
 ,
Mikaela J. Louie,
Anna La Torre,
Mai T. Vu,
Katherine L. Mitchell,
Karl J. Wahlin,
Vinod Jaskula-Ranga,
Yang Hu,
,
Mikaela J. Louie,
Anna La Torre,
Mai T. Vu,
Katherine L. Mitchell,
Karl J. Wahlin,
Vinod Jaskula-Ranga,
Yang Hu,
Edited by Carol Ann Mason, Columbia University, New York, NY, and approved November 10, 2020 (received for review March 20, 2020)
Author contributions: A.K.P., M.J.L., A.L.T., R.N.W., D.J.Z., and D.S.W. designed research; A.K.P., R.M.B., C.D.L., T.L., M.J.L., A.L.T., M.T.V., K.L.M., K.J.W., C.A.B., D.J.Z., and D.S.W. performed research; V.J.-R., Y.H., and X.D. contributed new reagents/analytic tools; A.K.P., R.M.B., C.D.L., M.J.L., A.L.T., H.A.-A., M.T.V., K.J.W., C.A.B., S.V., J.L.B., V.P.L., D.J.Z., and D.S.W. analyzed data; and A.K.P., C.D.L., H.A.-A., J.L.B., V.P.L., D.J.Z., and D.S.W. wrote the paper.
Axonal injury plays a major role in many neurodegenerative diseases. The dual leucine zipper kinase (DLK) signaling pathway is a key regulator of axonal injury-induced neuronal cell death; however, DLK also has an important role in promoting axonal outgrowth. Therefore, inhibiting DLK as a therapeutic approach for neurodegenerative diseases is limited to a neuroprotective outcome without axon regeneration, prohibiting restoration of function. In fact, there are currently no strategies that provide long-term neuroprotection and axonal regeneration after injury. Here, we identified the germinal cell kinase four (GCK-IV) family of kinases as targets to maximize neuroprotection while promoting axon regeneration, making it an attractive therapeutic approach for a subset of neurodegenerative diseases.
Axon injury is a hallmark of many neurodegenerative diseases, often resulting in neuronal cell death and functional impairment. Dual leucine zipper kinase (DLK) has emerged as a key mediator of this process. However, while DLK inhibition is robustly protective in a wide range of neurodegenerative disease models, it also inhibits axonal regeneration. Indeed, there are no genetic perturbations that are known to both improve long-term survival and promote regeneration. To identify such a neuroprotective target, we conducted a set of complementary high-throughput screens using a protein kinase inhibitor library in human stem cell-derived retinal ganglion cells (hRGCs). Overlapping compounds that promoted both neuroprotection and neurite outgrowth were bioinformatically deconvoluted to identify specific kinases that regulated neuronal death and axon regeneration. This work identified the role of germinal cell kinase four (GCK-IV) kinases in cell death and additionally revealed their unexpected activity in suppressing axon regeneration. Using an adeno-associated virus (AAV) approach, coupled with genome editing, we validated that GCK-IV kinase knockout improves neuronal survival, comparable to that of DLK knockout, while simultaneously promoting axon regeneration. Finally, we also found that GCK-IV kinase inhibition also prevented the attrition of RGCs in developing retinal organoid cultures without compromising axon outgrowth, addressing a major issue in the field of stem cell-derived retinas. Together, these results demonstrate a role for the GCK-IV kinases in dissociating the cell death and axonal outgrowth in neurons and their druggability provides for therapeutic options for neurodegenerative diseases.
Axon injury and the resultant neuronal cell death are key features of many neurodegenerative diseases. In a number of disease models, ranging from Alzheimer’s disease to glaucoma, dual leucine zipper kinase (DLK, MAP3K12) has been shown to be a key mediator of the injury response (1234567–8). Moreover, in both the peripheral nervous system (PNS) and central nervous system (CNS), the related molecule, leucine zipper kinase (LZK, MAP3K13), is emerging as an important comediator (3, 9, 10). Following neuronal injury, DLK activation and up-regulation is responsible for the retrograde transport of stress signals to the nucleus, resulting in changes in the activities of transcription factors like JUN and signal transducer and activator of transcription three (STAT3) (11, 12). In the PNS, the resulting gene expression changes promote axon regeneration and, in some cases, the restoration of function (13, 14). In contrast, in the less regeneration-permissive environment of the CNS, DLK-dependent alterations in gene expression often culminate in cell death. Although other axon injury pathways such as those dependent on importinß1 exist, the majority of axotomy-induced gene expression changes are dependent on DLK signaling (1, 11). Thus, inhibition of DLK (and LZK) has a durable and robust effect on survival, making DLK/LZK targeting an attractive neuroprotective strategy. However, inhibition of DLK drastically reduces CNS axon regeneration, even when regeneration is stimulated by knockdown of negative modulators such as phosphatase and tensin homolog (PTEN) (1). In chronic neurodegenerative diseases like glaucoma, there is not a substantial population of axotomized-but-not-yet-dead retinal ganglion cells (RGCs) at any given time that could be candidates for axon regeneration. Thus, the suppression of regeneration by DLK/LZK inhibition is largely irrelevant and should not hinder DLK/LZK inhibition as a neuroprotective strategy. In contrast, for more acute neurodegenerative diseases like traumatic optic neuropathy, there is a window of time in which there are many axotomized-but-not-yet-dead RGCs and, thus, a need to promote both RGC survival and axon regeneration. In this setting, DLK/LZK inhibition is unlikely to be a viable neuroprotective strategy, motivating the identification of novel targets that might promote survival while allowing for regeneration.
One strategy might be to inhibit a subset of the downstream transcription factors that are regulated by DLK/LZK, in an attempt to decouple the effects on regeneration and survival. However, this approach has not yet been successful. For example, targeted disruption of JUN can increase survival but, similar to DLK inhibition, impairs regeneration (15). Alternatively, one could move outside the DLK-JUN axis and target those regulators of axon regeneration that are highly survival-promoting. Interestingly, the most robust axon-promoting perturbations, such as knockout of Krüppel-like transcription factors (KLFs), PTEN, suppressor of cytokine signaling three (SOCS3), or signal transducer and activator of transcription 3 (STAT3), have only been shown to transiently promote survival (161718–19).
Taken together, these findings underscore the need to identify novel genes that can be targeted both to promote survival and to increase axon regeneration. Here, we describe the results of a chemical genetic screen in human stem cell-derived RGCs that identified the germinal cell kinase IV (GCK-IV) family as a suitable target. Interestingly, GCK-IV kinases have previously been implicated in the initiation of DLK activation (20). We demonstrate that GCK-IV kinase inhibition strongly promotes RGC survival in the optic nerve crush (ONC) model and synergizes, rather than retards, the axon regeneration triggered by knockdown of the regeneration repressor, PTEN. Finally, we demonstrate that inhibition of GCK-IV kinases can address a well-known problem with stem cell-derived retinal organoids—the attrition of RGCs over time—and promote the survival and neurite outgrowth of RGCs in mouse retinal organoids.
In order to model human CNS neurons, and specifically RGCs, we generated the SEC1 line of induced pluripotent stem cells (iPSCs), validated the presence of pluripotency markers (SI Appendix, Fig. S1 A and B), and used clustered regularly interspaced short palindromic repeat (CRISPR) genome editing to knock in a Thy1.2-P2A-tdTomato reporter at the BRN3B locus (21, 22). The cells were then differentiated toward a retinal fate (SI Appendix, Fig. S1C) in two-dimensional cultures and anti-Thy1.2 immunopurified to yield fluorescently labeled human stem cell-derived RGCs (hRGCs, SI Appendix, Fig. S1D). It has been previously shown that such an approach generates human RGCs with developmentally similar RGC gene expression patterns, functional characteristics of RGCs such as the ability to fire action potentials, and known RGC stress signaling mediators like DLK and LZK (21, 22). To identify kinases, beyond DLK and LZK, that are involved in axon injury and whose inhibition does not simultaneously impair axonal regeneration, we conducted two complementary small molecule-based high-throughput screens using the hRGCs. In the first screen, we searched for small molecules that were able to promote RGC survival following microtubule destabilization with colchicine, a pharmacologic intervention amenable to high-throughput screen assays, which has been shown to engage similar injury pathways as mechanical axon injury (4, 23, 24). In the second, we screened for small molecules that were able to promote, rather than retard, neurite outgrowth. For this screen, we chose to use the Glaxo-Smith-Kline Protein Kinase Inhibitor Set 1 (PKIS1) library, a collection of 366 PKIs that have each been profiled for their ability to inhibit a set of 224 protein kinase targets (25). Because different PKIs inhibit distinct but overlapping sets of kinases, this strategy allowed us to deconvolute out the key kinases that drive the patterns of compound activity in the various screens (26). Overall, this hRGC/PKI-based approach offered three advantages over our past small interfering RNA (siRNA)-based screens. First, small molecules often inhibit multiple structurally related kinases that could be functionally redundant and, therefore, missed by siRNA screens that target one gene at a time. Second, the degree of inhibition can be more complete with pharmacologic inhibition as opposed to siRNA-based knockdown. Finally, the use of stem cell-derived neurons allowed us to probe biology that is conserved in human cells.
For the survival screen, the colchicine challenge (30 nM) and arrayed library (3 µM) were added together on day 0. Survival was assayed, in duplicate, 72 h later using an ATP-based luminescent assay that has previously been shown to correlate well with conventional measures of viability (2, 9). For the neurite outgrowth screen, cells were plated in the presence of library compounds (3 µM) and imaged daily for 72 h using the fluorescence from the genetically encoded tdTomato reporter. We defined the results into three groups: those compounds that increased survival or neurite outgrowth more than three SD from controls were considered active, compounds that decreased survival or neurite outgrowth more than three SD from controls were considered opposing, and compounds in between were considered inactive (Fig. 1 A and B). The kinase inhibition spectra for both active and opposing compounds were then analyzed using the Identification of Drug TaRgets and Anti-targets by Cellular and Molecular Cross-referencing (idTRAX) approach (27, 28) to identify kinase groups whose inhibition promotes survival and/or neurite outgrowth. The top kinases whose inhibition predicts survival-promoting activity were tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE) 1 and 2, glycogen synthase kinase 3α/β (GSK-3α/β), cyclin-dependent kinases (CDKs), and casein kinase 1 while the kinases whose inhibition predicts neurite outgrowth included rho-dependent kinases (ROCKs) and ribosomal S6 kinases (RSKs; SI Appendix, Table S1). Validating the approach, ROCK inhibition has previously been shown to increase RGC axon regeneration while CDK inhibition has been shown to improve survival (29, 30). Interestingly, a set of three closely related kinases, misshapen like kinase 1 (MINK1), mitogen activated protein kinase kinase kinase kinase (MAP4K4) and Traf2 and Nck-interacting kinase (TNIK, also known as MAP4K7), collectively called the GCK-IV subfamily of kinases, were among the top of both lists (Fig. 1C). Consistent with this finding, examining compounds library-wide (based on known profiling) showed that both neurite-promoting and survival-promoting compounds had greater GCK-IV kinase inhibition compared to nonactive compounds (Fig. 1D) and that GCK-IV kinase-inhibiting compounds (Fig. 1E) had both greater survival- and neurite-promoting activity compared to compounds without GCK-IV kinase activity (Fig. 1F). Furthermore, we validated that cell survival was not confounding our neurite screens as the survival fold change among the most active compounds was equal to that of our opposing compounds (SI Appendix, Fig. S2). Finally, we tested mouse RGCs (mRGCs) to see if the phenotype extended across species and found that the top hRGC survival-promoting compounds (which all inhibited GCK-IV kinases) were active in mRGCs (Fig. 1G), while there was little-to-no activity for opposing compounds from the screen (Fig. 1H). Taken together, these results suggested that inhibition of GCK-IV kinases might be able to promote both CNS neuronal survival and neurite outgrowth, which was surprising given prior work which would suggest that GCK-IV kinases should closely phenocopy DLK and prevent neurite outgrowth.

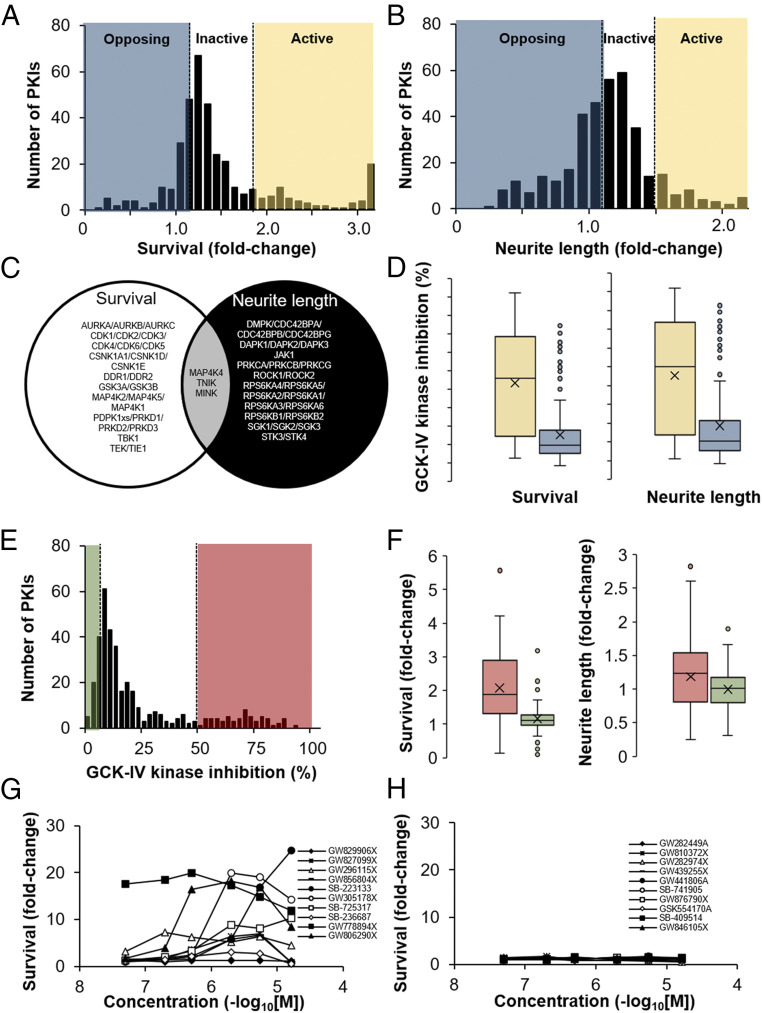
High-throughput screening of the PKIS library identifies the GCK-IV kinase family as mediators of RGC death and neurite outgrowth. (A) Histogram of the normalized hRGC survival generated by each of the PKIs (3 µM) when challenged with 30 nM colchicine. PKIs which increased survival more than 3 SDs were considered bioactive (yellow), while PKIs which generated less than 3 SDs of increased survival were considered as opposing activity (blue). (B) Histogram of the normalized neurite outgrowth of hRGCs generated by each of the PKIs (3 µM). PKIs which increased outgrowth more than 3 SDs were considered bioactive (yellow), while PKIs that generated less than 3 SDs of increased outgrowth were considered as opposing activity (blue). (C) Venn diagram of the top genes from the survival screen or neurite outgrowth screens identified by an idTRAX machine learning algorithm. (D) Box plot of GCK-IV kinase inhibition by neurite-promoting and survival-promoting compounds (yellow) compared to opposing activity compounds (blue). (E) Histogram of PKIs’ ability to inhibit GCK-IV kinases. PKIs which inhibit GCK-IV kinases more than 50% were considered active (red), while those compounds with less than 10% inhibition were considered inactive (green). (F) Box plot of the survival-promoting and neurite-promoting activity of the PKIs comparing compounds that had greater GCK-IV kinase inhibition (red) with those with less GCK-IV kinase activity (green). (G and H) Survival of mRGCs at 72 h, measured as the fold increase in CellTiter-Glo relative luciferase units (RLU) treated with the top active (G) or inactive (H) compounds from the PKIS library.
The PKIS library is composed of relatively nonselective compounds, a feature that is leveraged by the idTRAX approach to predict relevant kinase targets. Moreover, the library has not been fully characterized against all known kinases. For these two reasons, we considered the possibility that GCK-IV kinases were not the relevant target but were being nominated by the algorithm because they were pharmacologically linked to another unprofiled kinase (i.e., the same compounds that inhibit GCK-IV kinases might be inhibiting these other structurally similar kinases). To address this, we tested two unrelated GCK-IV kinase inhibitors, GNE-495 and PF-06260933, that appear highly selective when profiled against the kinome (31, 32). Using doses commensurate with the MAP4K4 IC50, we confirmed that that we could improve the survival and neurite outgrowth of both mouse and human RGCs in vitro (Fig. 2 A–E). As yet another validation, we used CRISPR technology to mutagenize the GCK-IV kinases in primary mouse RGCs. This approach takes advantage of the ability to transfect T7-transcribed guide RNAs (gRNAs) into primary mRGCs that are isolated from mice that express Streptococcus pyogenes Cas9 from the Rosa26 locus (33), using C57BL/6J WT mice as controls. In Cas9-expressing RGCs, but not WT controls, combined transfection of gRNAs targeting MINK1, TNIK, and MAP4K4 improved survival with no significant effect on survival when transfected individually (Fig. 2F). These results suggest redundancy among the three GCK-IV kinases. Finally, while GCK-IV kinase inhibition protected against spontaneous death (mRGCs) and colchicine-induced death (hRGCs), we wanted to test if other injury mechanisms were GCK-IV kinase-dependent. We challenged hRGCs with a variety of injury agents including vincristine (microtubule destabilizer), paclitaxel (microtubule stabilizer), rotenone (oxidative stress), cisplatin (DNA damaging agent), and carbonyl cyanide m-chlorophenyl hydrazine (CCCP, mitochondrial toxin) and found at least some degree of protection in all cases (SI Appendix, Fig. S3). These results thus confirm a broad neuroprotective role for GCK-IV kinase inhibition across species and injury mechanism.
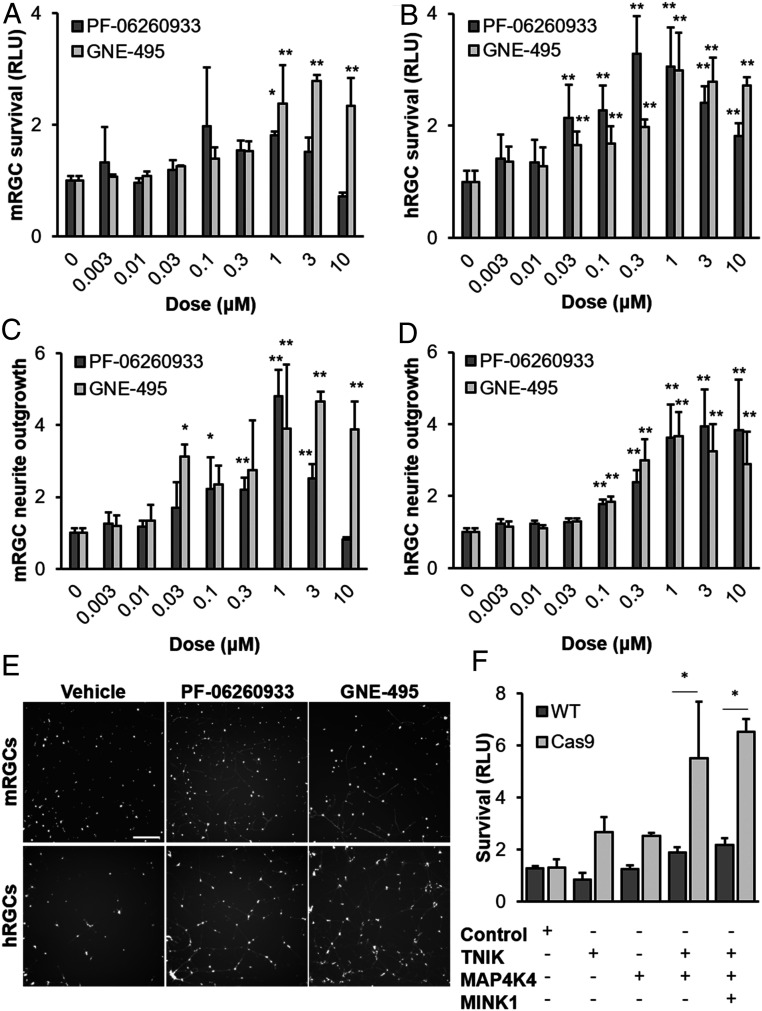
Inhibition of GCK-IV kinases promotes mouse and human RGC survival and neurite outgrowth. (A) Survival of primary mRGCs treated with either increasing doses of PF-06260933 or GNE-495, at 72 h (*P < 0.05, **P < 0.01, n = 4, ANOVA with Bonferroni correction and Dunnett’s test post hoc analysis, error bars: SD). (B) Survival of primary hRGCs treated with either increasing doses of PF-06260933 or GNE-495, 72 h after colchicine addition (*P < 0.05, **P < 0.01, n = 4, ANOVA with Bonferroni correction and Dunnett’s test post hoc analysis, error bars: SD). (C) Fold change in calcein-stained neurite length of mRGCs treated with increasing doses of PF-06260933 or GNE-495, at 72 h (*P < 0.05, **P < 0.01, n = 4, ANOVA with Bonferroni correction and Dunnett’s test post hoc analysis, error bars: SD). (D) Fold change in tdTomato-positive neurite length of hRGCs treated with increasing doses of PF-06260933 or GNE-495, at 72 h (*P < 0.05, **P < 0.01, n = 4, ANOVA with Bonferroni correction and Dunnett’s test post hoc analysis, error bars: SD). (E) Representative images of mRGCs and hRGCs in either vehicle, PF-06260933 (1 μM) or GNE-495 (1 μM), showing neurite outgrowth at 72 h. (Scale bar: 200 µm) (F) Survival of Cas9-expressing or WT mRGCs transfected with gRNA targeting the GCK-IV kinases, at 72 h. (*P < 0.05, n = 4, ANOVA with Bonferroni correction and Dunnett’s test post hoc analysis).
Based on the proregenerative and prosurvival effect seen in vitro with GCK-IV kinase inhibition, we next sought to validate the biology in vivo. Since generating a triple knockout of the GCK-IV kinase family would require extensive breeding, we developed an adeno-associated virus (AAV)/CRISPR-based method for rapid multigene targeting (SI Appendix, Fig. S4A). This approach relied on the natural ability of the compact mouse H1 promoter (170 bp) to drive transcription of a Pol II transcript (Parp2) in one direction and a Pol III transcript (Rpph1) in the other direction (34). Using this bidirectional promoter, we could simultaneously deliver gRNA (Pol III transcription) and nuclear-localized mCherry (Pol II transcription) in a rapidly expressing, self-complementary AAV2 vector, with Y444F/Y500F/Y730F triple-capsid mutations to improve RGC transduction (35). To test the efficacy of the system, we isolated primary RGCs from mice that constitutively express Cas9-P2A-EGFP and transduced them with increasing amounts of an AAV2 targeting Dlk. Using this approach, the scAAV2 vector was able to transduce nearly all RGCs (SI Appendix, Fig. S4B) and mutagenize nearly all of the Dlk loci in the population as evidenced by the loss of a BglII restriction site that is present within the target region (SI Appendix, Fig. S4C). To test the system in vivo, we intravitreally injected the scAAV2-DLK gRNA vector into Cas9-expressing or control mice and imaged the survival of RGCs 14 d after an ONC injury. Similar to prior results with a conventional knockout of DLK (1, 2, 36), the AAV/CRISPR-based disruption of DLK increased survival in Cas9-expressing mice compared to WT controls (SI Appendix, Fig. S4 D and E).
Having developed the AAV/CRISPR system, we next selected gRNAs that are specific for each of the GCK-IV kinases (37, 38), packaged triple capsid-mutant scAAV2, and demonstrated highly efficacious mutagenesis in primary mRGCs (Fig. 3A). We next turned to the ONC model of axon injury to further explore the role of the GCK-IV kinase family in the neuronal injury response in vivo, including cell death, axon regeneration, and distal axon (Wallerian) degeneration. A mixture of AAV targeting Map4k4, Mink1, and Tnik was intravitreally injected into Cas9-expressing (39) or control mice. After 2 wk, providing sufficient time for AAV life cycle completion, CRISPR knockout, and extant protein turnover, mice were subjected to an ONC injury or sham control. Then, after another 2 wk, the number of surviving RGCs was quantified using an RGC-specific marker, RNA-binding protein with multiple splicing (RBPMS). Consistent with the in vitro results, triple knockout retinas showed robust RGC protection (68 ± 2.73% surviving RGCs vs. 27 ± 1.25% for control retinas, P < 0.01), comparable to DLK knockout retinas (73 ± 6.15% surviving RGCs vs. 19 ± 1.34% for control retinas, P < 0.01; GCK-IV KO vs. DLK KO, P = 0.48; Fig. 3 B and C), although both GCK-IV KO and DLK KO remained significantly different from intact uninjured retinas (P < 0.05). Given that it is unlikely that we disrupt all six GCK-IV kinase alleles in all cells with our knockout strategy, it remains to be seen whether the incomplete protection implies the existence of other redundant death mediators. Importantly, when we extended the assay and measured survival at 10 wk after ONC, similar levels of protection were observed (SI Appendix, Fig. S5 A and B), suggesting that loss of GCK-IV kinases prevents rather than delays cell death.
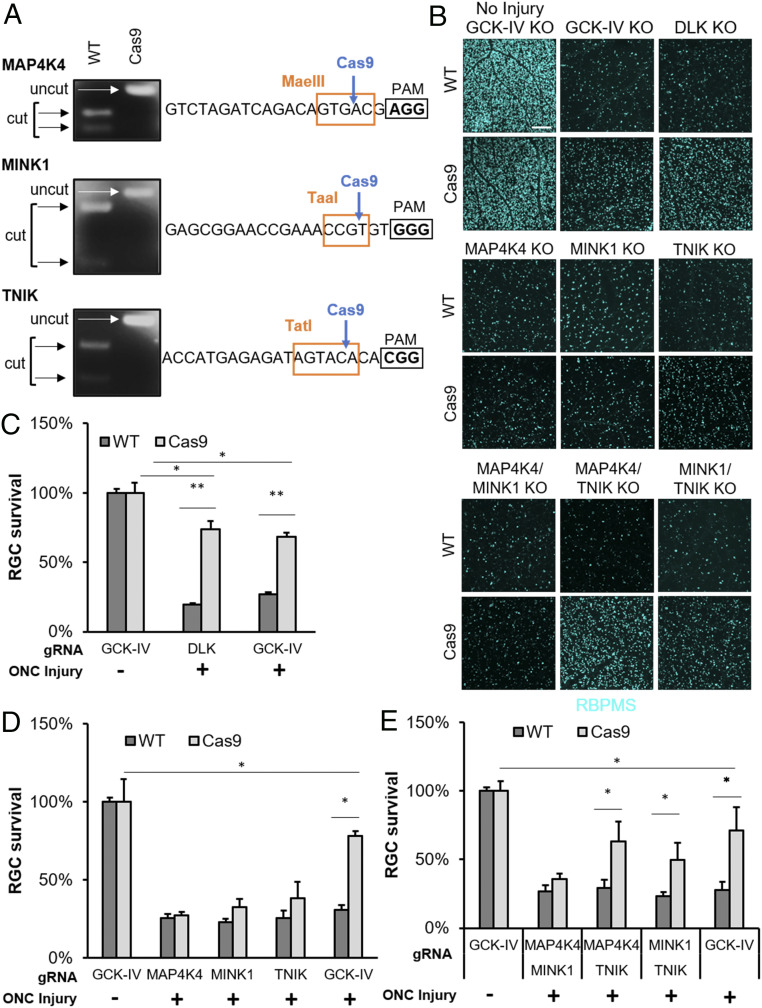
Targeted disruption of GCK-IV kinases is protective to RGCs in the mouse optic nerve injury model. (A) Restriction enzyme digestion of PCR products covering the gRNA target site from WT or Cas9-expressing mRGCs 9 d after transduction with scAAV2-expressing gRNAs against the GCK-IV kinases. (B) Representative immunofluorescence images of RBPMS-labeled retinal flatmounts 14 d following optic nerve crush of either WT or Cas9-expressing mice injected with AAV-delivering gRNA. (Scale bar: 200 µm.) (C–E) Quantification of RBPMS-positive cells by automated image analysis. (n = 4 per group, *P < 0.05, **P < 0.01, ANOVA with Bonferroni correction, error bars: SEM).
Given the modular design of the knockout strategy, we next asked if one or more of the GCK-IV kinases was primarily responsible for triple knockout phenotype. Control versus Cas9-expressing mice were then injected with each virus individually (Fig. 3D) or with pairwise combinations (Fig. 3E). While no knockout by itself showed a statistically significant increase in RGC survival, there was a trend for increased survival in the TNIK knockout retinas (38% survival vs. 25% in controls, P = 0.15). Moreover, pairwise knockouts of TNIK and either MAP4K4 or MINK1 did show a statistically significant increase in survival (63 ± 7.15% and 49 ± 6.24% survival, respectively, TNIK/MAP4K4, P < 0.05) with the TNIK/MAP4K4 double knockout promoting survival nearly as much as the triple knockout (63 ± 7.15% vs. 71 ± 8.75%, P < 0.05). Together, these results suggest that TNIK has the most prominent role in cell death, with a smaller and redundant contributions from MAP4K4 and MINK1.
Targeted disruption of GCK-IV kinases increases RGC survival, comparable to cells with a DLK knockout. Because initial results suggested that GCK-IV kinase inhibition also promotes axon growth, we next sought to compare the effect of GCK-IV kinase and DLK disruption on axon regeneration. Cas9-expressing or WT mice were injected with an AAV mixture expressing gRNAs targeting DLK or the three GCK-IV kinases and, after 2 wk, nerves were subjected to an ONC injury. After an additional 3 wk, cholera toxin subunit B (CTB) was intravitreally injected into the ipsilateral eye in order to trace axon regeneration. As expected, control optic nerves had sparse axons extending beyond the crush site while DLK knockouts had no detectable axons in the distal nerve segment (Fig. 4 A and B). Unlike the proneurite outgrowth effect seen in vitro, loss of the GCK-IV kinases did not significantly increase the amount of regeneration (Fig. 4C). As it has been previously demonstrated that PTEN loss can increase RGC axon regeneration in a DLK-dependent manner (1), we next tested the effect of the two knockouts in the setting of PTEN knockdown. AAV expressing a short hairpin RNA (shRNA) targeting PTEN effectively knocked down PTEN levels in primary RGC in a manner that was unaffected by GCK-IV kinase inhibition (SI Appendix, Fig. S6A). As expected, in the ONC model, PTEN shRNA-expressing AAV modestly increased axon regeneration in WT but not DLK knockout eyes (Fig. 4 D and E). Interestingly, while loss of the GCK-IV kinases had little effect on baseline regeneration, there was a synergistic increase in regeneration in the setting of low PTEN levels (Fig. 4F). Both in vitro and in vivo, PTEN knockdown had no effect on the amount of survival produced by GCK-IV kinase inhibition (SI Appendix, Fig. S6 B and C), highlighting the importance of PTEN knockdown’s proregenerative signal. Taken together, these results validate GCK-IV kinase inhibition as a strategy to improve neuronal survival after axon injury while retaining, and even enhancing, the ability to regenerate axons.
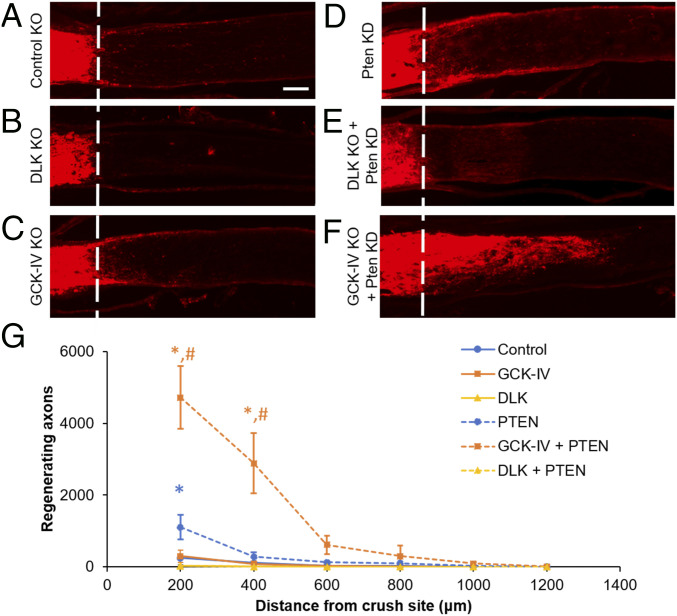
Targeted disruption of GCK-IV kinases synergizes to increase axon regeneration. (A–F) Representative longitudinal sections of optic nerves 3 wk following optic nerve crush injury (dashed line). Regenerating axons were labeled with intravitreal CTB 3 d prior to euthanasia for visualization. (G) Quantification of axon length in the optic nerve. Estimated axon number was quantified every 200 μm from the injury site (n = 4 per group, *P < 0.05 vs. control, #P < 0.05 vs. PTEN KD, ANOVA with Bonferroni correction, error bars: SEM). (Scale bar: 100 µm.)
In order to explore the mechanism by which GCK-IV kinases regulate survival and regeneration, we examined the state of DLK-JNK-JUN signaling in RGCs. GCK-IV kinases are MAP4Ks and, thus, reasonable candidates to phosphorylate and/or activate MAP3Ks like DLK and LZK. To test if GCK-IV kinases are upstream of DLK/LZK in mRGCs, we measured the effect of a GCK-IV kinase inhibitor on JUN phosphorylation, a known indicator of DLK pathway activation. Surprisingly, while a DLK inhibitor was able to totally suppress JUN phosphorylation in injured mRGCs, PF-06260933 had no effect, even at neuroprotective doses (Fig. 5A). However, PF-06260933 was able to left-shift the DLK inhibitor dose–response survival curve, suggesting that for a given amount of DLK activity, GCK-IV kinases increase the magnitude of the cell death response. This contrasts with prior findings in dorsal root ganglion cells in which GCK-IV kinases are involved in the activation of DLK (20). In human RGCs, we saw a similar left-shift of the dose–response curve, this time with a modest effect on JUN phosphorylation (Fig. 5B). In the ONC model, JUN is nearly fully activated in RGCs by 24 h. However, in the setting of DLK disruption, there is a marked attenuation of the both the intensity and percentage of JUN-positive cells (SI Appendix, Fig. S7). In contrast, GCK-IV kinase loss had no effect on either the intensity or percentage of JUN-positive cells (SI Appendix, Fig. S7). Two weeks after ONC, at which point selection should be enriching the virally transduced cells, we saw even more complete pathway suppression with the DLK knockout compared to the much more modest suppression of JUN intensity and nonsignificant trend in percentage of JUN-positive cells in the GCK-IV kinase knockout eyes (Fig. 5 C and D and SI Appendix, Fig. S8). As with the survival data, it appeared that TNIK was the primary driver of the effect as JUN suppression was only seen when TNIK knockout was combined with either MINK1 or MAP4K4 knockout and not when MAP4K4/MINK1 were dually-disrupted (Fig. 5 E and F). Taken together, these results suggest that the effect of GCK-IV kinase inhibition on RGC survival is not fully explained by its effect on DLK signaling although, in some settings, it does appear that GCK-IV kinases play a partial role in DLK activation.
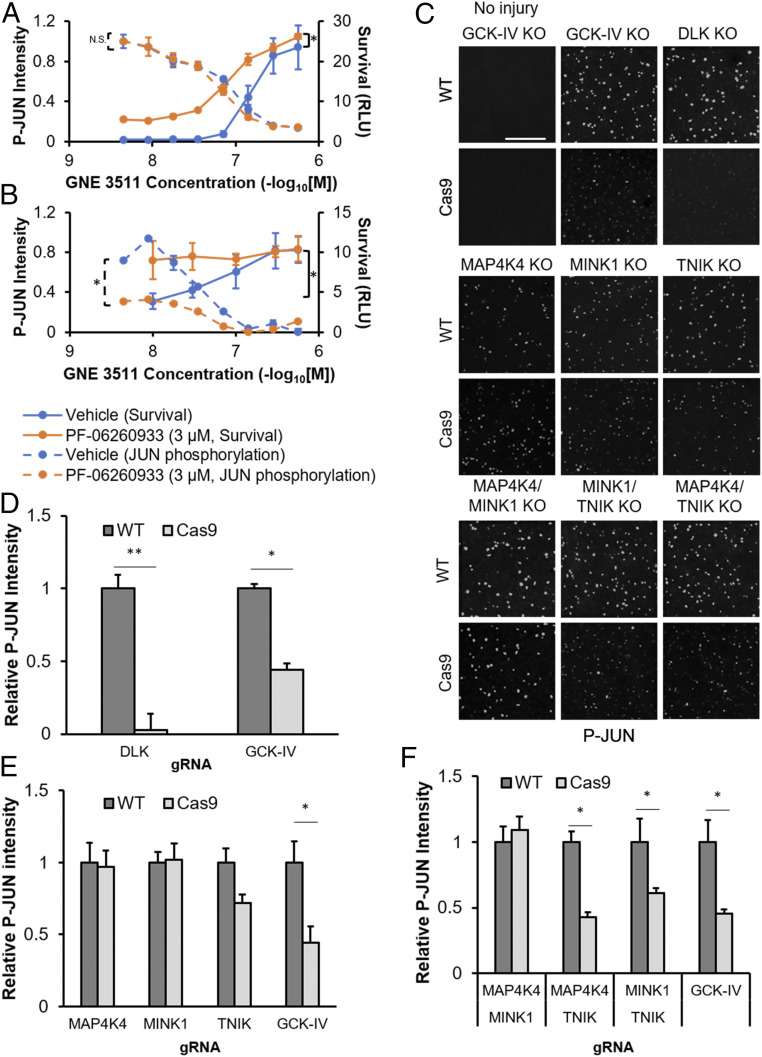
GCK-IV kinase inhibition affects RGC survival through DLK-dependent and DLK-independent mechanisms. (A) Survival and JUN phosphorylation of mRGCs, treated with increasing doses of the DLK inhibitor GNE-3511, in the presence of either vehicle or PF-06260933 (3 μM) 72 h after immunopanning (n = 3, *P < 0.05, paired t test). (B) Survival and JUN phosphorylation of hRGCs, treated with increasing doses of the DLK inhibitor GNE-3511, in the presence of either vehicle or PF-06260933 (3 μM) 24 h (JUN phosphorylation) or 72 h (survival) after 30 nM colchicine challenge (n = 3, *P < 0.05, paired t test). (C) Representative immunofluorescence images of phosphorylated JUN-labeled retinal flatmounts 2 wk after optic nerve crush in WT or Cas9-expressing mice transduced with AAV expressing gRNA against the GCK-IV kinases. (Scale bar: 200 µm.) (D–F) Quantification of phosphorylated JUN intensity by automated image analysis. (n = 4 per group, *P < 0.05, **P < 0.01, ANOVA with Mann–Whitney U test, error bars: SEM).
It has previously demonstrated that targeted disruption of DLK had little effect on distal axonal degeneration in the ONC model (36). To assess whether GCK-IV kinase inhibition was involved in the genetic program mediating axonal degeneration, we first examined axonal degeneration in hRGCs. Colchicine was added to hRGCs, after elaborating neurites, at doses from 0.3 to 3 µM, in the presence of the GCK-IV kinase inhibitor PF-06260933 or the vehicle control, and the length of fluorescently labeled axons was quantified at 72 h using an automated imaging/image analysis algorithm. At all doses of colchicine, GCK-IV kinase inhibition modestly decreased the amount of axon degeneration, suggesting a potential role for GCK-IV kinase signaling in the degeneration program (Fig. 6 A and B). To confirm these findings in vivo using a model of Wallerian degeneration, we turned back to the ONC model. Three-month-old Cas9-expressing or control mice were intravitreally injected with the mixture of AAV targeting the three GCK-IV kinases, combined with an AAV that expressed pancellular EGFP in order to trace axons. By using limiting doses of the GFP virus, individual axons could be imaged in the optic nerve. Nerves were then subjected to a crush injury, and the amount of distal axon degeneration 3 d later, evidenced by fragmentation and/or swellings, was semiquantitatively evaluated by masked observers. Consistent with the findings in vitro, targeted disruption of the GCK-IV kinases led to a modest delay in axon degeneration 3 d following nerve crush (degeneration score of 3.15 ± 0.31 vs. 2.1 ± 0.32, P < 0.05; Fig. 6 C and D), but was not maintained 6 d postinjury (SI Appendix, Fig. S9). Thus, whereas GCK-IV kinase inhibition leads to robust, sustained somal protection, the effect on axon degeneration is more modest and temporary.
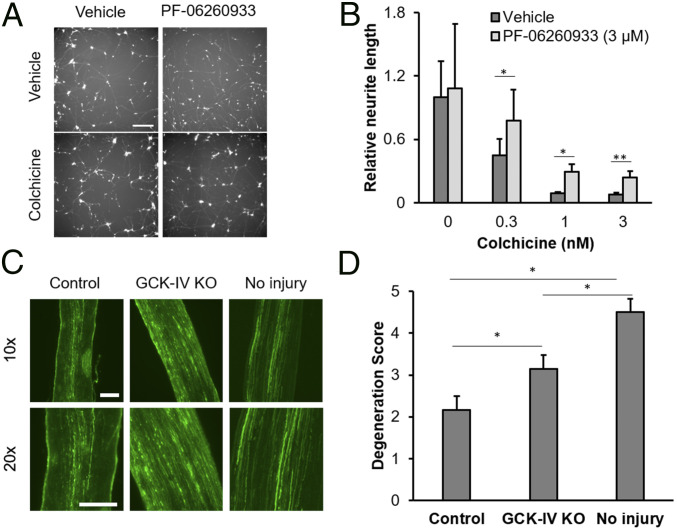
Inhibition of GCK-IV kinases attenuates axon degeneration in RGCs. (A) Representative images of hRGCs treated with either vehicle or PF-06260933 (3 µM), 72 h after colchicine (0.3 nM) challenge. (B) Quantification of intact neurite length in hRGCs, treated with vehicle or PF-06260933 (3 µM), 72 h following colchicine challenge (n = 3, *P < 0.05, **P < 0.01, ANOVA and Mann–Whitney U test, error bars: SD). (C) Representative longitudinal sections of optic nerves of from either WT or Cas9-expressing mice, transduced with AAV-expressing gRNA against GCK-IV kinases, 3 d after optic nerve crush. Eyes were coinjected with an AAV-expressing pancellular EGFP in order to label transduced axons. (Scale bars: 100 µm.) (D) Semiquantitative scoring (0 = totally degenerated, 5 = normal) of axon integrity in optic nerves from either WT or Cas9-expressing mice, 3 d after ONC or the sham control (n = 9 per group, *P < 0.05, Student’s t test, error bars: SD).
Multiple groups have demonstrated that RGCs have limited survival in both mouse and human retinal organoids. For example, by day 25 in mouse retinal organoids, most RGCs are either dead or in the process of dying (404142–43). Having demonstrated the ability of GCK-IV kinase knockouts to promote RGC survival without disrupting axon growth, we asked whether a GCK-IV kinase inhibitor could improve RGC survival and morphology in retinal organoids. Mouse embryonic stem cells were differentiated as described before (44, 45) and, on day 13, either PF-06260933 or vehicle control was added to the media. After 30 d, organoids were collected, sectioned, and stained for RBPMS and β-III-tubulin, two markers of RGCs. As expected, vehicle-treated organoids had few remaining RGCs (Fig. 7A). In contrast, organoids treated with the GCK-IV kinase inhibitor had significantly more surviving RGCs, including those with clearly visible neurites (Fig. 7B). Quantification of RGCs showed a 3.7-fold increase (P < 0.01), as well as a statistically significant increase in neurite density, in organoids developed in PF-06260933 versus vehicle (Fig. 7C). Given the three-dimensional (3D) nature of the organoids, it was not possible to quantify the number per cell nor length of the individual neurites. The fluorescence intensity of the RGC markers was 3.5-fold higher in PF-06260933–treated cells, suggesting that GCK-IV kinase inhibition may help maintain the overall health of target cells in addition to preventing their death. These results also indicate that in addition to coupling regeneration with survival in adult neurons, GCK-IV kinase inhibition is also neuroprotective to developmentally immature neurons.
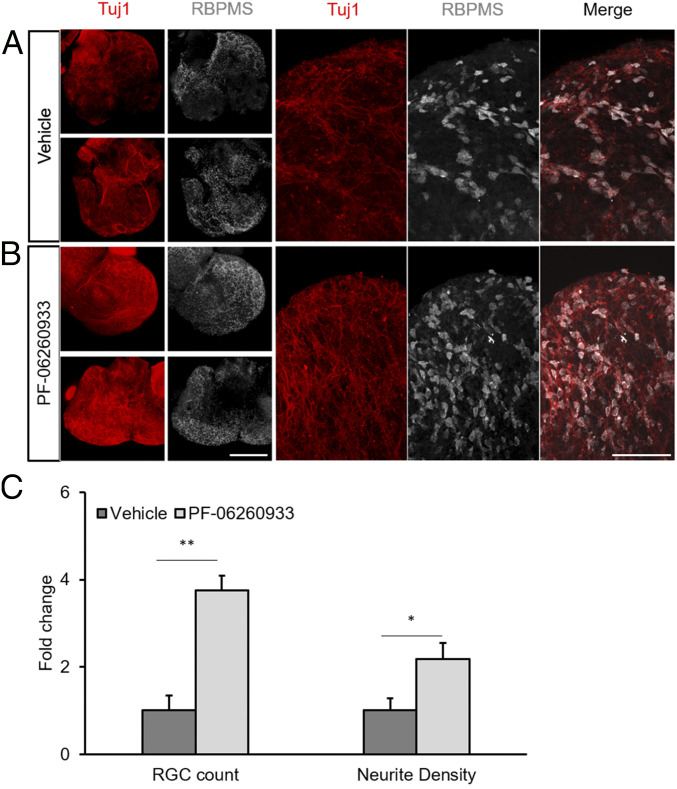
GCK-IV kinase inhibition increases RGC survival and promotes neurite outgrowth in mouse retinal organoids. miPSCs were differentiated into retinal organoids for 30 d in either vehicle (DMSO) or PF-06260933 (200 µM). Retinal organoids were fixed and immunolabeled for RBPMS to identify RGCs and Tuj1 to label neurites. (A and B) Representative sections through organoids showing increased RGC survival and neurite elaboration. (Scale bar: 100 μm.) (C) Quantification of RBPMS-positive cells for RGC counts and intensity using automated image analysis (n = 6–7 per group, *P < 0.05, **P < 0.01, Student’s t test, error bars: SD).
DLK and downstream pathway members like JUN have been shown to be key mediators of neuronal cell death and, while targeted disruption of DLK leads to robust increases in CNS neuron survival following axon injury, it invariably decreases, and in some cases abolishes, axon regeneration (123–4, 9, 11, 46). For some neuroprotective applications, especially chronic neurodegenerative diseases, this incompatibility with regeneration may not limit the use of DLK inhibitors if they can prevent future loss of connectivity. However, for other situations in which connectivity has already been lost and axonal regeneration is required, the effect of DLK inhibition on regeneration presents a serious limitation. This is unfortunate given that DLK inhibition leads to robust, sustained survival and blocks the majority of injury-associated gene expression changes, thus making it a particularly attractive neuroprotective intervention (1, 2). In this study, we present data demonstrating that GCK-IV kinase inhibition can partially suppress DLK pathway activation, leading to similar levels of sustained, robust survival. Additionally, in contrast to DLK inhibition, GCK-IV kinase inhibition enhances rather than inhibits axon regeneration.
GCK-IV kinases like MAP4K4 have been implicated in neuronal cell death signaling in models of amyotrophic lateral sclerosis, and recently it was shown that GCK-IV kinases are involved in the initial activation of DLK (20, 47, 48). Thus, the identification of GCK-IV kinases as targets with functions distinct from DLK expands our thinking about the roles of these kinases in neurons. One possibility is that GCK-IV kinases are among one of several upstream activators of DLK and that GCK-IV kinase inhibition leads to a state of partial DLK activation, enough to permit regeneration but insufficient to trigger cell death. Consistent with this finding, we saw evidence of partial DLK inhibition in vivo (after 14 d) with intermediate levels of phosphorylated JUN in GCK-IV kinase knockout RGCs compared to controls. However, it is unlikely that this entirely explains the difference. Mice that have partial DLK pathway inhibition via a heterozygous null allele of Dlk also have similar intermediate levels of JUN phosphorylation following axonal injury but show only modest and temporary improvements in RGC survival after ONC (46). Moreover, it is unknown if heterozygous loss of DLK impairs axonal regeneration. Thus, we favor a model in which GCK-IV kinases have a second activity, independent of DLK, which modulates the response to a cell death signal. Consistent with this rheostat model, we demonstrate that for a given amount of DLK activity (as measured by JUN phosphorylation) in primary mouse RGCs in vitro or 24 h after ONC in vivo, a change in GCK-IV kinase signaling leads to differences in neuronal cell death. The specific mechanism by which GCK-IV kinase inhibition allows cells with active DLK signaling to survive remains to be determined. This role in cell death also fits with the growing literature suggesting that MAP4K4 inhibition is protective in systems like myocardium, where DLK has not been detected (4950–51). Indeed, the fact that GCK-IV kinases repress neurite outgrowth, as opposed to the role of DLK in promoting outgrowth, further suggests pleiotropic effects beyond a simple upstream initiator of DLK.
Prior studies looking at neuroprotection and cardioprotection with GCK-IV kinase inhibition have focused on the role of MAP4K4 (20, 4748–49). Using our CRISPR/AAV system and combinatorial knockouts, we found that TNIK was the primary GCK-IV kinase responsible for the phenotype, with lesser contributions from MAP4K4 and, possibly, MINK1. RNA transcriptome analysis of rodent RGCs showed that messenger RNAs for all three GCK-IV kinases are abundantly expressed (52). While the three GCK-IV kinases have closely related kinase domains, they may have different mechanisms of action, including interacting with different partners, owing to differences in other protein domains. MINK1 and TNIK were shown to differentially act on the regulation of AMPA receptors and postsynaptic neuronal processing with MINK1 signaling through a Rap2-mediated pathway and TNIK instead acting in a Wnt/GSK3β signaling-dependent manner (53, 54). Furthermore, there is evidence of TNIK playing a key role in TNFα-dependent NFκB and JNK signaling (55), which have been linked to RGC death and glaucoma (56, 57).
The mechanism by which GCK-IV kinase inhibition improves axonal regeneration is unclear. Past work on MAP4K4 knockout cells has suggested that MAP4K4 may be involved in cytoskeletal dynamics and focal adhesion turnover (58), both known to have important roles in axon outgrowth (59). Moreover, an siRNA-based screen in SH-SY5Y cells identified MAP4K4 knockout as promoting neurite outgrowth in vitro (60). Interestingly, we saw increases in outgrowth in RGCs in vitro, but GCK-IV kinase inhibition did not, by itself, promote significant axon regeneration in the ONC assay. Rather, it synergized with loss of PTEN, a known regulator of axon regeneration, to increase regeneration in this assay. We speculate that at least one mechanism by which GCK-IV kinase inhibition leads to increased regeneration is the increase in the number of long-term surviving, PTEN shRNA-expressing cells after axotomy, although the degree to which this contributes to this effect is not yet clear. In contrast to DLK inhibition, the permissive nature of GCK-IV kinase inhibition allows these surviving cells to regrow axons, capturing most of the neuroprotective benefit without compromising regeneration. An alternate, or possibly complementary, explanation may be that in our study, although we have significant PTEN knockdown, we are not altering the AKT pathway to the same degree as knockouts shown in other studies. This may explain why we see more subdued axon regeneration in the context of PTEN knockdown alone, whereas the addition of a GCK-IV kinase knockout shifts the AKT pathway to the levels required to induce robust regeneration. A number of promising proregenerative perturbations, beyond PTEN knockdown/knockout, have been demonstrated to promote RGC axon regeneration in vivo, albeit with modest effects on survival (61, 62). The modular nature of the AAV/CRISPR system should allow for different permutations of these to be evaluated in the context of the prosurvival benefit of GCK-IV kinase inhibition. Furthermore, the GCK-IV kinases are druggable targets, which allow for new combinatorial therapeutic possibilities with other proregenerative strategies to maximize survival and the potential for regrowth of axons leading to restored function.
All studies with de-identified patient samples were carried out in accordance with the declaration of Helsinki and with the approval of the institutional review board (IRB) of the University of California San Diego with IRB and ESCRO approval (#S07370 and #160431XF). Blood was collected from a patient and reprogrammed in iPSCs. iPSC cells derived from PBMC reprogramming were genetically edited and differentiated into hRGCs as previously described in Sluch et al. (22) with slight modifications. Please see SI Appendix, Supplemental Methods for more details.
Mouse induced-pluripotent stem cells (miPSCs) were derived from day 13.5 embyros and differentiated into retinal organoids as previously described by La Torre et al (45). Please see SI Appendix, Supplemental Methods for more details.
The PKIS1 library was obtained from Glaxo-Smith Kline. This library contains 366 protein kinase inhibitors. Each inhibitor was profiled in its ability to inhibit a set of 224 kinase targets (26).
hRGCs were dissociated and plated into 384 plates containing a library of compounds. Plates were assayed at 72 h for survival using Cell Titer Glo and a luminescent plate reader or neurite outgrowth by quantifying endogenous fluorescence. Please see SI Appendix, Supplemental Methods for more details.
Target deconvolution analysis was performed as previously described by Al-Ali et al. using the idTRAX machine learning-based approach (27). Please see SI Appendix, Supplemental Methods for more details.
All procedures involving mice were carried out following the rules of the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee at the University of California San Diego. All experiments were conducted in Streptococcus pyogenes Cas9 expressing mice (Jax Labs Stock no. 027650) (39) and C57/BL6J control mice (Jax Labs Stock no. 000664). All procedures were conducted on mice postnatal day (P)0–2 or 10–14 wk old.
Primary mouse RGCs were isolated from P0 to P2 mouse pup retinas using the protocol described by Welsbie et al (9). Please see SI Appendix, Supplemental Methods for more details.
In vitro RGC viability was assayed using Cell Titer Glo. Please see SI Appendix, Supplemental Methods for more details.
The Broad Institute GPP sgRNA Designer tool was used to identify candidate sgRNA sequences that maximized on to off target Streptococcus pyogenes Cas9-based DNA mutagenesis (37, 38). The candidate protospacer sequences were subcloned into a self-complementary AAV construct expressing nuclear GFP and sgRNA, both driven by the bidirectional H1 promoter. AAV plasmids were sent to the University of Florida Ocular Gene Therapy Core for packaging into AAV serotype 2 with Y444F/Y500F/Y730F triple-capsid mutations. Virus was diluted to a titer of 1 × 1012 viral particles/mL before use.
Intravitreal injections were performed as previously described by Welsbie et al (9). Please see SI Appendix, Supplemental Methods for details.
Optic nerve injury was performed on mice 2–4 wk following intravitreal injection of AAV as previously described by Welsbie et al (9). Please see SI Appendix, Supplemental Methods for more details.
Mouse primary RGCs were live stained with Calcein, AM dye and imaged with an Image Xpress imager (Molecular Devices). Human IPSC-derived RGCs and mouse primary RGCs were fixed with PFA and labeled with anti P-C-JUN imaged using the Image Xpress imager. Mouse eyes were enucleated and fixed in PFA. Retinas were carefully extracted, labeled with anti-RBPMS and anti-P-C-JUN. Cell number and signal intensity were quantified by MetaXpress image analysis software (Molecular Devices). Please see SI Appendix, Supplemental Methods for more details.
Mouse eyes were injected with AAVs expressing gRNA and an AAV-expressing pancellular GFP. Mice were perfused with PFA 3 d following optic nerve injury. Optic nerves were carefully extracted and cryosectioned longitudinally and then imaged using a fluorescent microscope. Images of distal axons in the optic nerve were then assessed by a panel of investigators for the level of degeneration scored from 0 to 5, with 0 being most degenerated. Please see SI Appendix, Supplemental Methods for more details.
Axons were labeled with cholera toxin subunit B (CTB) by intravitreal injection 2 d prior to animal euthanasia. Animals were perfused using PFA, and eyes and optic nerves were carefully dissected and cryosectioned longitudinally. Axon growth was quantified by counting the number of CTB-labeled axons that extended past every 200-µm division from the end of the injury site. Please see SI Appendix, Supplemental Methods for more details.
Statistical analysis was conducted using ANOVA with Bonferroni correction, Mann–Whitney U test, Dunnetts test, and Student’s t test, using GraphPad Prism and Microsoft Excel. P values less than 0.05 were considered statistically significant.
We acknowledge funding from the NIH (including R01EY029342 and core grant P30EY022589), Research to Prevent Blindness, E. Matilda Ziegler Foundation, Brightfocus Foundation, Fight for Sight Foundation, the Richard C. Atkinson Laboratory for Regenerative Medicine, and the Glaucoma Research Foundation Catalyst for the Cure. We also thank the Tushinsky family for their generous funding.
All study data are included in the article and supporting information.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
52
53
54
55
56
57
58
59
60
 Inhibition of GCK-IV kinases dissociates cell death and axon regeneration in CNS neurons
Inhibition of GCK-IV kinases dissociates cell death and axon regeneration in CNS neurons
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
