
Edited by Cyrille Costentin, Universite Paris Diderot (Paris 7), Paris, France, and accepted by Editorial Board Member Tobin J. Marks October 8, 2020 (received for review March 19, 2020)
Author contributions: S.J.S., J.-Y.L., and T.D.C. designed research; S.J.S., S.P., and J.G.L. performed research; S.J.S. contributed new reagents/analytic tools; S.J.S., S.P., J.Y., and D.-W.H. analyzed data; S.J.S. and T.D.C. wrote the paper; and J.Y. and D.-W.H. visualized and illustrated the figures.
The electrochemical reaction at the insulator is extraordinary. Despite its counterintuitiveness, it is made possible by using a silicon oxide dielectric electrode as the cathode. In this study, we use such a dielectric electrode to enable a version of electroorganic reaction. Interestingly, the oxidized products are produced at the cathode in the case of anthracene and its derivatives. Besides, normal reduction also occurred in the case of nitrobenzene. We suggest the electrochemically generated hydrogen species, supposedly the hydrogen atom, is responsible for this phenomenon. This is the first case to use such a reagent in the mild electrochemical system, and this reaction may be applied to a synthetic strategy for organic molecules.
The faradaic reaction at the insulator is counterintuitive. For this reason, electroorganic reactions at the dielectric layer have been scarcely investigated despite their interesting aspects and opportunities. In particular, the cathodic reaction at a silicon oxide surface under a negative potential bias remains unexplored. In this study, we utilize defective 200-nm-thick n+-Si/SiO2 as a dielectric electrode for electrolysis in an H-type divided cell to demonstrate the cathodic electroorganic reaction of anthracene and its derivatives. Intriguingly, the oxidized products are generated at the cathode. The experiments under various conditions provide consistent evidence supporting that the electrochemically generated hydrogen species, supposedly the hydrogen atom, is responsible for this phenomenon. The electrogenerated hydrogen species at the dielectric layer suggests a synthetic strategy for organic molecules.
Electrosynthesis is a classical method in which the reactant is activated by the faradaic reaction to trigger an electroorganic reaction, and it often involves electrochemical promotion of oxidative addition (1, 2), C-H activation (23–4), or C-C coupling (2, 5, 6). Such organic reactions generally require chemical additives such as oxidant/reductant, acid/base, and catalysts. Electrosynthesis substitutes such complicated combinations by employing electrodes to supply or abstract electrons. Electrosynthesis has been ceaselessly evolving to contribute to economic and sustainable strategy (2). Particularly in the last decade, intense studies were conducted to make substantial progress (12345–6). However, considerable attention was focused on electroorganic reactions rather than the electrode material. The electrode used in electrosynthesis is deemed to be a component to complete the circuit and deliver/collect electrons. As a consequence, only limited options of chemically inert conductors have been available such as carbon or boron-doped diamonds (2, 5).
In this regard, there is an unusual approach that deserves attention, i.e., electrochemistry at an insulator thin film (789101112–13). Electrochemistry at an insulator, in which few charge carriers are available, is counterintuitive and thereby rarely found in the literature. The previous reports on this unique electrochemical reaction fall into three categories. The first are the reactions triggered by static electricity generated by contact electrification of the dielectric materials. Kelvin-probe microscopy showed a mosaic of positive and negative charges (8) that could drive the electrochemical reduction of metal ions and metal complexes (78–9). The second is a faradaic reaction at the glass as demonstrated on nanometer-thick glass-covered Pt nanoelectrode (10). Voltammetric investigation of the ruthenium redox couple in this system revealed the behavior of the hydrogel-like state of glass in a strongly acidic medium.
The third category is the electrochemistry at the thin film of thermal SiO2 (1112–13). The thermal SiO2 layer has a highly dielectric structure with minimal defects, and with the underlying conductive substrate, it constitutes an electrolyte–oxide–semiconductor (EOS) system. Reportedly, hydrogen atoms can be produced at the highly n-doped Si (n+-Si, considered metallic)/thermal oxide interface, being responsible for faradaic current in voltammetry. Protons selectively permeate along a steep voltage gradient across the oxide layer and are reduced at the interface between the oxide and n+-Si. In addition, silanol defects in thermal SiO2 are subject to electrochemical reduction, leading to asymmetric voltammetric behavior in acidic aqueous solution (only reduction observed). This thermal SiO2 layer permits the selective migration of protons as well as the diffusion of hydrogen atoms which could react with the nearby chemical environment as shown from metal electrodeposition (11, 13). Thus, the thermal SiO2 layer serves as a “chemical electrode” in which electrochemical reaction generates reactive species being stored in the insulating layer before intermolecular reaction. In addition, we might be able to monitor the progress of the whole reaction by simply examining the faradaic current and controlling the reaction by adjusting the applied voltage.
Electroorganic reactions potentially benefit from thermal SiO2 film on n+-Si. The thermal SiO2 surface where the electroorganic reaction takes place is much more resistive to chemical fouling, e.g., platinum poisoning (14, 15), than most electrocatalysts. Highly reactive hydrogen atoms need extremely negative potential to cause a catastrophic increase in background current at conventional conductive electrodes (−2.30 V vs. normal hydrogen electrode) (16, 17). A thin insulator film, across which the electric potential gradient is widely distributed, prevents vigorous electrolysis.
In this study, we introduced a defective 200-nm-thick n+-Si/SiO2 as a dielectric electrode, which has chemical defects such as dangling bonds or atom vacancies but a negligible number of pinholes and cracks. The electrochemical behavior and cathodic electroorganic reactions of various organic compounds, especially anthracene, which is one of the basic polycyclic aromatic hydrocarbons (PAHs), were investigated at this electrode. We performed electrolysis on the dielectric electrode in an H-type divided cell and analyzed the products. Surprisingly, the oxidized products were generated at the cathode, indicating that the n+-Si/SiO2 dielectric electrode does not allow electron tunneling through itself but creates reactive chemical species acting as a chemical electrode.
The defective 200-nm n+-Si/SiO2 electrode was prepared, and its cross-section was characterized by transmission electron microscopy (TEM; Fig. 1A). There are no observable physical defects such as pinholes or cracks, implying to inherently possess chemical defects. Fig. 1B shows the typical electrochemical behavior observed from the defective 200-nm n+-Si/SiO2 electrode in this study. Cathodic current starts to flow around −1 V in the acidic medium. Current density and onset potential sensitively respond to proton concentration. Anodic current, on the other hand, is negligible in the wide range of high positive bias. This is unchanged wherever potential scan begins and regardless of linear sweep or cyclic voltammetry (the inset of Fig. 1C). Thus, the defective 200-nm n+-Si/SiO2 electrode behaves as a pure insulator in the positive potential region, and the precedential cathodic reaction has a negligible influence on the anodic reaction in voltammetry. Fig. 1C shows the voltammetric responses in the presence of redox couple that undergoes outer-sphere electron transfer in aqueous media. In the absence of an acid, only a small cathodic current is observed, indicating negligible electron tunneling across the SiO2 layer. This is analogous to the electrochemical behavior of the previously reported 6-nm-thick n+-Si/SiO2 EOS system (11). In the presence of an acid, redox couple coexisting in the solution does not cause any notable difference in the voltammograms. This behavior is also observed in aprotic solvents such as acetonitrile (MeCN) containing perchloric acid (HClO4) and tetrabutylammonium perchlorate (TBAP) electrolyte (Fig. 2 A and B). Compared to the aqueous solution, the current density decreases, whereas the onset potential appears to be invariant. The lower current density stems from less dissociation of the acids in an aprotic solvent (18). The influence of the redox couple on the proton-driven cathodic current in an acidic solution is insignificant (11).


Characterization of the prepared defective 200-nm n+-Si/SiO2 electrode by (A) TEM cross-section, (B) linear sweep voltammograms of various acid concentrations, and (C) various redox couples and acid composition in aqueous media. The inset in C shows the cyclic voltammogram in a wide potential range. All electrochemical measurements were in N2 atmosphere with a scan rate of 10 mV/s.
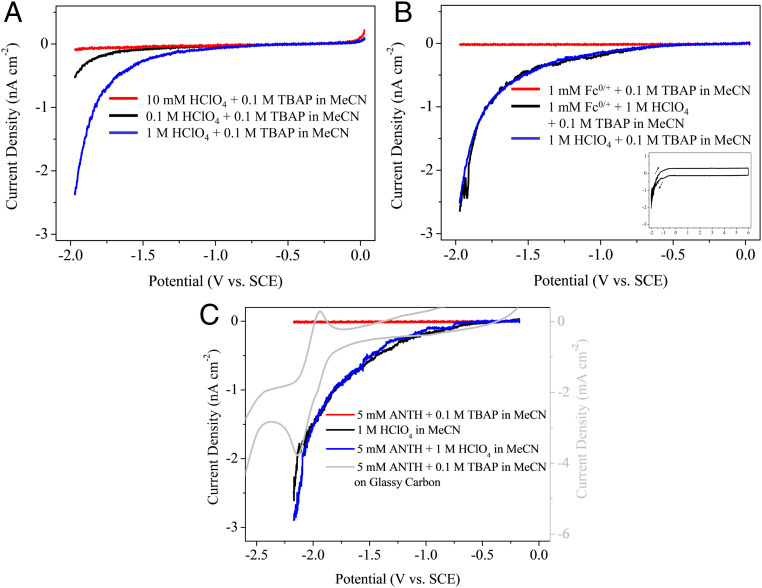
Representative electrochemical behavior of the defective 200-nm n+-Si/SiO2 electrode. All of the measurements were in N2 atmosphere with a scan rate of 10 mV/s. Linear sweep voltammograms of (A) various acid concentrations and (B) Fc0/+ redox couple in MeCN. The inset in B shows the cyclic voltammogram in a wide potential range. (C) Linear sweep voltammograms of ANTH in acidic MeCN. Cyclic voltammogram of ANTH on glassy carbon electrode is for comparison (scan rate: 100 mV/s).
The reactant for the cathodic electroorganic reaction on the defective thermal SiO2 in this study is anthracene (ANTH, 1a), one of the basic building block molecules of PAHs. Chemical conversion and degradation of PAHs have been attracting research interest because of the health risks they pose in our daily lives (19, 20). Fig. 2C shows that the presence of 1a has a negligible effect on the voltammetric behavior of the defective thermal SiO2. Importantly, the onset potential of the dielectric electrode in acidic organic media is much more positive than the reduction potential of 1a as shown in the voltammogram obtained with glassy carbon (Fig. 2C; Ec1, 1/2o = −2.02 V. The full voltammogram is in SI Appendix, section 3.1). This indicates that 1a does not directly uptake an electron from the cathode but undergoes a chemical reaction with something generated electrochemically at the electrode. Electrolysis of 1a in the acidic organic medium in H-type divided cell (SI Appendix, Fig. S1 A–C) under N2 atmosphere at −1.67 V, which is less negative than its reduction potential, provides more interesting information about the electroorganic reactions occurring at the defective thermal SiO2. Both high-performance liquid chromatograph mass spectroscopy (LC-MS) and proton nuclear magnetic resonance (1H-NMR) analyses unambiguously confirmed that anthraquinone (AQ, 2a) is produced by electrolysis (SI Appendix, Fig. S2). Compound 2a is obviously not the electrochemically reduced form of 1a but the oxidized product at the cathode. Various sets of control experiments confirmed that the reaction is truly the electrochemical reaction (details in SI Appendix, section 2).
The electroorganic reactions of 1a on defective 200-nm n+-Si/SiO2 electrode in various solution compositions are summarized in Table 1. Entries 1–3 show the electrolysis results for three different concentrations of protons while the concentration of the reactant 1a is fixed. The electroorganic reaction strongly depends on the proton concentration. Both amounts of consumed (1a) and generated (2a) compounds are sensitively proportional to the proton concentration. Selectivity (the percentage yield of 2a from 1a) is inversely proportional to the proton concentration, indicating that a lower concentration of protons promotes a more specific reaction resulting in 2a. Note that the Faraday efficiency is lower than 10% in general for the 1 M acid case, and it sensitively improves up to 37% for lower proton concentration (SI Appendix, Table S1).

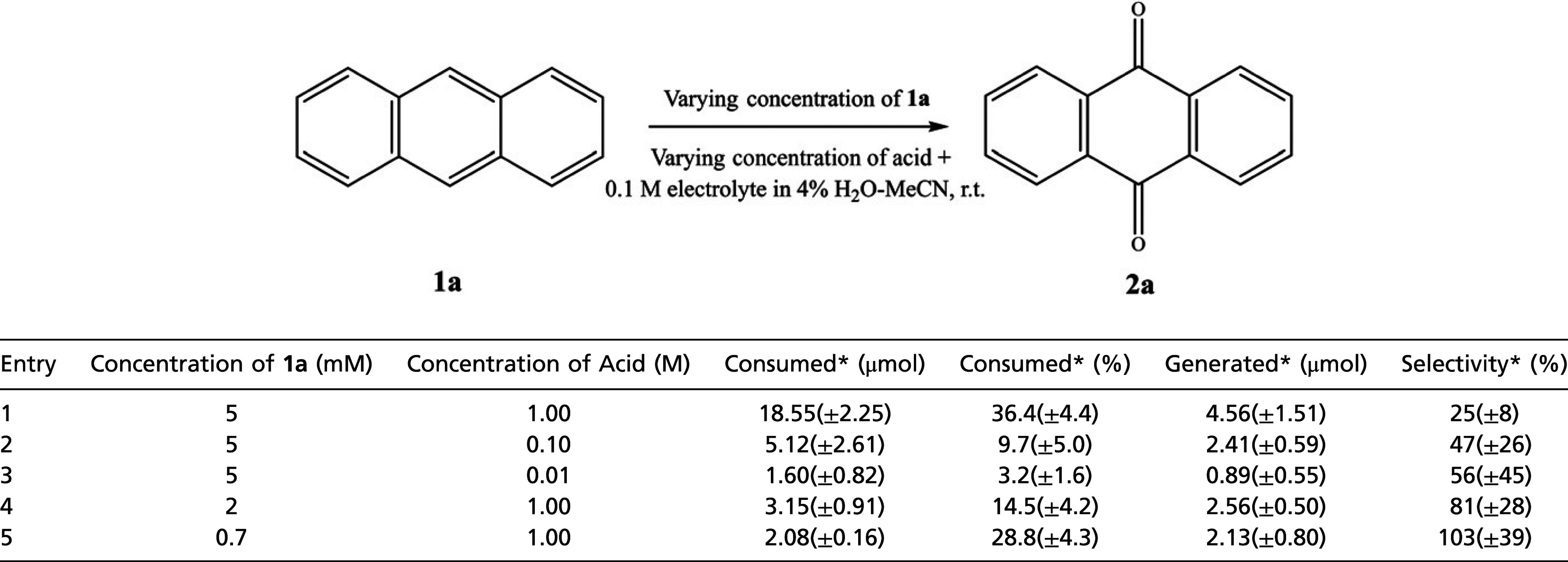 |
The reaction was carried out for 10 mL volume of the solution. Perchloric acid was used as the acid, and the water content was 4 vol% in MeCN.
* Quantification done by LC-MS. Extended version of the table with Faraday efficiency is in SI Appendix, Table. S1. Selectivity calculated as the percentage of the generated amount of the product over the consumed amount of the reactant. r.t., room temperature.
On the other hand, entries 1, 4, and 5 reveal the effect of the concentration of 1a at a fixed proton concentration. A low concentration of 1a favors the selective generation of 2a. It is obvious that a higher concentration of reactants or protons results in a more indiscriminate reaction. Since 1a cannot permeate the SiO2 film, protons should uptake electrons from the electrode producing reactive hydrogen species. It is plausible that 1a in the solution encounters the electrochemically generated species on the dielectric film surface. Such a heterogeneous process may not be too selective because of the highly unstable hydrogen species.
Table 2 shows the summary of several electroorganic reactions on defective 200-nm n+-Si/SiO2 electrodes. All of the chemical derivatives of 1a (1b, 1c, and 1d) are converted into the oxidized forms as a consequence of reductive electrolysis. Product analysis revealed that the oxidation selectively occurs at the preferred acene site. We also performed electrolysis of other PAHs such as phenanthrene (Phen, 3a) and pyrene (Py, 4a), but 3a hardly reacted. Although a significant amount of 4a was consumed, no identifiable products were found. Not only oxidation at dielectric electrode as the cathode but also reduction of organic reactant occurred as shown by the electroorganic reaction from nitrobenzene to aniline (Table 2). In addition, we could electrodeposit various metals directly on the dielectric electrode as reported in the previous works (11, 13).
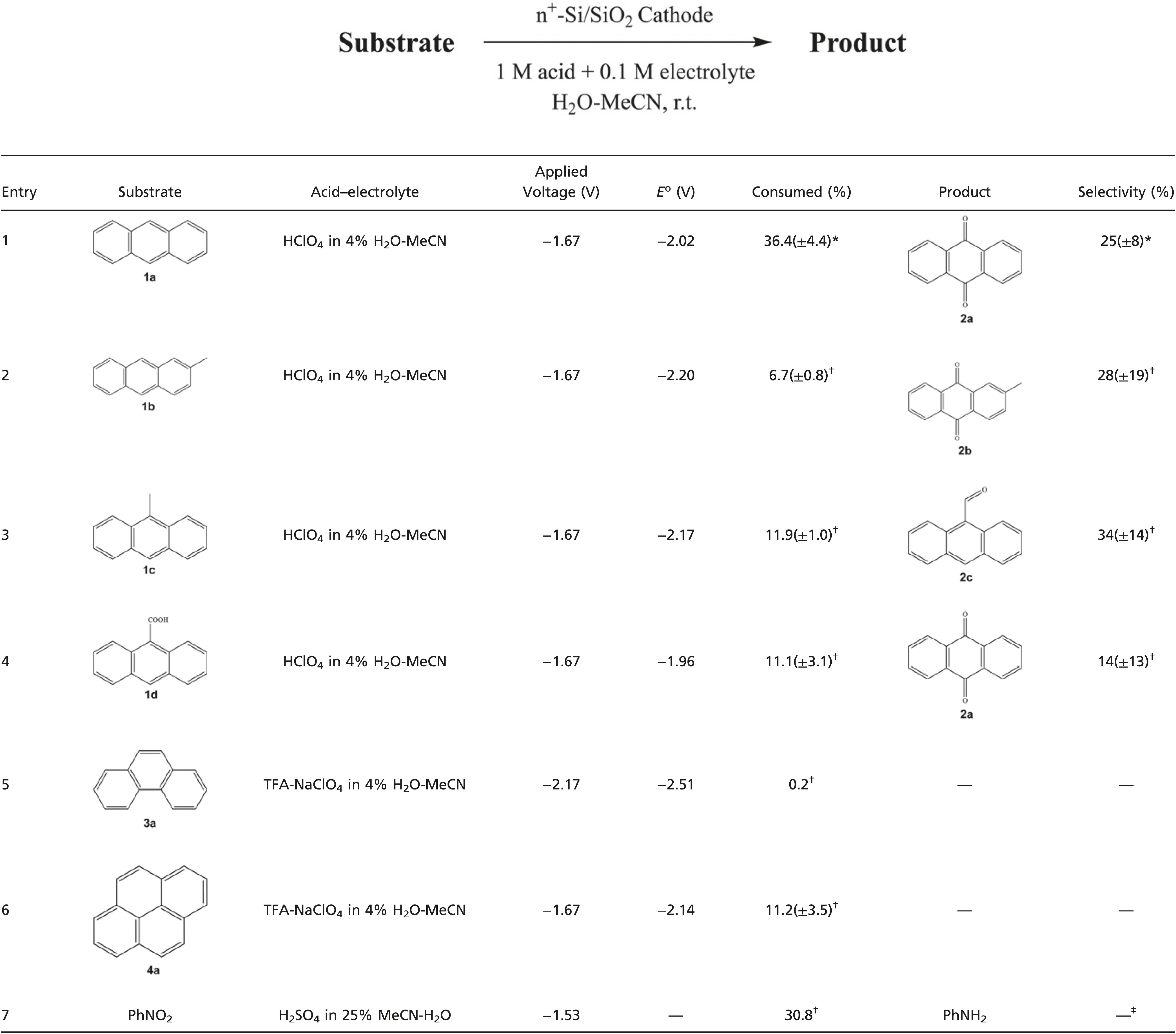 |
The 5 mM substrate, 1 mM biphenyl (BP, internal standard), and 1 M acid–0.1 M electrolyte in (vol/vol) H2O-MeCN at room temperature in N2 atmosphere. Only the identified products are stated. (V) were measured from the cyclic voltammetry on glassy carbon electrode; see SI Appendix, section 3.1. r.t., room temperature; TFA, trifluoroacetic acid; NaClO4, sodium perchlorate; PhNO2, nitrobenzene; PhNH2, aniline.
* Quantification done by LC-MS.
† Quantification done by UV-VIS.
‡ It was difficult to determine the product quantity due to spectral overlap between substrate and product. The product was identified by GC-MS measurement and MS library comparison. Selectivity calculated as the percentage of the generated amount of the product over the consumed amount of the reactant.
During electrolysis at defective 200-nm n+-Si/SiO2, faradaic current gradually elevates where the current density increases to the order of 100 μA∙cm−2 (SI Appendix, Fig. S3A). The charge passed is ∼50 C that causes a scalable change in both the substrate and the product of subμmol for a given electroorganic reaction. Such high current is discussed in detail on the basis of the surface analysis and electrodeposition in SI Appendix, section 1.3 and Figs. S3–S5. In short, both the proton permeation and chemical defects are supposedly responsible for the quasi-reversible change (or damage) in film properties.
Furthermore, 200-nm-thick n+-Si/SiO2 electrode can be fabricated as well by tetraethylorthosilicate chemical vapor deposition and plasma-enhanced chemical vapor deposition on n+-Si with the same thickness. The electrolysis of 1a was conducted on these SiO2 for comparison (SI Appendix, Table S2). It indicates that the defective 200-nm n+-Si/SiO2 electrode was a specifically appropriate candidate to study the electroorganic reaction as compared to the tested oxide films. The detailed discussion is described in SI Appendix.
The observations in the proposed system guide us to the reaction mechanism of the electroorganic reactions, which is illustrated in Fig. 3. The electrochemically generated species at the cathode is assumed to be a hydrogen atom as a few reports suggested based on indirect evidence including Raman spectroscopic results (11, 21, 22). It is predicted that protons and supposedly hydrogen atoms in the thermal oxide cathode move in different ways according to the literature (11, 12). The protons are migrated through the permeation of SiO2 by a strong field across the dielectric oxide layer. At the interface of n+-Si and thermal oxide, protons are supposedly reduced to hydrogen atoms that are neutral in charge and thus not driven by the electric field. Electrogenerated hydrogen species can either diffuse out of the oxide film or be transported by redox relay with protons inside the oxide. Reportedly, the resistance of the oxide film decreases under similar conditions as this study (12).

Schematic illustration of the proposed mechanism of the ANTH (1a) reaction on defective 200-nm n+-Si/SiO2 electrode.
This proposed mechanism agrees quite well with the result in Table 2. The compound 1a is converted to 2a supposedly by the preferential reaction of the C-H bond in the central ring of acene over peripheral acene (20). A similar reaction occurs from 1b to 2b because 1b has C-H bonds at the 9- and 10-sites like 1a (see SI Appendix, section 1.4 for miscellaneous discussion on the site reactivity). In the case of 1c, the C-H bond at 9-substituted methyl (benzylic acene) is more labile than the 10-site (central ring of acene); thereby HAA takes place preferentially at the 9-site. The reaction of 1d is distinct in that the carboxylic group at the 9- site leaves to yield 2a. Since the 9-COOH site is more reactive than other peripheral C-Hs, decarboxylation by hydrogen atom takes place to produce the intermediate similar to 1a followed by the oxidation to 2a, as described earlier (the interpretation of 1d’s cyclic voltammogram in SI Appendix, section 3.1). We believe that such decarboxylation is similar to the Kolbe pathway which generates neutral radical intermediate from carboxylic acids (2, 29). For other PAHs of 3a, nearly no reaction occurred. On the other hand, 4a was consumed, but we failed to identify the products, presumably due to either the decomposition of the reactant or the loss by work-up. The intriguing feature of the reaction of PAHs is that the amounts of the PAHs electrolyzed in this study linearly correlate with the reduction potentials () rather than BDE except for 1d (SI Appendix, Fig. S6). The easier to electrochemically reduce, the greater the consumption of PAHs. The reason for this is unclear, yet we could infer it as described in SI Appendix. Overall, it is certain that the observed reactions of PAHs are governed by the electrochemical reaction.
Nitrobenzene undergoes reduction in the same system, unlike anthracene derivatives as shown in Table 2. According to the suggested mechanism, hydrogen atom induces a slow and site-specific reaction probably because of its insufficient capability for HAA. The C-H bond in nitrobenzene is phenyl-H which is an unfavorable site as found in selective reaction at only the 9- and 10-sites (central ring of acene) in the anthracene derivatives. Rather, a hydrogen atom should be oxidized to a proton by losing an electron that is taken by the nitro group, simply the hydrogenation.
The electroorganic reaction on the dielectric electrode implies two significant findings. First, this dielectric electrode may serve as a hydrogen atom, providing a platform without hydrogen atom transfer catalysts. Second, the oxidation reaction occurs by the mediation of electrogenerated hydrogen atom. This is unprecedented, although the oxidation reaction by a hydrogen atom generated by radiolysis in harsh conditions was reported previously (16, 30).
Concerning the experimental evidence of the proposed mechanism, 2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), a renowned known radical capture agent (31, 32), was used to capture the neutral charge radical intermediate (SI Appendix, Fig. S7). The captured intermediate species found by LC-MS indicated [M + H] as m/z = 489.5 where M = ANTH − 2 H + 2 TEMPO (SI Appendix, Fig. S7). We experimented with 1 mM acid organic solution because TEMPO is unstable in a strongly acidic solution (<pH 3) and undergoes disproportionation. The resulting products were too small in amount to be characterized by NMR. As listed in Table 1, the consumption of 1a on the dielectric electrode becomes very inefficient in a weak acidic solution, which is an unfavorable condition to capture the target intermediate.
The role of moisture in such an electroorganic reaction has been addressed (Table 3). Because the added acids contain moisture in themselves, the solution is not dry. The effect of acid is minor as shown in entries 2 and 3 of Table 3. When water was added to be 4% (vol/vol), both 1a consumption and 2a generation increased remarkably. However, excess water, e.g., 10% of water content, reduced 2a production as well as 1a consumption. Therefore, the electroorganic reaction of 1a at the defective n+-Si/SiO2 electrode definitely involves water, while the reaction is inhibited in the presence of too much or no water. It must be mentioned that the role of other reactive oxygen species such as O2 was also considered. The reaction was carried out in N2 atmosphere so the possibility of O2 could be discarded. In addition, the separate experiment was carried out in O2 atmosphere, but the extent of the reaction was less than that of N2 atmosphere (SI Appendix, Table. S3)
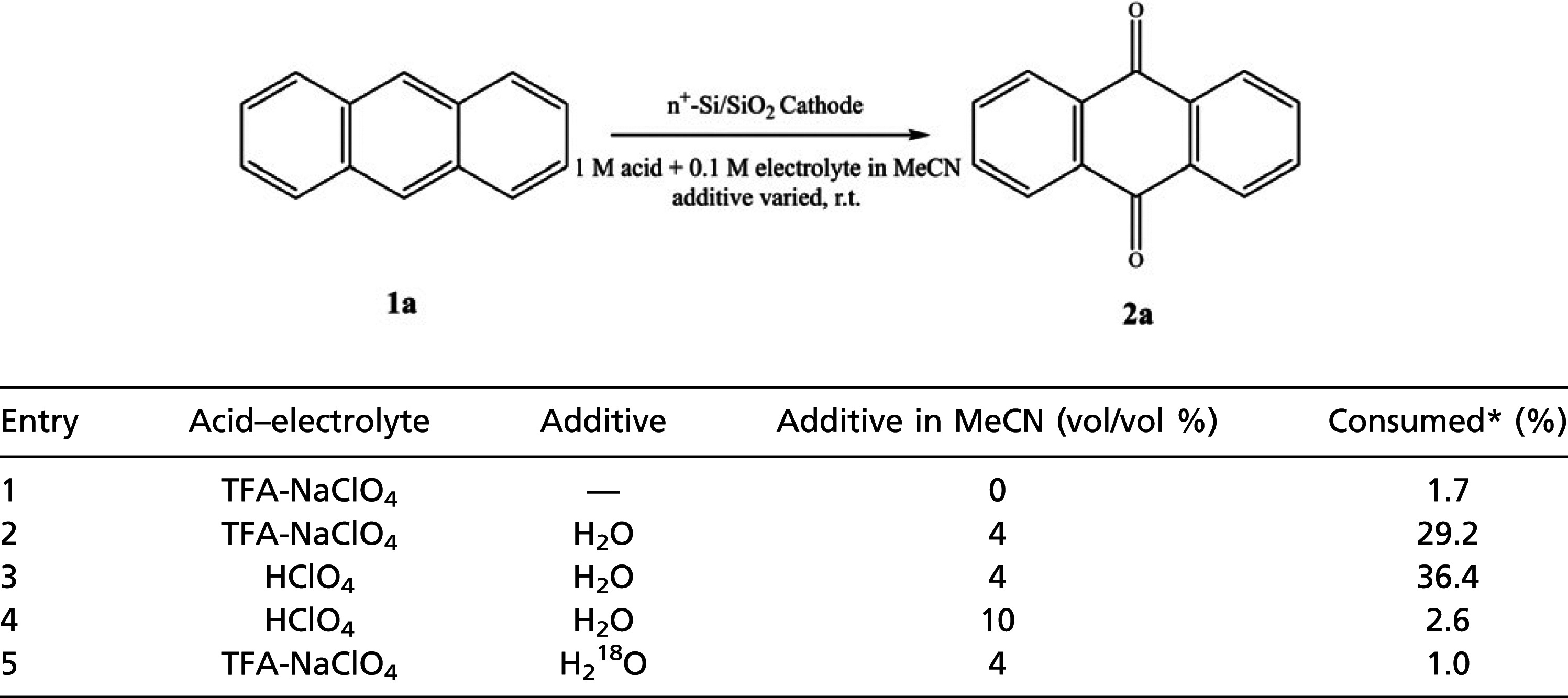 |
* Quantification done by LC-MS.
When H218O is added instead of water, both the consumption of 1a and the generation of 2a are significantly suppressed. In addition, LC-MS analysis shows the percent ratio of the observed m/z peaks for 2a, which is equivalent to [M + H], as (C14H916O2, 209): (C14H916O18O, 211): (C14H918O2, 213) = 62.9: 26.2: 6.6. The produced 2a has minor content of 18O, indicating that only a limited portion of oxygen atoms comes from additive H218O. Considering that the solution contains moisture even before H218O addition, we can infer that the proposed hydrogen atom abstracted neutral radical intermediate reacts with H2O preferentially rather than H218O. As found in the oxygen evolution at metal complexes (33), 18O and 16O should have different bond strength, which is generally stronger for heavier atoms. Therefore, 18O-H is stronger than 16O-H. Although the difference in BDE only barely justifies the specific reaction of H2O, many other factors affecting the dissociation of O-H bonds such as molecular orientation, thermodynamic stability, and free energy change of both the intermediate and additives need to be considered. Furthermore, this phenomenon may be explained by thermodynamic isotope effect (34, 35). In addition, we must take into account the solubility of 1a in acetonitrile. Although the addition of oxygen sources, e.g., water, promote the overall reaction, too much could cause a solubility problem; 1a will start to precipitate in 10% water.
In summary, we prepared a defective n+-Si/SiO2 electrode and investigated the cathodic electroorganic reactions in the EOS system. The dielectric electrode exhibits similar electrochemical behavior to the previously reported 6-nm-thick thermal SiO2. It gives low faradaic current upon negative bias in acidic organic media, which depends only on the proton concentration of the solution. The presence of redox couples, obeying outer-sphere electron transfer, has a minor effect on current density. The prolonged electrolysis of organic reactants at negative potential showed that cathodic current can yield not only the reduced products but also oxidized products. Based on the analysis of the electrode itself and products of electrolysis, we propose that the observed phenomenon involves electrochemically generated species, probably a hydrogen atom, produced by proton reduction in the SiO2 film. The mechanism for the cathodic electroorganic reaction was proposed and consistently supported by the experimental results of this study. Introduction of a dielectric film to the electrode creates a unique chemical electrode, suggesting a strategy for synthetic chemistry. We believe that this system paves a way for utilizing the creative combination of electrochemistry and organic synthesis. In addition, we are still looking for more practical and valuable reactions in the EOS system as this work was demonstrated for a simple reaction to prove the concept of such a system.
N-type, arsenic-doped, <100> oriented Si (n+-Si) wafers with resistivity as low as 0.005 Ω cm were obtained from STC. Highly n-doped Si with thermally oxidized SiO2 (n+-Si/SiO2) was produced via oxidation n+-Si wafers under the oxygen environment at 850 °C. n+-Si was cleaned by standard SPM and SC-1,2 cleaning consecutively. Then, the target 200-nm-thick thermal SiO2 was prepared at 850 °C in a furnace by blowing O2. The native oxide was not removed prior to the thermal oxidation. Note that not all of the defective thermal SiO2 show faradaic current upon negative bias. The electrodes were tested by voltammetry prior to electrolysis to discard the total insulator (roughly one out of four were discarded). This is deemed to be ascribed to the inhomogeneous defect density of the SiO2 layer. Because of the significant deviation in current density and onset potential between the prepared dielectric electrodes, Figs. 1 and 2 were obtained in an identical electrode. Nonetheless, we made sure of the overall trends through a sufficiently large number of electrodes
ANTH (≥99.0% GC), AQ (97%), BP (99.5%), 2-methylanthracene (2-Me-ANTH, 97%), 9-methylanthracene (9-Me-ANTH, 98%), 9-anthracenecarboxylic acid (9-COOH-ANTH, 99%), phenanthrene (Phen, ≥98.0%), pyrene (Py, 98%), perchloric acid (HClO4, 70%), TFA (99%), sodium perchlorate (NaClO4, ≥98%), TBAP (≥99.0%), sodium chloride (NaCl, ≥99.0%), sodium sulfate (Na2SO4, ≥99.0%), chloroform-d (CDCl3, ≥99.8 atom % D, 0.5 wt % Ag foil as stabilizer, 0.03 vol % TMS), and water–18O (H218O, 97 atom %) were purchased from Sigma Aldrich. Nitrobenzene (PhNO2, 98.0%) was purchased from TCI chemicals. Sodium hydroxide (NaOH, 98.0%), MeCN (99.5%), sulfuric acid (H2SO4, 98%), benzene (99.5%), and dichloromethane (MC, 99.5%) were purchased from Dae Jung. All chemicals were used without further purification.
The wafer samples were degreased by sonication in acetone and isopropanol and rinsed with methanol or deionized water. For back contact, the backside of the wafer sample was scratched with a diamond knife to remove the air-formed SiO2 followed by smearing with a eutectic of Ga-In (≥99.99% trace metal basis from Sigma Aldrich). Electrochemical characterization was performed by a conventional three-electrode cell connected to electrochemical analyzers (CHI 660 and 750, CH Instrument). Pt wire (diameter of 0.5 mm) was employed as the counter electrode. The reference electrodes were Ag/Ag+ (10 mM AgNO3 + 0.1 M TBAP in MeCN, BAS Inc.) and Ag/AgCl (3 M NaCl, BAS Inc.). The former was employed for MeCN-rich solution and the latter for a water-rich solution. All potentials refer to the saturated calomel reference electrode (SCE). In short, Ag/Ag+ was +0.327 V versus SCE for nondistilled acetonitrile.
Electrolysis was performed in a homemade H-type divided cell with working and counter electrode (WE and CE) chambers separated by Nafion 117 proton exchange membrane (0.007-inch thickness, Sigma Aldrich). It was carried out in 10 mL each in chambers for 2.7 d. The WE chamber was stirred 250 rpm and purged with N2 gas (99.999%) throughout the reaction. Typical electrolysis solution in the WE chamber composed of 5 mM of starting compound with 1 M acid dissolved MeCN. The CE chamber was filled with acid–electrolyte solution. Note that the potential drift of Ag/Ag+ reference electrode occurs upon prolonged electrolysis. The drift is −17 ± 9 mV which is not significant considering the typical applied voltage for electrolysis.
We performed UV-visible spectroscopy measurements using a UV-visible spectrometer (UV-VIS; Cary 60 G6860A, Agilent Technologies) to check spectrum change prior to collection. Liquid aliquots from electrolysis were collected for work-up to remove excessive acids prior to chemical analyses. An equivalent volume of benzene was mixed with collected liquid aliquots and two equivalent volumes of saturated aqueous NaCl solution to separate the aqueous electrolyte layer and the organic solvent layer. The organic layer was extracted from the mixture and subsequently dried using anhydrous Na2SO4. We analyzed the products using a high-performance liquid chromatography mass spectrometer (LC-MS/MS; LCMS-8050, Shimadzu). For quantification, BP was chosen as an internal standard and put into the WE chamber prior to electrolysis. We obtained quantitative results based on standard calibration. The detailed method for quantitative analyses is described in SI Appendix. The measurement of 1H-NMR was obtained by 300 MHz (AVANCE DPX-300, Bruker).
TEM samples of n+-Si/SiO2 were prepared with focused ion beam (FIB; SMI3050SE, SII Nanotechnology) gun of field emission scanning electron microscopy after sequential deposition of carbon and Au for sample protection. The sampling area for TEM was selected to have cross-sections perpendicular to the wafer. High-resoution (HR) images were taken by HR-TEM (JEM-2100F, JEOL) operated at 200 kV. Electron energy loss spectrum (EELS) was obtained by Cs-STEM (JEM-ARM200F, Cold field emission gun, JEOL).
S.J.S. thanks the Posco TJ Park Foundation for a Cheong-Am Science Fellowship. We thank National Center for Inter-University Research Facilities, National Instrumentation Centre for Environmental Management, Research Institute of Advanced Material, and Biocenter in Gyeonggi-do Business and Science Accelerator for the FIB, electron microscopies, EELS, LC-MS, and GC-MS. Additionally, we thank Inter-university Semiconductor Research Centre for fabricating n+-Si/SiO2. This research was supported by a National Research Foundation of Korea grant funded by the Korean government (Ministry of Science and ICT) (No. 2017R1E1A1A01074236).
All study data are included in the article and SI Appendix.
December 30, 2020: The tables have been updated to match the PDF display.
1
2
3
4
5
6
8
9
10
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
30
31
32
33
34
 Cathodic electroorganic reaction on silicon oxide dielectric electrode
Cathodic electroorganic reaction on silicon oxide dielectric electrode
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

