGE-tet system also allows us to combine different tTA lines with tetO-GFP-2A-bPAC mice to achieve other cell-type–specific or conditional regulation of cAMP pathways. For example, we crossed Iba1-tTA mice with tetO-GFP-2A-bPAC mice and generated Iba1-tTA::tetO-GFP-2A-bPAC (Iba1-bPAC) mice in which GFP and PAC are expressed specifically in microglia (SI Appendix, Fig. S10).
In this study, we used Mlc1-bPAC mice to elucidate the impact of astrocytic cAMP increase on synaptic plasticity and memory. We found that the astrocytic cAMP increase was sufficient to induce LTP in hippocampal slices and c-Fos expression in hippocampal neurons in vivo (Fig. 3). In behavioral tests, we used an optic fiber to deliver blue light stimulation directly into the CA1 area of the hippocampus (Fig. 1D); pCREB expression in astrocytes was higher in the CA1 area than in adjacent hippocampal areas (Fig. 1 E and F). However, it should be noted that when performing blue light stimulation, especially using a blue LED attached to the skull (methods shown in SI Appendix, Fig. S5, which were not used for behavioral tests in the present study), a possible brain region–dependent effect of astrocytic cAMP on memory formation and retention cannot be ruled out. We found that the astrocytic cAMP increase coupled to memory formation resulted in more persistent memory (Fig. 2). Since astrocytic cAMP is sufficient to induce LTP in hippocampal neurons, it is possible that the astrocytic cAMP increase during memory formation induces LTP in a larger neuronal population, encoding a new memory into a long-lasting one. For example, the astrocytic Ca2+ increase during memory formation has been shown to enhance memory performance by directly inducing LTP of the surrounding neurons (5). In contrast, the astrocytic cAMP increase uncoupled to memory formation (1 d after training) impaired memory retention (Fig. 2). Previous research has shown that artificially induced long-term depression in neurons that encode an existing memory impairs the retrieval of memory (38). Impaired memory performance is also observed when neurons encoding the existing memory are reactivated to a novel context or recruited by a new memory (39, 40). Thus, the results in our current study might be caused by astrocytic cAMP–induced synaptic plasticity in neurons encoding the existing memory.
One interesting observation is the dual role of increased cAMP in memory formation and retention. Previously, knockdown of astrocytic β2 adrenergic receptors was shown to impair memory performance, while knockout of astrocytic adenosine A2A receptors has been shown to improve memory performance (41, 42). Although both receptors activate the cAMP pathway, the results are contradictory. This may be caused by the different time frames during which β2 adrenergic receptors and A2A receptors are activated (43, 44). Using Mlc1-bPAC mice, we were able to show that increasing cAMP in different memory phases (i.e., memory formation and memory retention) resulted in improved or impaired memory performance.
Finally, we determined that astrocytic cAMP increase–dependent synaptic plasticity and memory changes are mediated through ANLS and NMDA receptor activation in neurons (Fig. 4). Lactate release from astrocytes can be triggered by glycogenolysis, which is known to be triggered by astrocytic cAMP in vitro (45). Indeed, an astrocytic cAMP increase in acute brain slices has been shown to induce lactate release (4). Lactate is not only necessary for memory and synaptic plasticity but also able to induce c-Fos expression in neurons by activating NMDA receptors (30). Although ANLS is known to be triggered by cAMP and to activate NMDA receptors in vitro, we show that a cAMP–ANLS–NMDA receptor signaling pathway exists in vivo and modulates memory.
In conclusion, the use of the newly developed Mlc1-bPAC mice revealed that an astrocytic cAMP increase during memory formation or retention can either facilitate memory formation or impair memory retention. We also found that astrocytic cAMP drives NMDA activation and the ANLS pathway to induce synaptic plasticity. We consider this pathway to be the mechanism underlying the alteration of memory. Although we have shown that increasing astrocytic cAMP is sufficient to modulate memory formation and retention, further studies such as in vivo astrocytic cAMP imaging will be needed to determine the time-dependent role of cAMP levels in memory modulation and characterize the astrocyte-specific suppression of cAMP levels in vivo to conclude that increased cAMP in astrocytes is essential for these memory processes. Nevertheless, the development of an astrocyte-specific PAC optogenetic method is important and provides a valuable tool to study the role of these cells in synaptic modulation and behavior. In summary, our research has shed light on the importance and potential role of astrocytic cAMP in memory.
Animal experiments were performed with the approval of the animal experiment ethics committee at the University of Tokyo (approval number: 24–10) and according to the University of Tokyo’s guidelines for the care and use of laboratory animals.
The generation of tetO-GFP-2A-bPAC was achieved by knock-in of tetO-GFP-2A-bPAC insertion. For high expression of GFP-2A-bPAC, the insertion site was targeted downstream of the beta-actin gene polyadenylation signal, 800 bp downstream of the Actb polyA signal (22). We constructed the plasmid containing the tetO-GFP-2A-bPAC cassette with the Neo selection marker flanked on both sides by flippase recognition target (FRT) sites, in which the following elements were connected in tandem: tetO sequence, rabbit beta globin intron, GFP-2A-bPAC complementary DNA (cDNA), SV40 polyadenylation signal, and FRT-flanked PGK-EM7-Neo. The cassette was inserted 30 bp downstream of the Actb polyA signal sequence by homologous recombination. Germline-transmitted offspring were established as tetO-GFP-2A-bPAC-Neo knock-in mice. tetO-GFP-2A-bPAC-Neo mice were crossed with ROSA-Flpe mice, and FRT-flanked Neo selection markers were removed. tetO-bPAC-2A-GFP mice (tetO-bPAC mice) were subsequently generated.
For generation of the double-transgenic mouse lines Mlc1-tTA::tetO-GFP-2A-bPAC and Iba1-tTA::tetO-GFP-2A-bPAC, the tetO-GFP-2A-bPAC mice were crossed with Mlc1-tTA and Iba1-tTA mice, respectively. The tTA lines were generated by a coauthor (K.F.T.) as reported in a previous study (21).
For some experiments, doxycycline (Wako) was mixed in food pellets (0.0057%).
Astrocyte cultures were prepared as previously described with minor modifications from P1 mice (46). The cortices were dissected out in cooled (4 °C) Hank’s balanced salt solution (HBSS) and minced. Tissues were incubated with 0.25% trypsin (Nacalai) and 0.01% DNase I (Roche Diagnostics) in HBSS at 37 °C for 40 min. FBS was added to stop trypsinization, and tissues were centrifuged for 10 min at 1,200 rpm (Kubota KN-70). The tissues were then dissociated into a single-cell suspension in culture medium containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Nacalai) in Dulbecco's modified Eagle medium (DMEM; Gibco, GlutaMAX). Single cells were filtered through a 100-µm–pore cell strainer and plated in a poly-D-lysine (Sigma)-coated T75 flask. The flask was placed in a 37 °C, 5% CO2 incubator (Panasonic, MCO-18AC). The medium was changed 2 d after plating of the mixed cortical cells and every 3 d thereafter.
To acquire astrocyte cultures, confluent glial cultures were vigorously shaken for 5 min, and the culture medium was removed. The cultures were rinsed twice with 10 mL of phosphate-buffered saline (PBS) and mildly trypsinized with 0.05% trypsin in DMEM for 30 min. The astrocyte layer was detached after mild trypsinization and collected with supernatant discarded through centrifugation. The cells were then incubated with 0.25% trypsin-EDTA (Nacalai) at 37 °C. Detached cells were collected and centrifuged for 5 min at 1,000 rpm. After the supernatant was discarded, the cells were dissociated into a single-cell suspension in culture medium and plated on poly-D-lysine–coated 12-mm glass coverslips at a density of 1.5 × 105 cells/well. Secondary cultures were placed in a 37 °C, 5% CO2 incubator, and the medium was replaced 2 d after plating and every 3 d thereafter.
Animals were perfused transcardially with cold PBS followed by 4% paraformaldehyde in 0.1 M PBS, and the brains were quickly removed and placed in 4% paraformaldehyde (Wako) for 4 h. For c-Fos immunostaining, the mice were fixed 90 min after light stimulation. For pCREB immunostaining, the mice were fixed 15 min after light stimulation.
For the detection of GFP expression in the whole brain and experiments using optical fibers to deliver blue light stimulation, brain sections were made by a microtome. The fixed brain was immersed in 20% and then 30% sucrose in PBS overnight and consecutively frozen by dry ice. Sagittal or coronal brain sections with a 50-μm thickness were made. For pCREB immunostaining after blue LED stimulation, sections with a thickness of 100 μm were made using a vibratome.
Brain sections were permeabilized, and nonspecific staining was blocked with 10% goat serum (Funakoshi) in 0.3% Triton X-100 (TaKaRa) in PBS. For immunohistochemical detection of GFP, GFAP, S100β, pCREB, NeuN, Iba1, and c-Fos, we used primary antibodies including chicken anti-GFP antibody (1:1,000; Abcam), rabbit anti-GFAP antibody (1:500; Sigma-Aldrich), anti-S100β antibody (1:250; Sigma-Aldrich), rabbit anti-pCREB antibody (1:250; Abcam), mouse anti-NeuN antibody (1:500, EMD Millipore), rabbit anti-Iba1 antibody (1:500; Wako), mouse anti-c-Fos antibody (1:500; EnCor), and rabbit anti-Arc antibody (1:1,000; Synaptic Systems). Sections were incubated overnight at 4 °C with primary antibody. After extensive washes in PBS, the sections were incubated with the appropriate secondary antibody conjugated with Alexa Fluor dyes (1:500; Invitrogen). After incubation overnight at 4 °C with secondary antibody, the sections were extensively washed with PBS and mounted. For Nissl staining, NeuroTrace 435/455 Blue Fluorescent (1:100; Invitrogen) was used. Nissl was mixed with secondary antibodies, and the brain sections were incubated with Nissl overnight.
Cultured cells were fixed with 4% paraformaldehyde for 30 min at 37 °C. The samples were then incubated with blocking solution (5% goat serum and 0.1% Triton X-100 in PBS) for 1 h at room temperature. The samples were subsequently incubated with primary antibodies overnight at 4 °C. Antibodies against GFP, GFAP, S100β, pCREB, NeuN, Iba1, and c-Fos were used as described above. The samples were washed with PBS and incubated with secondary antibodies (Alexa Fluor dyes) for 6 h.
For nuclear staining with Hoechst 33342 (1:1,000; Invitrogen), brain sections or cell cultures were incubated with Hoechst in PBS for 15 min and washed with PBS.
Astrocyte cultures were used for the cAMP assay by EIA. After approximately 1 wk, the secondary cultures became confluent and were subjected to stimulation by blue light or reagents affecting cAMP levels. Intracellular cAMP levels of cultured astrocytes were then assayed using an enzyme immunoassay kit according to the manufacturer’s instructions (GE Healthcare).
In order to assess intracellular cAMP levels, astrocyte cultures were stimulated with blue light for 10 min or 30 min. Blue LEDs (20,000 mcd, wavelength 465 to 475 nm; Rhos) were used for light stimulation. Four LED lamps were connected in a parallel circuit with a 3-V direct current power source. The intensity of the light output from the lamp was ∼2.0 mW. Each LED lamp was installed at the four corners of the lid of a 24-well plate ∼3 mm above the surface of the culture medium. Pulsed blue light stimulation (1 s/5 s, 1 s duration with 4 s interval) was applied to cell cultures.
For the positive control, forskolin at 10 μM (Sigma-Aldrich) or isoproterenol at 10 μM (Wako) was added to the astrocyte cultures. In some cultures, 1 μM doxycycline was added to the culture medium to inhibit the expression of GFP-2A-bPAC. Five microliters of 100 μM doxycycline was added to 500 μL of culture medium every 1.5 d to maintain the concentration of doxycycline. The culture medium was replaced every 3 d.
A genetically encoded red fluorescent cAMP sensor, cADDis (Montana Molecule), was used (24, 25). Baculovirus-carrying cADDis was added to the astrocyte cultures. Cells were incubated with mixed solution including the virus, 2 mM sodium butyrate (Wako), and Opti-MEM (Gibco) for 6 h in a 37 °C, 5% CO2 incubator and cultured with 10% FBS DMEM with 1 mM sodium butyrate. The cADDis sensor was expressed under the cytomegalovirus promoter. cAMP imaging was carried out 2 or 3 d after transfection.
Glass coverslips with cultured astrocytes were incubated in artificial cerebrospinal fluid (ACSF) containing 127 mM NaCl, 1.6 mM KCl, 1.24 mM KH2PO4, 1.3 mM MgSO4, 2.4 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose for >30 min for stabilization. Light stimulation was applied using a high-powered LED illumination system LEX2 (peak wavelength 465 nm; Brainvision, Inc.). Every light stimulus lasted for 1 s. For a series of stimuli, the interval of stimulus was 1 min. The first blue light stimulation was applied 30 s into every imaging session. The red fluorescence of the cAMP sensor was captured at 2 Hz using a cooled electron-multiplying charge-coupled device camera (iXonEM+ DV897; Andor Technology) through a water-immersion objective lens (×40, 0.8 numerical aperture [NA], Nikon) and a Nipkow disk confocal laser scanner (CSU-10/X1; Yokogawa Electric). GFP expression was assessed using a blue laser after every imaging session. The interval between each imaging session was >20 min.
For the analysis of cAMP imaging, the fluorescence intensity of the cAMP sensor and GFP-expressing astrocytes was analyzed by ImageJ software (NIH). The fluorescence intensity of each cell was determined by its average intensity with the average intensity of the background subtracted. The fluorescence intensity before light stimulation, F0, was defined as the average intensity of 30 s before light stimulation of each cell. The intensity change of each cell was defined as ΔF/F0 = (Ft – F0)/F0, where Ft is the cellular fluorescence intensity at time t.
Stereotaxic surgery was performed on Mlc1-bPAC or Mlc1-tTA mice to enable in vivo blue light stimulation. Mice were anesthetized using pentobarbital (Tokyo Chemical Industry, 40 mg/kg, i.p.) and xylazine (Sigma, 10 mg/kg, i.p.) and aligned in a stereotaxic apparatus (Narishige). Blue LED lamps (3 mm × 3 mm; Bio Research Center, Inc.) were attached to the skull next to the central line (the LED center was positioned at approximately antero-posterior axis [AP]: −1.5 mm, lateral-medial axis [LM]: ±1.5 mm). Optical fibers (200 μm core diameter, 0.48 NA, Thorlabs) epoxied to ceramic zirconia ferrules (Thorlabs) were implanted bilaterally above the hippocampal CA1 regions (AP: −2.0 mm, LM: ±1.5 mm, dorsal-ventral axis [DV]: −1.0 mm). A drill (Argofile Japan, SBH35nST-B) was used to penetrate the skull to allow the fibers in. The optical fibers were secured to the skull using an instant adhesive (Loctite 454) and a mixture of acrylic and dental cement (Provinice). Carbon powder (Strem Chemicals) was added to the cement to make it opaque.
For intrahippocampal injection of reagents, cannulas (EiCom, CX type) were installed in addition to optical fibers. In these surgeries, the mice were anesthetized through isoflurane (Pfizer) inhalation (1 to 1.5%; BioMachinery TK-7). Guide cannulas were implanted near optical fibers targeting the hippocampus (AP: −2.0 mm, LM: ±1.8 mm, DV: −1.5 mm). Dummy cannulas were inserted into guide cannulas to keep the pathway clean.
For CREB phosphorylation induction in the Mlc1-bPAC mouse cortex and hippocampus under anesthesia, blue LEDs were used. Blue LED was unilaterally attached to the mouse skull (see Stereotaxic Surgery) and connected to a stimulator. Thirty minutes of pulsed blue light stimulation (3.5 mW; 1 s/5 s, 1 s duration with 4 s interval) was applied while the mice were aligned on the stereotaxic apparatus. Three- to five-month-old Mlc1-bPAC mice were used.
Blue light stimulation in awake mice was used to induce CREB phosphorylation, followed by c-Fos immunostaining, lactate assays and behavioral tests. Blue light stimulation was given to the bilateral hippocampus using plastic optical fibers. The optical fibers were bilaterally implanted above the hippocampal CA1 region through surgeries (see Stereotaxic Surgery). During stimulation, pulsed blue light stimulation (1 s/5 s, 1 s duration with 4 s interval, 1.8 to 2.2 mW) was applied using a 473-nm laser generator (COME2-LB473/100s/Lucir) with optical cables (Thorlabs) connected to the implanted optical fibers guiding the blue light laser to mouse hippocampi. For pCREB and c-Fos immunostaining, 2- to 3-mo-old Mlc1-bPAC or Mlc1-tTA mice underwent surgery for fiber implantation. After surgery, the mice recovered for at least 2 wk. Before blue light stimulation, the mice were handled for at least six consecutive days. The mice were also familiarized with the light chamber for blue light stimulation 10 min/d for 2 d immediately before blue light stimulation. The light chamber was a small chamber (16.7 cm in width, 23.1 cm in length, and 34.5 cm in depth with an open top) with tall and opaque walls. The mice in the no-light group underwent surgery, handling, and habituation as well. They were placed in the light chamber with optical cables connected to the implanted optical fibers, but there was no laser input. For NMDA receptor blockade, the mice were intraperitoneally injected with MK801 (Tocris; 0.1 mg/kg or 1 mg/kg) or saline 30 min before blue light stimulation. For MCT blockade, α-cyano-4-hydroxycinnamate (4-cin, Sigma; 100 μM in 0.1% DMSO) or vehicle (saline in 0.1% DMSO) was injected intrahippocampally with a syringe pump (Muromachi) at 100 nl/min for 3 min. After injection, the injectors were kept in place for an additional minute. The animals were exposed to blue light 5 min after intrahippocampal injection.
The lactate level in the hippocampus of the Mlc1-bPAC mice after blue light stimulation was measured by a lactate EIA kit (Abcam) according to the manufacturer’s instructions. After light stimulation, the Mlc1-bPAC mice were immediately anesthetized by isoflurane and decapitated. Hippocampi were dissected at 4 °C, weighed and processed according to the manufacturer’s instructions.
For all behavioral tests (object–location memory test, Morris water maze, open field test), Mlc1-bPAC mice and their littermates Mlc1-tTA mice were used. Each behavioral subject received surgery for optical fiber implantation into the bilateral hippocampus when they were 2 to 3 mo old, recovered for at least 2 wk, and was habituated to human handling for at least six consecutive days.
An object chamber and the light chamber mentioned above were used in the object–location memory test. The object chamber was a square, transparent plastic box (30 cm in width, 30 cm in length, and 34.5 cm in depth with an open top) and was placed in a stable environment with limited changes during behavioral sessions. Mice were able to see the surroundings through the transparent walls. A light chamber was used for blue light stimulation. The mice were habituated to these two chambers 10 min per day for two days. During training, the mice explored the object chamber for 10 min with two identical objects (45 mm × 55 mm × 65 mm cube or other sets of objects with similar sizes) placed around the corners (40 to 45 mm away from the walls). Blue light stimulation (1 s/5 s, pulsed) or no light stimulation was applied at different times with different lengths according to the time courses of each experiment.
For the memory tests, the mice were placed in the object chamber again for 5 min with the same objects that they had explored during training. However, one object was relocated while the other remained at the same position. A camera was installed above the center of the object chamber to monitor the instantaneous position of the mouse during training and testing. The time the mice spent exploring the objects during training and testing was measured. Exploring time was defined as the time the mice spent sniffing or touching the objects; however, the time the mice spent chewing or climbing the objects or accidentally touching the objects during grooming was excluded. Memory was assessed by the discrimination score as follows:
where ta is the time the mice spent exploring the relocated object and tb is the time the mice spent exploring the other object. Retraining of the same subjects was carried out using a different and novel set of objects.
Acute slices were prepared from the hippocampi of Mlc1-tTA and Mlc1-bPAC mice (postnatal days 35 to 42). The mice were anesthetized with isoflurane and subsequently decapitated. The brains were removed and placed in ice-cold oxygenated (95% O2/5% CO2) ACSF. The brains were sliced horizontally at a thickness of 400 µm using a vibratome (VT1200S, Leica) in ice-cold, oxygenated modified ACSF that consisted of 222.1 mM sucrose, 27 mM NaHCO3, 1.4 mM NaH2PO4, 2.5 mM KCl, 1.0 mM CaCl2, 7.0 mM MgSO4, and 0.5 mM ascorbic acid as previously described (47). Slices were maintained for 30 min at 35 °C in oxygenated ACSF and then incubated for at least 30 min at room temperature before use. The slices were transferred to a recording chamber continuously perfused with ACSF at 33 to 35 °C. The stimuli were delivered through a tungsten electrode, and the fEPSPs were recorded using glass pipettes (1 to 2 MΩ) filled with ACSF. The electrodes were carefully placed in the CA1 stratum radiatum. A 50-μs pulse was given every 30 s. The stimulus intensity was set to result in an fEPSP slope of ∼30 to 50% of the maximum fEPSP slope, ranging between 0.7 and 1.0 mA. Electrophysiological data were acquired using a MultiClamp 700B amplifier and a Digidata 1440 digitizer controlled by pCLAMP 10 software (Molecular Devices). Blue light stimulation was applied at a power of 1.5 mW, pulse width of 1 s, and frequency of 0.2 Hz (1 s/5 s).
Images of the fixed samples were acquired using an FV1200 or FV1000 confocal scanning microscope with 10×, 20×, 40× and 100× objective lenses (Olympus).
pCREB fluorescence intensity was calculated as the mean fluorescence intensity in the nuclei (stained with Hoechst) with the average background fluorescence intensity subtracted. The pCREB-positive rate in the GFP-positive astrocytes was determined by the number of pCREB GFP double-positive cells and the number of GFP-positive cells. Whether a cell is pCREB positive or negative was determined by eye.
The density of the c-Fos–positive cells was determined as follows: c-Fos–positive cells were counted in two 150 μm × 300 μm regions in the hippocampal CA1, CA3, and dentate gyrus granule cell layers. The area of the cell layers was measured and used to determine the density of the c-Fos–positive cells.
All statistical analyses were performed using SigmaPlot 12 software. An unpaired Student’s t test was used to determine the statistical significance between two groups. ANOVA was used to determine the statistical significance among three or more groups. In the in vitro electrophysiology experiment, two-way ANOVA was used. In other experiments involving three or more groups, one-way ANOVA was used. Post hoc analysis of ANOVA was performed using Tukey’s method to determine the statistical significance between each group.
This work was supported in part by a Japan Society for the Promotion of Science (JSPS) International Research Fellowship (P19411 to Z.Z.), a Grant-in-Aid for challenging Exploratory Research (17K19440 to R.K.) and a Grant-in-Aid for Transformative Research Areas (A) “Glia decoding” (20H05897 to R.K.) from JSPS, by Japan Science and Technology Agency (JST) Precursory Research for Embryonic Science and Technology (PRESTO) (JPMJPR18H4 to R.K.), and by JST Exploratory Research for Advanced Technology (ERATO) (JPMJER1801 to Y.I.). We thank Nonprofit Organization for Biotechnology Research and Development for providing technical assistance.
Raw data files data have been deposited in Google Drive (https://drive.google.com/drive/folders/1fFc-lNVYpdIxxItjTONjYg_XrUK3-5fS?usp=sharing).
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
25
26
27
28
29
30
31
32
33
34
35
36
37
40
41
42
43
44
45
46
 Astrocytic cAMP modulates memory via synaptic plasticity
Astrocytic cAMP modulates memory via synaptic plasticity
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

