
 ,
Abir Yamak,
Jamieson Whitcomb,
Aizhu Lu,
Mathieu Joyal,
Hiba Komati,
Wenbin Liang,
Céline Fiset,
,
Abir Yamak,
Jamieson Whitcomb,
Aizhu Lu,
Mathieu Joyal,
Hiba Komati,
Wenbin Liang,
Céline Fiset,
Edited by Christine E. Seidman, Howard Hughes Medical Institute, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, and approved October 19, 2020 (received for review April 16, 2020)
Author contributions: L.G. and M.N. designed research; L.G., A.Y., J.W., A.L., and M.J. performed research; W.L. and C.F. contributed new reagents/analytic tools; L.G., A.Y., J.W., A.L., M.J., H.K., W.L., C.F., and M.N. analyzed data and interpreted results; L.G. and M.N. wrote the paper.
1A.Y. and J.W. contributed equally to this work.
GATA6 is a key regulator of sinus node (SAN) development. Loss of one Gata6 allele disrupts patterning and size of the SAN. GATA6 plays cell autonomous as well as non-cell autonomous functions in several cell types required for proper SAN development and function. Haploinsufficiency of Gata6 in mice leads to electrophysiological alterations and increases susceptibility to develop arrhythmias. GATA6 is critical for pacemaker cell differentiation and cardiac conduction.
The sinus node (SAN) is the primary pacemaker of the human heart, and abnormalities in its structure or function cause sick sinus syndrome, the most common reason for electronic pacemaker implantation. Here we report that transcription factor GATA6, whose mutations in humans are linked to arrhythmia, is highly expressed in the SAN and its haploinsufficiency in mice results in hypoplastic SANs and rhythm abnormalities. Cell-specific deletion reveals a requirement for GATA6 in various SAN lineages. Mechanistically, GATA6 directly activates key regulators of the SAN genetic program in conduction and nonconduction cells, such as TBX3 and EDN1, respectively. The data identify GATA6 as an important regulator of the SAN and provide a molecular basis for understanding the conduction abnormalities associated with GATA6 mutations in humans. They also suggest that GATA6 may be a potential modifier of the cardiac pacemaker.
The cardiac conduction system (CCS) consists of muscle cells and conducting fibers that ensure the initiation of the impulse and its propagation throughout the heart. The CCS is composed of the sinus node (SAN), known as the pacemaker of the heart since it generates the electrical impulse, and the atrioventricular node (AVN), the His or atrioventricular bundle (AV bundle), and the Purkinje fibers, which propagate the signal to the ventricular myocytes (1). Despite remarkable progress in the past decade, the cellular origins of the CCS components remain incompletely elucidated. The SAN is thought to be composed mainly of secondary heart field cells derived from ISL1- and TBX18-expressing cells (2), while the AVN and His bundle are thought to segregate from precursors of the working myocardium and are positive for transcription factors TBX2 and NKX2.5 (34–5).
Abnormalities affecting the generation or propagation of the electrical impulses lead to conduction defects ranging from benign to fatal arrhythmias. Impaired impulse generation in the SAN causes sick sinus syndrome (SSS), manifesting as sinus pause/arrest or bradycardia. Blockage of the impulse conduction to the AVN or Purkinje fibers results in atrioventricular block. In humans, atrial fibrillation (AF), a condition involving a rapid and irregular cardiac rhythm, is the most common type of arrhythmia, affecting 2.7 million individuals in the United States alone (6). Its frequency increases with aging, and it occurs in isolation or in association with other complications such as dementia, stroke, and heart failure (7). Atrial fibrillation can be triggered by alterations in atrial electrophysiology or calcium handling, genetic factors, and contractile and structural remodeling in the heart that increase with aging. Human genetic studies as well as analysis of genetically modified mouse models have identified several genes encoding critical cardiac transcription factors (TFs) that contribute to rhythm regulation disturbances. For example, mutations in the GATA family of TFs (GATA4/5/6), the T-box factor TBX5, and the homeodomain protein NKX2.5 have been reported in several cases of familial AF (8, 9). The mutations frequently lead to loss of function, but gain of function mutations have also been found. Analysis of mice lacking a Tbx5 or an Nkx2.5 allele confirmed that decreased levels of these TFs is sufficient to produce conduction abnormalities, including AV block (10). Decreased levels of another T-box family member, TBX3 in different regions of the developing heart, also leads to lethal arrythmias (11). TBX5 and NKX2.5 cooperatively activate transcription of several genes important for cardiac conduction, including connexin 40 (Cx40) and the DNA-binding protein inhibitor Id2 (10, 12). In cardiomyocytes, TBX5 and NKX2.5 also interact with GATA4 (13, 14); mutations in GATA4 are found in association with human AF, and GATA4 was shown to regulate conduction genes such as Cx40 and Cx30.2 (15).
A role for GATA6 in CCS regulation was first suggested by the finding that an enhancer region upstream of the Gata6 promoter is specifically active in the AV conduction system (16). The presence of GATA6 in the ventricular CCS was subsequently confirmed, and its contribution to AV development and function was inferred from analysis of mice with myocardial-specific deletion of its carboxyl zinc finger domain using Mlc2v-cre (17). In this model, a truncated GATA6 protein containing the N-terminal activation domain and the first zinc finger can still be produced and retains the ability to bind GATA sites and interact with known GATA cofactors. Of note, the mutant protein was restricted to MLC2V-expressing myocytes, while intact GATA6 is expressed in atrial cells and in other cell types that may contribute to the proximal CCS.
To determine the contribution of GATA6 to the CCS, we analyzed CCS structure and function in mice haploinsufficient for Gata6. We found that loss of one Gata6 allele leads to electrophysiological alterations and increased susceptibility to arrhythmias. Furthermore, hypocellularity and reduction in HCN4+/TBX3+ conduction cells marked the SAN of Gata6+/− mice. Cell-specific deletion of Gata6 revealed its requirement in different cell types that contribute to SAN development and CCS function, namely, conduction, second heart field (SHF), and endothelial cells. The results support a key function for GATA6 in pacemaker cell differentiation and in cardiac conduction. They also suggest that GATA6 may be an arrhythmia-causing or -modifying gene.
A surface electrocardiogram (ECG) was performed on Gata6+/− and control littermates to assess whether these mice display any electrophysiological abnormalities. The ECG profiling showed significant increases in both PR segment and QT interval in the Gata6+/− group indicative of problems at the sinus node and ventricular levels (Fig. 1 A and B). Arrhythmia analysis using the Data Sciences International/Ponemah platform revealed the presence of premature atrial contractions (PACs) in three out of nine Gata6+/− mice (33%) but in none of the 17 wild-type (WT) mice analyzed (Fisher test, P =0.0323); PAC can lead to AF when occurring in the atrial vulnerable period (18) (Fig. 1C). To determine whether Gata6+/− mice were more prone to arrhythmia, programmed electrical stimulation (PES) was performed on adult Gata6+/+ and Gata6+/− hearts. Ventricular arrhythmia was induced in one out of eight (12.5%) control hearts but in four out of six (66.7%) Gata6+/− hearts (Fig. 1 D and E). Together, these results suggest that loss of one Gata6 allele leads to CCS abnormalities, atrial and ventricular, at baseline and in response to stimuli.

![Adult Gata6+/− mice display ECG abnormalities. (A) Electrocardiogram profile showing the different interval measurements for Gata6+/+ and Gata6+/− mice. HR: heart rate; FC: cardiac frequency. Values are mean + SEM. *P < 0.05. (B) Representative ECG showing prolonged QT in Gata6+/− mice. (Scale bar: 100 ms.) (C) Representative ECG showing the presence of PAC in Gata6+/− mice following telemetry measurements (Gata6+/+ [n = 17] and Gata6+/− [n = 9]). (D and E) Ex vivo heart ECG recording for Gata6+/+ (n = 8) and Gata6+/− (n = 6) mice following isoproterenol perfusion at 0.1 µM showing arrhythmia development. *P < 0.05. (Scale bar: 300 ms.)](/dataresources/secured/content-1765821162311-5a5b5be9-f6ce-46cd-9b92-da3e0affac16/assets/pnas.2007322118fig01.jpg)
Adult Gata6+/− mice display ECG abnormalities. (A) Electrocardiogram profile showing the different interval measurements for Gata6+/+ and Gata6+/− mice. HR: heart rate; FC: cardiac frequency. Values are mean + SEM. *P < 0.05. (B) Representative ECG showing prolonged QT in Gata6+/− mice. (Scale bar: 100 ms.) (C) Representative ECG showing the presence of PAC in Gata6+/− mice following telemetry measurements (Gata6+/+ [n = 17] and Gata6+/− [n = 9]). (D and E) Ex vivo heart ECG recording for Gata6+/+ (n = 8) and Gata6+/− (n = 6) mice following isoproterenol perfusion at 0.1 µM showing arrhythmia development. *P < 0.05. (Scale bar: 300 ms.)
Several GATA6 mutations have been reported in individuals with familial AF (SI Appendix, Fig. S1). Of note, both loss and gain of function mutations have been identified, suggesting that GATA6 may be involved in CCS regulation. Structural defects such as atrial dilation and impaired ventricular function are common in patients with chronic AF (19). We previously reported that adult mice with 50% reduction in GATA6 have reduced left ventricular function as assessed by echocardiography, and several display a dilated right atrium (RA) and right ventricle (20). As shown in Fig. 2 A and B, these structural defects are evident at birth. Further examination of Masson trichrome–stained sections revealed a hypoplastic SAN with fewer myocytes in all Gata6+/− neonates compared to control littermates (Fig. 2C). To determine whether this phenotype is associated with differentiation defects, expression of key atrial and CCS markers and regulators, including NKX2.5, TBX5, ANP, and HCN4, was examined. Maintenance of the SAN and atrial identities involves transcriptional repression of atrial and SAN genes, respectively. For example, TBX3 represses the ANP gene in the SAN, while in the adjacent atrial myocardium, NKX2.5 activates ANP and represses HCN4 and TBX3, as evidenced by ectopic expression of TBX3 and HCN4 in the embryonic myocardium of Nkx2.5 null mice (1). In Gata6+/− mice, NKX2.5 and TBX5 were up-regulated in the SAN relative to control littermates (Fig. 2 D, Top and Bottom). Interestingly, ANP immunostaining was decreased in the atria but increased in the SAN of Gata6+/− neonates (Fig. 2 D, Middle). In addition to these compartment-specific alterations, qRT-PCR analysis revealed decreased levels of Kcnj3 (encoding Kir3.1, a key component of the acetylcholine-sensitive K+ channel) (21), Gja1 (Cx43), and atrial markers NKX2.5 and NPPA (encoding ANP) as well as EDN1 (encoding endothelin 1) and its B-type receptor EDNRB in embryonic Gata6+/− hearts as early as E11.5 (Fig. 2E). Changes in several transcript levels persisted in adult Gata6+/− hearts; notably, levels for HCN4 were 46% of those in control mice, NKX2.5 were 27% of those of Gata6+/+ mice, and GATA6 were at 47% of control littermates (P < 0.05). These results suggest that GATA6 haploinsufficiency (confirmed by Western blot analysis as shown in SI Appendix, Fig. S2) leads to profound genetic reprogramming in atrial and CCS cells.
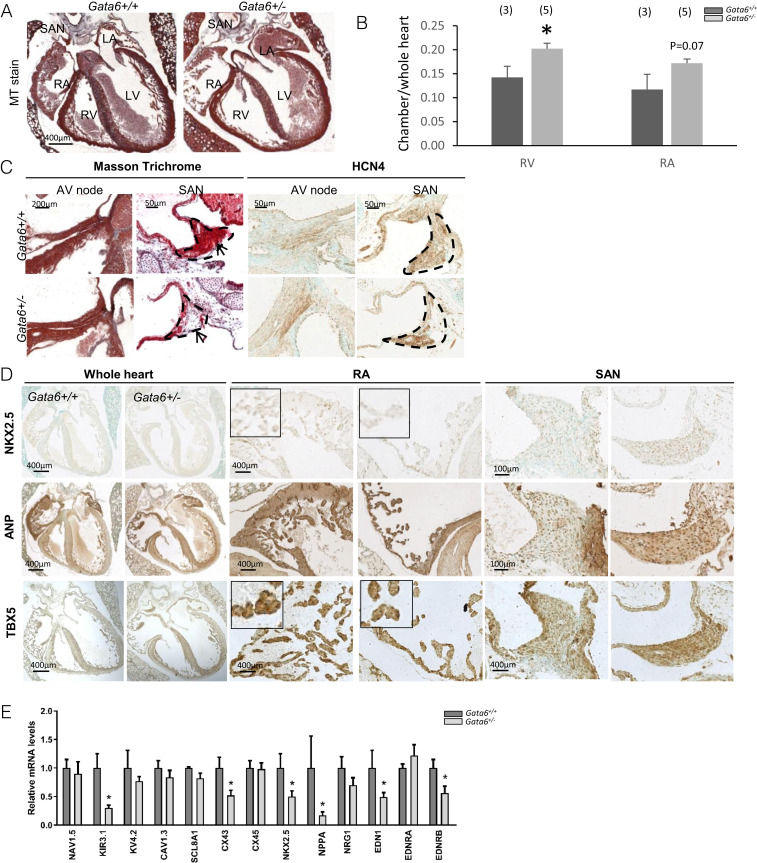
GATA6 is expressed in the sinus node. (A) Masson trichrome (MT) staining showing enlarged right atria and ventricle in Gata6+/− newborn mice. (Scale bar: 400 μm.) (B) Quantification graph showing enlarged surface area of RA and RV Gata6+/− when compared to control mice. RV: right ventricle; LV: left ventricle; LA: left atrium. *P < 0.05. (C) Masson trichrome staining on newborn Gata6+/+ and Gata6+/− mice showing hypoplastic SAN in Gata6+/− mice evident in examination of three consecutive sections (n = 5 to 9). Notice the reduction in HCN4 positive cells in SAN of Gata6+/− mice. Arrows point to the SAN. (Scale bars: 200 μm, 50 μm.) (D) Immunohistochemistry in the SAN and RA of newborn Gata6+/+ and Gata6+/− mice showing gene expression changes expression of NKX2.5 (Top), ANP (Middle), and TBX5 (Bottom). Note how expression of atrial markers NKX2.5, TBX5, and ANP is decreased in atria but increased in SAN of Gata6+/− mice. The micrographs shown are representative of results obtained from n = 5 to 9 mice of each genotype. (Scale bars: 400 μm, 100 μm.) (E) qRT-PCR on RNA extracted from E11.5 hearts showing altered expression of important conduction system regulators; note the significant decrease in the expression of KIR3.1, NKX2.5, CX43, NPPA, and EDN1 (corrected to RPS16, n = 5 to 8 per group). *P < 0.05.
GATA6 is known to be present in several cardiovascular cells, including atrial and ventricular myocytes, as well as some endothelial, neural crest, and smooth muscle cells (22, 23). GATA6 transcripts were also reported in the distal CCS, including the AV node, His bundle, and Purkinje fibers (17). Given the SAN phenotype and the electrophysiology described above, we checked for GATA6 expression in the SAN using immunofluorescence. GATA6 immunostaining was evident in the SAN as early as E14.5 (Fig. 3 A and B), where it localized with HCN4-expressing cells (Fig. 3 A, last two panels of Top row). Interestingly, GATA6 expression colocalized with TBX3- and ISL1-expressing cells but was also found in SAN cells that did not express these proteins (Fig. 3 A, last two panels of Middle and Bottom rows).
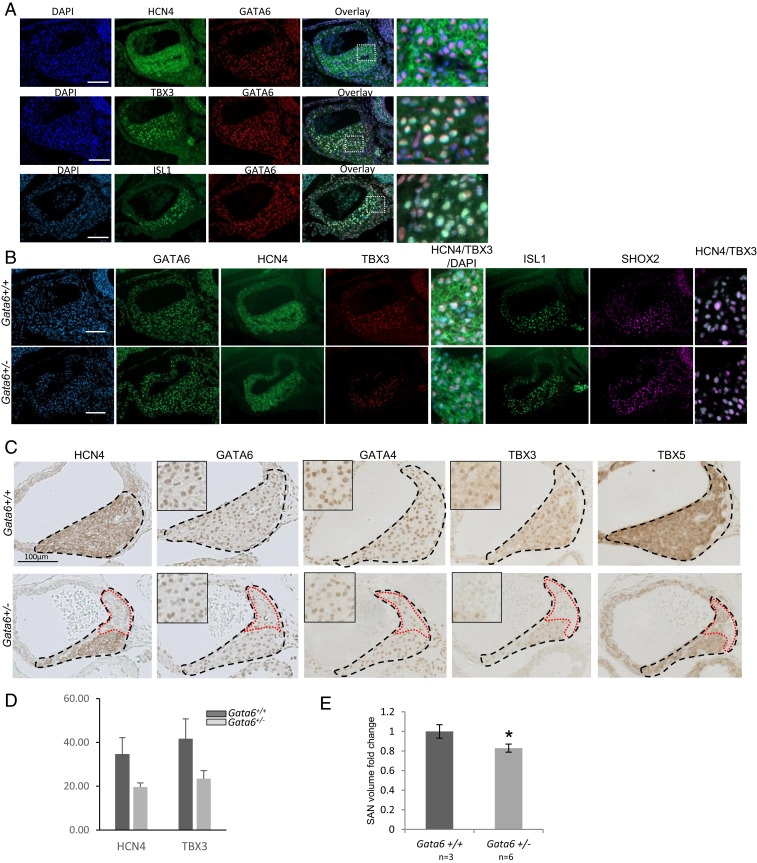
Cellular and molecular changes in the SAN of Gata6+/−. (A) Immunofluorescence showing the colocalization of GATA6 with staining HCN4, TBX3, and ISL1 in the SAN of E14.5 Gata6+/+ control embryos. The last column is an image zoom. (Scale bar: 100 μm.) (B) Staining for GATA6, HCN4, TBX3, ISL1, and SHOX2 comparing Gata6+/− to Gata6+/+ controls. Note the colocalization of HCN4/TBX3 and SHOX2/ISL1 within cells of the SAN (zoomed-in images). (Scale bar: 100 μm.) (C) Immunohistochemistry staining on SAN from E14.5 Gata6+/+ and Gata6+/− embryos showing expression of HCN4, GATA6, GATA4, TBX3, and TBX5. Note the decreased expression of GATA6, GATA4, and TBX5 and the lack of HCN4+ and TBX3+ cells in SAN of Gata6+/− mice. The micrographs are representative of similar findings in four to five embryos for each genotype. (Scale bar: 100 μm.) (D) Quantification graph showing the number of positive cells for HCN4 and TBX3 within the SAN region in both Gata6+/− and control mice. (E) Three-dimensional reconstruction of the SAN from E14.5 embryos showing a smaller SAN in Gata6+/− mice. *P < 0.05.
Next, we analyzed genetic changes at the cellular level during early stages of embryonic SAN development (E14.5). Alterations in the pattern of expression of several markers, including TBX5, TBX3, ISL1, GATA4 and GATA6, were observed in E14.5 Gata6+/− embryos. Staining intensity for HCN4, TBX3, TBX5, and ISL1 was lower in Gata6+/− mice (Fig. 3 B and C), and there were fewer HCN4+ and TBX3+ cells in Gata6+/− SAN, especially in the head region (Fig. 3 C and D). Furthermore, three-dimensional reconstruction confirmed that Gata6+/− SANs are significantly smaller than controls (Fig. 3E).
Given the critical role of TBX3 in pacemaker differentiation and its altered expression pattern in Gata6+/− SAN, we checked whether GATA6 is a direct transcriptional activator of TBX3. In silico analysis identified four putative GATA binding sites on the TBX3 promoter (Fig. 4A). Cotransfection in NIH 3T3 cells of a GATA6-expressing vector and a TBX3–luciferase construct lead to robust dose-dependent activation that was dependent on the presence of at least the proximal GATA site (Fig. 4 B and C). GATA6 activation of the TBX3 promoter was consistently higher than that of two other GATA-dependent promoters, the Nppa promoter, a known GATA6 target (Fig. 4D), and EDN1(Fig. 4E). Chromatin immunoprecipitation demonstrated that GATA6 binds to both distal and proximal GATA sites of the TBX3 promoter (Fig. 4F). These sites were also able to compete for GATA6 binding with the well-characterized GATA elements from the Nppa promoter (Fig. 4 G and H). Thus, TBX3 appears to be a transcriptional target for GATA6. Given that TBX3 was shown to directly bind and modulate the Gata6 promoter (24), our results raise the intriguing possibility that GATA6 and TBX3 may be part of a forward reinforcing feedback loop that promotes/maintains pacemaker cell identity. Last, expression of endothelin 1 (EDN1), a secreted peptide hormone known to modulate cardiac innervation and pacemaker cell differentiation, was also decreased in Gata6+/− hearts (25, 26) (Fig. 2E), and GATA6 significantly upregulated the EDN1–luciferase promoter in transfection assays (Fig. 4E). Together the results suggest that GATA6 may act in several cell types to coordinately regulate genes critical for normal SAN development.
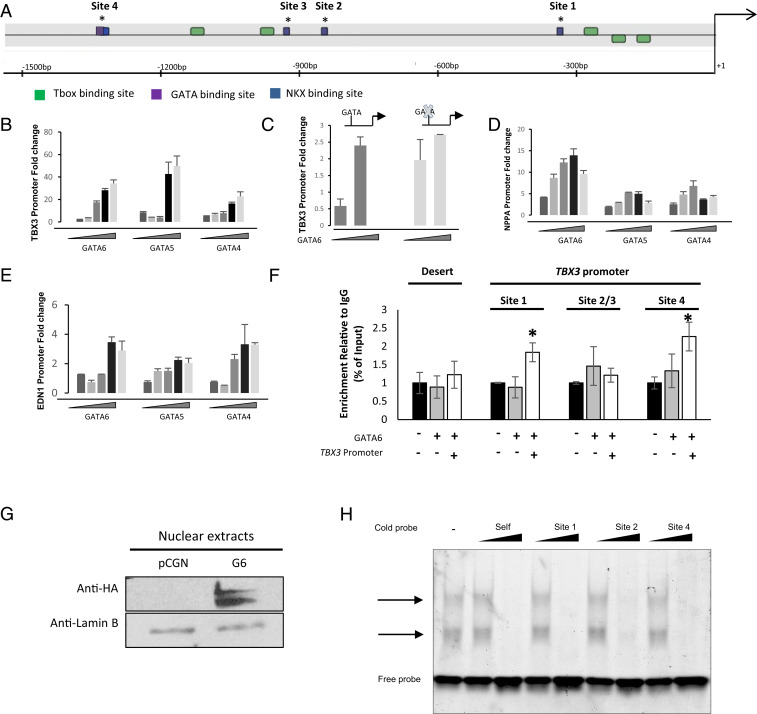
GATA6 transcriptional regulation of TBX3 promoter. (A) Schematic representation of TBX3 promoter showing the different GATA, T-box, and Nkx-binding sites. bp, base pair. *, indicates the position of the different GATA sites. (B–E) Increasing amounts of GATA4/5/6 expression vectors are transiently cotransfected with the luciferase reporter under the control of the TBX3, NPPA, and EDN1 promoters in NIH 3T3 cells (25, 50, 100, 250, and 500 ng of expression vector). The pGL3-TBX3-One GATA site is a 5′ deletion, leaving only one GATA site, and the p-GL3-TBX3-No GATA site promoter has a mutation in this site. Relative luciferase activities are represented as fold changes. The data are a representative of three independent experiments done in duplicates. Values are mean + SEM. *P < 0.05. (F) Chromatin immunoprecipitation of GATA6 occupancy on the different GATA binding sites on the TBX3 promoter. A chromosome 20 gene desert was used as a negative gene, and an IgG antibody was used as an immunoprecipitation negative control. Results are expressed as the fold enrichment of GATA binding compared to IgG binding. The results are reported as the mean ± SEM, *P ≥ 0.01 compared to IgG. IgG, Immunoglobulin G. (G) Western blot analysis of transfected, HA-tagged GATA6 using the HA antibody. The panel shows the expression of recombinant HA-GATA6 WT in nuclear extracts of AD293 cells. (H) Immunofluorescent electrophoretic mobility shift assays performed using nuclear extracts from AD293 cells overexpressing WT GATA6. The binding of the NPPA probe to GATA6 is indicated by an arrow. A cold probe of increasing concentrations of the different GATA binding sites on the TBX3 promoter was added for competition.
The SAN is a unique structure within the heart whose cellular basis is incompletely understood. The SAN tissue is composed of both pacemaking myocytes (mainly ISL1+ SHF cells) and nonpacemaking cells (Fig. 5A), with the latter including endothelial cells and fibroblasts, as well as transitional cells. We used mouse genetics to determine the cellular basis for the GATA6-dependent SAN phenotype. Gata6 was deleted from endothelial cells and from SHF-derived myocardial conduction cells by crossing Gata6Fl/Fl mice with Tie2cre and Isl1cre mice, respectively. Genotyping of embryos from timed matings indicated high perinatal lethality when both Gata6 alleles were conditionally deleted from either TIE2+ or ISL1+ cells. with decreased viability evident as early as E14.5 (Fig. 5B).
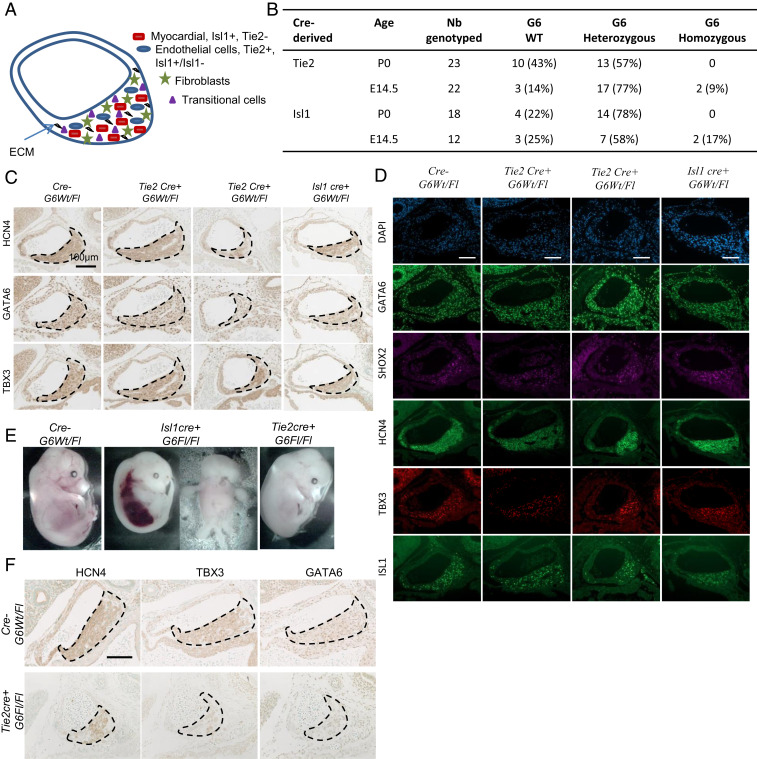
Cellular basis of the GATA6 role in SAN formation. (A) Schematic representation of the different cell types contributing to the formation of the SAN. ECM: extracellular matrix. (B) Frequency of genotypes obtained from the various cre lines. Embryonic and perinatal lethality of cre+ homozygous G6 is demonstrated by the reduced frequencies of those embryos at various developmental stages. These results were obtained based on crossing two heterozygotes with each other for each of the cre lines. (C) Immunohistochemistry staining for HCN4, GATA6, and TBX3 in the SAN of E14.5 embryo control Gata6+/+ (cre- G6Wt/Fl) and cre+Gata6Wt/Fl mice. Note the decreased staining of HCN4, TBX3, and GATA6 in Isl1cre+Gata6Fl/Fl and some Tie2cre+Gata6Fl/Fl SAN. The micrographs are representative of similar findings in four to five embryos for each genotype. (Scale bar: 100 μm.) (D) Staining for GATA6, SHOX2, HCN4, TBX3, and ISL1 comparing control Gata6+/+ (cre- G6Wt/Fl) and cre+Gata6Wt/Fl mice. (Scale bar: 100 μm.) (E) Representation of E14.5 embryos with homozygous Gata6 deletion in specific cell types vs. their control Gata6+/+ (cre- G6Wt/Fl). (F) Immunohistochemistry staining for HCN4 (Left), GATA6 (Middle), and TBX3 (Right) in the SAN of E14.5 embryo control (Top) and Tie2cre+Gata6Fl/Fl mice (Bottom). Note the decreased staining of HCN4, TBX3, and GATA6 in cre+Gata6Fl/Fl SAN. The micrographs are representative of similar findings in four to five embryos for each genotype where applicable. (Scale bar: 100 μm.)
Next, we examined whether cell-specific loss of a single Gata6 allele is sufficient to recapitulate the Gata6+/− phenotype. Loss of one Gata6 allele from ISL1+ cells led to smaller SANs (Fig. 5 C and D, fourth column vs. first column) and decreased expression of HCN4 (Fig. 5 C, Top) and TBX3 (Fig. 5 C, Bottom) as observed in Gata6+/− mice. Loss of Gata6 from TIE2+ cells led to a partially penetrant phenotype with smaller SAN size and decreased marker gene expression observed in 50% of animals (Fig. 5 C and D, second and third columns vs. first column). Thus, loss of GATA6 from either ISL1- or TIE2-expressing cells leads to structural and genetic changes in the SAN. Examination of the homozygous embryos from the Tie2- and Isl1-cre crosses revealed variable defects, including abnormal vascularization (Fig. 5E). Histological analysis of Tie2cre+ G6Fl/Fl embryos revealed a smaller hypocellular SAN, recapitulating the phenotype observed in the Gata6+/− line (Fig. 5F). The SAN of Tie2cre+ G6Fl/Fl line was characterized by a decreased number of HCN4- and TBX3-expressing cells as well as a lower cellular level of both markers (Fig. 5F). The results indicate that lack of Gata6 in endothelial cells leads to hypoplastic SAN and decreased expression of key SAN markers. Unfortunately, analysis of the SAN in Isl1cre+ G6Fl/Fl embryos was not possible due to the general severity of their phenotype.
Since cell-specific deletion of one Gata6 allele did not compromise survival, we examined heart electrophysiology in adult mice from all of the lines generated. A surface ECG showed distinct alterations in the various lines which differentially recapitulated some of those present in Gata6+/− mice: Tie2cre+ G6Wt/Fl mice displayed a prolonged QT interval indicative of defects in the ventricular CCS, while Isl1cre+ G6Wt/Fl showed increased P wave duration and prolonged PR segment, suggesting slowed conduction at SAN, atrial tissue, and/or AVN (Fig. 6A). The structural and functional defects observed in all genotypes studied are summarized in Table 1. Last, to determine whether loss of GATA6 from ISL1 or TIE2 cells increases susceptibility to arrhythmia, PES was performed on adult Isl1cre+ G6Wt/Fl and Tie2cre+ G6Wt/Fl hearts. As shown in Fig. 7, arrhythmias were observed in 40% (two out of five) Isl1cre+ G6Wt/Fl hearts but not in hearts fromTie2cre+ G6Wt/Fl. Last, we examined the effect of removing GATA6 from HCN4+ cells to directly evaluate its cell-autonomous role within conduction cells. Mice lacking one Gata6 allele from HCN4+ cells were born at the expected Mendelian ratio (Fig. 8A) and displayed a SAN phenotype but no visible structural heart defects (Fig. 8C). As shown in Fig. 8 and Table 1, half of these mice had a hypoplastic SAN characterized by a decreased number of TBX3, ISL1, and SHOX2 positive cells (Fig. 8B). Immunofluorescence on consecutive tissue sections (Fig. 8 D and E) clearly shows the decreased presence of biomarkers of conduction cells, including SHOX2, ISL1, TBX3, and HCN4, and the smaller SANs of mice with loss of one Gata6 allele in conduction cells. ECG on young adult mice (60 to 70 d old) showed prolonged PR similar to that seen in mice with heterozygote deletion of Gata6 in ISL1+ cells or in all cells (heterozygote mice) (Fig. 8F).Taken together, the results indicate that GATA6 is required in various SAN cell types for normal SAN formation and that it plays cell autonomous as well as non-cell autonomous functions in SAN development and cardiac electrophysiology.
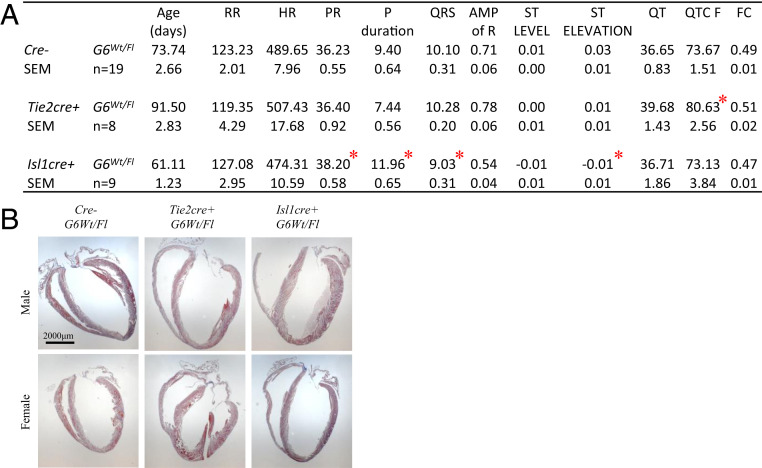
Conduction defects in conditionally deleted Gata6 adult mice. (A) Electrocardiogram profile showing the different interval measurements for each of the conditional knockout mice. Values are mean + SEM. *P < 0.05. (B) Masson trichrome staining on adult heart from Tie2cre+ and Isl1cre+ G6Wt/Fl mice. (Scale bar: 2,000 μm.)

| Genotype | Hypoplastic SAN, % | Functional changes |
| Gata6+/+ | 0 (0/9) | None (n = 33) |
| Gata6+/− | 100 (6/6) | Prolonged PR and QT intervals, ST elevation (n = 15) |
| Tie2cre+ G6Wt/Fl | 50 (4/8) | Prolonged QT interval (n = 8) |
| Isl1cre+ G6Wt/Fl | 67 (2/3) | Prolonged PR interval and P duration, ST elevation (n = 9) |
| Hcn4cre+ G6Wl/Fl | 50 (3/6) | Prolonged PR and amplitude of R (n = 7) |
SAN structure was assessed at E14.5. Electrophysiological defects were assessed at 60 to 115 d.
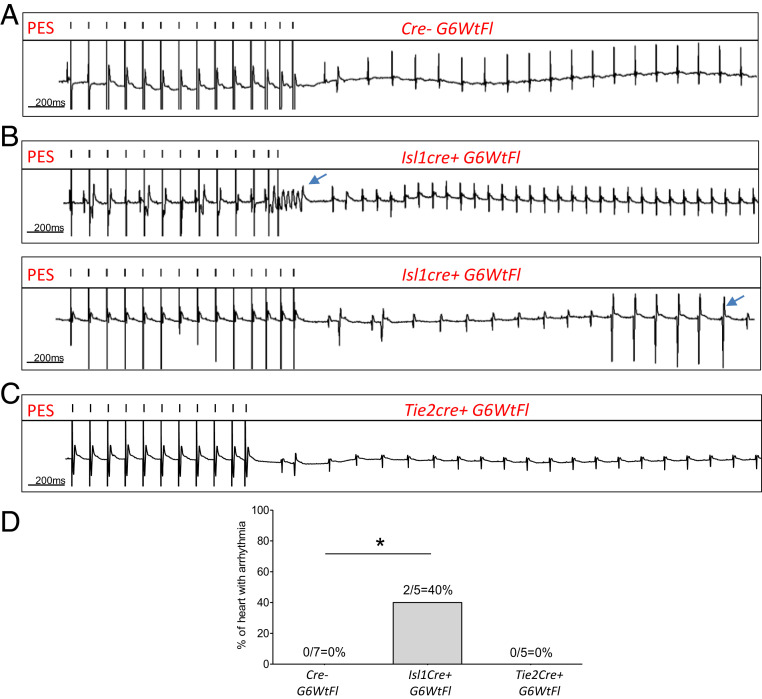
Isl1cre+ G6Wt/Fl mice recapitulate the phenotype of Gata6+/‒. (A–D) Ex vivo heart ECG recording following isoproterenol perfusion at 0.1 µM showing arrhythmia development in Isl1cre+ G6Wt/Fl mice when compared to control littermates. *P < 0.05.
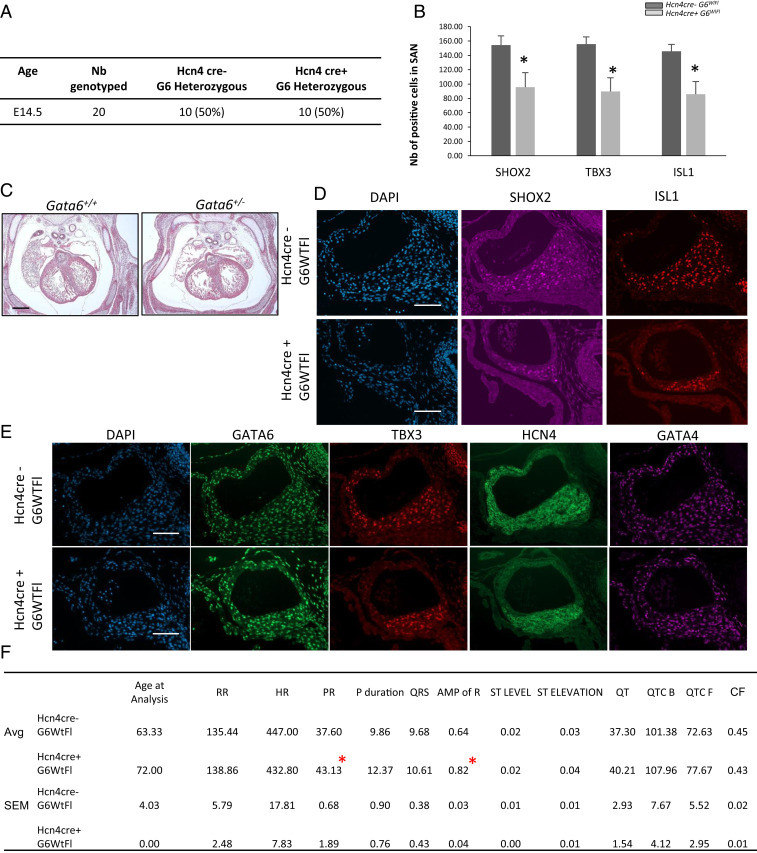
Structural conduction defects in heterozygous Hcn4 conditionally deleted Gata6 embryos. (A) Frequency of genotypes obtained from crossing Hcn4cre+ with the G6FLFL line. No changes in the expected Mendelian ration was observed. (B) Quantification graph showing the number of positive cells for Shox2, TBX3, and ISL1 within the SAN region in both Hcn4cre+G6Wt/Fl and control mice. *P < 0.05. (C) Masson trichrome staining on E14.5 embryos from Gata6+/+ and Gata6+/− mice showing the absence of structural defects. (D and E) Immunofluorescence staining for HCN4, GATA6, GATA4, SHOX2, TBX3, and ISL1 in the hearts of E14.5 Hcn4cre+ G6wtFl embryos vs. control (Hcn4cre- G6wtFl). (Scale bar: 100 μm.) (F) Electrocardiogram profile showing the different interval measurements for each of the conditional knockout mice. Values are mean + SEM. *P < 0.05.
The sinus node is the normal pacemaker of the heart, and its dysfunction ultimately leads to SSS, a very important clinical problem and the major indication for electronic pacemaker implantation. SSS is generally a disease of aging, but it can be present in younger individuals as a genetic disease or in association with heart failure. Given the aging population and the rising frequency of heart failure, the incidence of SSS is predicted to steadily increase (27). Over the past decades, there have been significant advances in our understanding of SAN development and function, but the cellular and molecular pathways underlying SAN structure in health and disease are incompletely elucidated (28). Studies of the genetic and structural basis of SAN pathophysiology (reviewed in ref. 29) revealed, among other things, that transient NOTCH activation or loss of transcription factors Tbx3 or Pitx2 leads to SSS in mice (3031–32).
In humans, most known SSS-associated mutations involve genes encoding ion channels or structural proteins, but variants near transcription factors important for cardiac development have also been associated with heart rate and atrial rhythm variabilities (3334–35). It is also increasingly evident that SAN structure and cellular remodeling promote sinus node dysfunction. Adequate SAN volume is required for proper electrical coupling to adjacent tissues, and SAN hypoplasia in newborns is linked to arrhythmias (36). In fact, loss of cells in the SAN and structural changes therein are features in patients with SSS (37, 38). Notwithstanding the important insights gained in recent years, the biological processes that contribute to the architecture of the SAN during development remain poorly understood.
Here we show that transcription factor GATA6 is present throughout the SAN and that mice with a mutated Gata6 allele have a hypoplastic SAN and significant disruption in the normal genetic program as evidenced by decreased levels of key pacemaker cell regulators such as TBX3 and TBX5 and up-regulated atrial genes like Nkx2.5 and Nppa. In addition, patterning of the SAN appears to be disrupted, as evidenced by the loss of HCN4+ pacemaker cells predominantly in the head region. It was previously suggested that the head and tail regions represent separate regulatory domains with specific genetic programs and contributions to pacemaking (39, 40). Moreover, TBX18-expressing mesenchymal cells were shown to contribute differentially to the head region (39). In Gata6+/− SANs, TBX18 levels are down-regulated, whereas PITX2 and NKX2.5 expression is up-regulated (Fig. 2D). These changes are consistent with a role for GATA6 in the differentiation of pacemaker myocytes. Hypoplastic SANs were also observed when one copy of Gata6 was specifically deleted either from ISL1+ secondary heart field cells or from TIE2+ endothelial cells. However, the patterning defect in the head region was not reproduced in these lines, raising the possibility that some pacemaker cells in the SAN head region may have a distinct embryonic origin. This would be consistent with the finding in chicken embryos of SAN progenitors in a region posterior to the ISL1+ domain referred to as the tertiary heart field (41).
We suggest that within the SAN, GATA6 plays cell-specific roles in regulating pacemaker cell differentiation. In ISL1+ myocytes and directly within HCN4+ conduction cells, GATA6 functions as a key activator of the genetic program required for differentiation into pacemaker myocytes, at least in part, by acting upstream of several SAN transcriptional regulators, notably TBX3, TBX5, and TBX18. In endothelial cells, GATA6 could regulate pacemaker myocyte differentiation and possibly survival/proliferation indirectly, through its effect on paracrine factors, like EDN1, known to contribute to SAN cell differentiation (26). Our transcript analysis reveals decreased levels of EDN1, as well as one of its receptor, EDNRB, in Gata6+/− hearts as early as E11.5 (Fig. 2E), suggesting that modulation of EDN1 may be one mechanism by which GATA6 contributes to paracrine regulation of pacemaker cell differentiation.
In addition to its role in the formation of the SAN, GATA6 appears to modulate cardiac conduction by affecting several conduction segments. Interestingly, the main two defects observed in Gata6 heterozygote mice, prolonged QT and PR, can be dissociated using cell-specific deletion. Notably, loss of Gata6 from HCN4+ conduction cells leads to prolonged PR but normal QT indicative of delayed SAN conduction. This is also observed in mice with deletion of Gata6 in ISL1+ cells that give rise to conduction cells of the SAN. Conversely, Gata6 deletion from the endothelial lineage that contributes to the Purkinje fibers results in prolonged QT and normal PR, reflecting repolarization defects. As mentioned earlier, sick sinus syndrome generally affects older individuals and can be difficult to diagnose due to its nonspecific presentation, including the ECG findings. It is therefore possible to expect further rhythm abnormalities in older Gata6 heterozygote mice. Nonetheless, the presence of a fully penetrant hypoplastic SAN phenotype in embryos and conduction abnormalities in young Gata6 heterozygote mice suggest that GATA6 acts in multiple cell types to coordinately regulate SAN development and cardiac electrical activity.
Mutations in GATA6 and two other members of the GATA family of transcription factors, notably GATA4 and GATA5, have been associated with human atrial fibrillation (42). Additionally, GATA4 was shown to regulate several atrioventricular canal enhancers, and loss of one Gata4 allele in mice leads to alterations in the proximal component of the cardiac conduction system, essentially at the atrioventricular node. Our data show that GATA4 is abundantly expressed in the SAN (Figs. 3 and 8) within GATA6+ cells. Gata4 heterozygote mice have shorter PR, whereas Gata6 heterozygote mice have longer PR, suggesting they each play distinct roles in the proximal CCS. Our preliminary analysis of SANs in Gata4 heterozygote mice did not reveal obvious changes in SAN size and structure. Whether this reflects a possible compensatory role of GATA6 and whether the two GATA proteins play opposing or partially overlapping roles in the SAN and in other CCS components deserve to be investigated. In vitro, GATA proteins have the ability to bind through their highly homologous zinc finger domain to similar DNA elements and to activate similar promoter targets. The two proteins differ in their N and C activation domains which play important roles in their mechanisms of action. This includes, at least in part, interactions with other activators and coactivators such as TBX proteins and NKX2.5. It is noteworthy that GATA6 mutations associated with human arrhythmias are found mostly in the N- and C-terminal domains. The mechanism(s) by which these amino acid changes cause atrial fibrillation will need to be clarified and may involve modified interactions with one or more coregulators. The findings presented here provide a molecular framework to explore how GATA6 mutations cause atrial fibrillation in humans and raise the intriguing possibility that GATA6 may be a genetic modifier of cardiac conduction disease, including sick sinus syndrome.
Mouse handling and experimentation were performed in accordance with the guidelines of the Canadian Council on Animal Care and the NIH (43). Experiments were approved by the Animal Care Committee of the University of Ottawa (protocol number BMI-1973). Gata6 heterozygous (Gata6+/−, C57BL/6) mice were previously described (20). Cell-specific knockout mice were obtained by crossing the Gata6Fl/Fl line with the respective cre lines (20, 44). Embryos and adult heart tissues were fixed with 4% paraformaldehyde in phosphate-buffer saline (PBS), paraffin embedded, sectioned at 4 μm intervals, and processed. Masson trichrome staining was performed by the histology service of the University of Ottawa. The opening of the aortic valve on the left ventricle was used as a reference to stain SAN in all animals, ensuring the same plane in the heart.
Details of the rest of the methods used are included in SI Appendix.
We are grateful to Megan Fortier, Janie Beauregard, and Nathalie Ethier for technical support; Hélène Touchette for secretarial help; and members of the M.N. laboratory for discussions and helpful suggestions. We thank Dr. Sharon Prince for sharing the TBX3-Luc reporter construct and Dr. Farah Sheikh for sharing the HCN4-ERTcre mice and acknowledge the invaluable support of the University of Ottawa Histology and Animal Physiology cores. This work was funded by a Canadian Institutes of Health Research (CIHR) Foundation grant (CIHR Grant 353388) to M.N. and a Natural Sciences and Engineering Research Council of Canada grant (Grant RGPIN-2017-05353) to C.F. L.G. was the recipient of the M. E. Abbott Scholarship from the Bicuspid Aortic Foundation and a graduate excellence scholarship from the University of Ottawa. J.W. received CIHR and University of Ottawa PhD scholarships.
All study data are included in the article and SI Appendix.
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
30
31
32
33
34
35
36
37
38
39
40
41
42
43
 GATA6 is a regulator of sinus node development and heart rhythm
GATA6 is a regulator of sinus node development and heart rhythm
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
