
These authors shared co-first authorship.
Increasing lines of evidence identified that dexmedetomidine (DEX) exerted protective effects against sepsis-stimulated acute lung injury via anti-inflammation, anti-oxidation and anti-apoptosis. However, the mechanisms remain unclear. Herein, we investigated whether DEX afforded lung protection by regulating the process of mitochondrial dynamics through the HIF-1a/HO-1 pathway in vivo and in vitro. Using C57BL/6J mice exposed to lipopolysaccharide, it was initially observed that preemptive administration of DEX (50μg/kg) alleviated lung pathologic injury, reduced oxidative stress indices (OSI), improved mitochondrial dysfunction, upregulated the expression of HIF-1α and HO-1, accompanied by shifting the dynamic course of mitochondria into fusion. Moreover, HO-1-knockout mice or HO-1 siRNA transfected NR8383 cells were pretreated with HIF-1α stabilizer DMOG and DEX to validate the effect of HIF-1a/HO-1 pathway on DEX-mediated mitochondrial dynamics in a model of endotoxin-induced lung injury. We found that pretreatment with DEX and DMOG distinctly relieved lung injury, decreased the levels of mitochondrial ROS and mtDNA, reduced OSI, increased nuclear accumulation of HIF-1a and HO-1 protein in wild type mice but not HO-1 KO mice. Similar observations were recapitulated in NC siRNA transfected NR8383 cells after LPS stimulation but not HO-1 siRNA transfected cells. Concertedly, DEX reversed the impaired mitochondrial morphology in LPS stimulated-wild type mice or NC siRNA transfected NR8383 cells, upregulated the expression of mitochondrial fusion protein, while downregulated the expression of fission protein in HIF-1a/HO-1 dependent pathway. Altogether, our data both in vivo and in vitro certified that DEX treatment ameliorated endotoxin-induced acute lung injury by preserving the dynamic equilibrium of mitochondrial fusion/fission through the regulation of HIF-1a/HO-1 signaling pathway.
Sepsis is a complex life-threatening clinical disorder that arises from multiple organ dysfunctions due to a dysregulated host response towards an infection, for which efficient targeted therapies are lacking [1]. The lung is the most vulnerable organ during sepsis with mortality rates ranging from 34.9% to 46.1% [2]. Aberrant oxidative stress and mitochondrial dysfunction are emerging as key process in the pathophysiology of septic lung injury [3]. During sepsis, elevated reactive oxygen species (ROS) mainly arise from mitochondria can result in loss of mitochondrial DNA integrity, ATP deprivation, cell apoptosis and trigger a cytosolic oxidative burst [4]. Intriguingly, as the balance of mitochondrial dynamics is a prerequisite for functional integrity [5], thus, strategies that preserving mitochondrial dynamic equilibrium in septic settings are critical to reserve cell homeostasis and might lead to a better prognosis.
Dexmedetomidine (DEX), as a highly selective and potent α2 adrenergic agonist, is commonly used in the intensive care unit and operating room for its sedation and analgesia. Clinically, DEX provided relevant benefits by improving oxygenation and lung mechanics with fewer pulmonary complications in moderate COPD patients undergoing lung cancer surgery [6]. Besides, increasing lines of evidence have demonstrated that DEX conferred anti-inflammatory, anti-apoptotic and antioxidant properties to attenuate the lung injury following ischemia/reperfusion, lipopolysaccharide, and ventilation in animal models [[7], [8], [9]]. Additionally, DEX was reported to possess cytoprotective effects against endotoxin-induced lung injury by reducing oxidative stress, mitochondrial damage and mitochondria-dependent apoptosis [10,11]. Recently, it has been found that DEX was endowed with regulation of mitochondrial fusion/fission to mitigate sepsis related lung injury [12]. However, the underlying molecular mechanism of action is still ill-defined.
Hypoxia inducible factor-1α (HIF-1α), the major functional subunit of HIF-1, serves as one of pivotal cellular targets of endogenous ROS, which further regulates the expression of stress response gene involved in inflammation, metabolism, oxygen delivery and cell survival [13]. Of note, activated or stabilized HIF-1α in response to hypoxia or enhanced ROS levels behaved as lung protection by inducing its downstream gene, heme oxygenase-1 (HO-1) [14,15]. The importance of HO-1 and its yielding the heme-breakdown byproducts carbon monoxide, biliverdin and free iron in mediating antioxidant and anti-inflammatory effects has been well characterized both in vivo and in vitro models of cellular stress and organ injury [[16], [17], [18]]. In this regard, HIF-1α/HO-1 pathway has been documented to be crucial for alleviating LPS-induced lung injury [19]. In addition, our previous study has certified the endogenous defensive role of HO-1 system against septic lung injury through modulating mitochondrial dynamics [5,20]. Herein, we speculated that the lung protective effect mediated by HIF-1α/HO-1 pathway might be related to the regulation of mitochondrial fusion/fission balance.
Collectively, by using the widely accepted septic lung injury model of LPS exposed to mice, the current study was designed to illuminate whether DEX treatment alleviated endotoxin-induced acute lung injury by preserving the dynamic equilibrium of mitochondrial fusion/fission. And then, further studies were undertaken using HO-1-knockout mice or HO-1 or HIF-1α siRNA transfected NR8383 cells to investigate whether HIF-1a/HO-1 signaling pathway was involved in the beneficial ability of DEX to mitigate septic lung injury.
All animal studies were conducted according to the National Institutes of Health Guidelines with pre-approval of the Animal Ethical and Welfare Committee of Tianjin NanKai Hospital (Approval Document No. NKYY-DWLL-2019-028). Well characterized HO-1 conditional knockout (-/-) mice on a C57BL/6J background (HO-1fl/fl/CAG-CreERT2) were provided by Beijing Biocytogen Co., Ltd, and HO-1 conditional knockout was induced by Tamoxifen dissolved in corn oil, which specifically operated as our preliminary study [21]. Eight-week-old male C57BL/6J mice weighing 20–25g were obtained from the Laboratory Animal Center of Tianjin NanKai Hospital and housed 3–5 per cage under standardized conditions (22–24 °C, 60–65% of humidity, 12h light/dark cycle) with access to chow and water ad libitum. All mice were anesthetized with 2% sodium pentobarbital 50mg/kg intraperitoneally for induction, and 1%~3% isoflurane inhalation for maintenance. The adequacy depth of anesthesia was monitored by the absence of reflex response to foot squeeze. Then, mice were assigned randomly into four groups (n = 6/per group): sham group, LPS group, LPS plus DEX group, and DEX group. For the endotoxin-induced acute lung injury in vivo, caudal vein injection of LPS (E.coli-L2630, Sigma, USA) 15mg/kg diluted in 2ml saline was performed as described previously. And, the mice in LPS plus DEX group were administered 50ug/kg DEX (H20110085, Jiangsu Nhwa Pharmaceutical Co., LTD) intraperitoneally 30min prior to LPS treatment. Moreover, LPS plus DEX group was further randomized into subgroups in the setting of HO-1-/- or wild-type (WT) mice to receive dimethyloxalylglycine (DMOG, 50mg/kg, ip, D1070, Frontier Scientific, Inc. USA), a competitive inhibitor of prolyl hydroxylase (PHD) to activate HIF-1a. Subsequently, after 12h of monitoring, the mice were euthanized by exsanguination with overdose of sodium pentobarbital and every effort was made to minimize animals suffering. Blood samples were immediately collected through cardiac puncture and centrifuged 3000g for 30 min at 4 °C to obtain serum. Meanwhile, the lung tissues were excised for histological analysis or snap-frozen in liquid nitrogen, stored at −80 °C for further biochemical detection. The experimental schedule was shown in Table 1.

| Grouping | Treatment |
|---|---|
| Effects of Dexmedetomidine (DEX) on LPS-induced ALI in C57BL/6J mice (n=6/per group) | |
| Sham | Sham operation plus normal saline |
| LPS | Caudal vein injection of 15mg/kg LPS diluted in 2ml saline for 12h |
| LPS + DEX | 50ug/kg DEX was pretreated intraperitoneally (i.p.) 30min prior to LPS challenge |
| DEX | Sham operation plus 50ug/kg DEX |
| Effects of HIF-1a/HO-1 pathway on DEX-mediated mitochondrial dynamics during septic lung injury in HO-1 knockout and WT mice (n=6/each group) | |
| HO-1 KO + LPS + DEX + DMOG | 50mg/kg DMOG i.p. 30min before DEX prior to LPS in HO-1 knockout mice |
| WT + LPS + DEX + DMOG | 50mg/kg DMOG i.p. 30min before DEX prior to LPS in WT mice |
| Effects of HIF-1a/HO-1 pathway on DEX-mediated mitochondrial dynamics during LPS-induced oxidative injury in HO-1siRNA and NC siRNA-transfected NR8383 cells | |
| NR8383 HO-1siRNA + LPS + DEX + DMOG | 100μM DMOG pre-incubated 1h before 10μM DEX 2h prior to 10μg/ml LPS for 24h in HO-1siRNA-transfected NR8383 macrophages |
| NR8383+LPS + DEX + DMOG | 100μM DMOG pre-incubated 1h before 10μM DEX 2h prior to 10μg/ml LPS for 24h in NC siRNA-transfected NR8383 macrophages |
Abbreviations: DEX, dexmedetomidine; LPS, lipopolysaccharide; ALI, acute lung injury; HO-1, heme oxygenase-1; HIF-1a, hypoxia inducible factor-1α; DOMG, dimethyloxalylglycine; KO, knockout; HO-1 siRNA, HO-1 small interfering RNA (siRNA); NC siRNA, negative control siRNA; NR8383, rat alveolar macrophage cell line NR8383.
The collected lung tissues were immersed in 10% neutral buffered formalin, followed by routinely dehydration, clearing, paraffin-embedded and sectioned into 4μm slices. For pathological analysis, sections were sequentially stained with hematoxylin and eosin (H&E) and 10 randomly non-overlapping fields were taken for per animals under a light microscope ( × 200). A semi-quantitative scoring system was used to evaluate the lung injury based on the following categories: neutrophil infiltration, pulmonary edema, disorganization of lung parenchyma and hemorrhage [22]. Briefly, the grading scale of 0 = none; 1+ = light injury (25%); 2+ = moderate injury (50%); 3+ = severe injury (75%); 4+ = very severe injury (almost 100%) to score the degree of lesion in all samples, as previously described [5,14]. The lung injury was scored by two pathologists blinded to the experimental setup.
Rat alveolar macrophage cell line NR8383 was purchased from American Type Culture Collection (CRL-2192, ATCC, Manassas, VA, USA) and was cultured in Ham’s F12K medium supplemented with 2 mM l-glutamine, 1.5g/L sodium bicarbonate and 15% heat inactivated fetal bovine serum under a humidified atmosphere of 5% CO2 at 37 °C. The medium was renewal two to three times weekly. Cells at passages two to three were used in the experiments. NR8383 cells were seeded onto 96-well plates at a starting density of 5×104/ml and grown to 80~90% confluence for the below experiments. To mimic in vitro sepsis model, cells were incubated with 10μg/ml LPS for 24h in the presence of appointed concentrations of DMOG (100μmol/L) for 1h ahead of DEX (10μmol/L) preincubated for 2h [23,24]. Subsequently, cultured cells were harvested for protein and RNA expression analysis, and supernatants were collected for ELISA assay.
Cell Counting Kit-8 (CCK-8, E1CK-000208-10, EnoGene, Nanjing, China) assay was operated
to determine the viability of NR8383 cells exposed to LPS plus DEX or DMOG. Briefly, 10μl CCK-8 solution was added to each well, followed by incubation for 2.5 h at 37 °C. After mixing well, the OD absorbance at 450nm was recorded by using a microplate reader (Bio-Rad, Hercules, CA, USA).
HO-1, HIF-1α small interfering RNA (siRNA) and negative control siRNA (NC siRNA) were designed by Shanghai GenePharma Co, Ltd. (China). For the transfection experiment, NR8383 cells at 5×105/ml were seeded into 6-well plates. Based on the manufacturer’s protocols, transient transfection of siRNA mimics (5μl) were performed in cells using 7.5μl Lipofectamine® 3000 (No. L3000075, Invitrogen, USA) for 6h. Then, the medium was aspirated and replaced with 2ml fresh Ham’s F12K nutrient mixture containing 20% FBS for 24h. After incubation, NR8383 cells were harvested for western blotting to evaluate the efficiency of siRNA silencing, and the culture supernatant was collected to detect the levels of TOS and TAS.
An isolated mitochondrial fraction from mice lung tissues was performed as previously delineated with slight modifications [25]. The final mitochondrial pellet was gently resuspended in homogenization medium and centrifuged 12000g for 10 min at 4 °C. Mitochondrial ROS was measured by using 2’,7’-dichlorofluorescein diacetate (DCFH-DA) probe for 30 min at 37 °C in a humidified 5% CO2 atmosphere. The fluorescence signal was measured respectively at an excitation/emission wavelength of 488nm/520nm by a microplate reader.
Oxidative stress index (OSI) was calculated as the ratio between total oxidant status (TOS) and total antioxidant level (TAS) levels. The TOS and TAS concentrations of serum or cell supernatant were respectively measured by a commercial kit (Immundiagnostik AG, Cat No: KC5100 and KC5200, Bensheim, Germany) with a microplate spectrophotometer following the manufacturers’ instructions. For TOS, the assay was calibrated with hydrogen peroxide (H2O2), and the results were stated as μmol H2O2 equivalent/L. For TAS, the assay was calibrated with Trolox, and the results were expressed as μmol Trolox equivalent/L [26].
The concentrations of glutathione peroxidase (GSH), glutathione disulfide (GSSG) and the ratio of GSH/GSSG in lung tissues were determined by the commercial available assay kits (A061-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) complied with the instructions of the manufacturers. The levels of GSH and GSSG were expressed as nmol/mg protein. Besides, the formation of hydrogen peroxide (H2O2) in the lung tissues were measured by fluorescence methods using the Amplex Red H2O2 assay kits (ab102500, Abcam, UK). As described in detail of Messier EM, et al., 25μM Amplex Red reacted with H2O2 in the presence of 0.2U/ml horseradish peroxidase with a 1:1 stoichiometry to form resorufin [27]. Absorbance was measured using an excitation filter of 530nm and an emission filter of 590nm, and H2O2 production was normalized to protein contents.
The copy number of mitochondrial DNA (mtDNA) was assessed by ratio of mtDNA to nuclear encoded β-actin using real time PCR as described by Amaral A, et al [28]. DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions. Briefly, RT-PCR was performed using the Applied Biosystems 7300 real-time PCR system using 10μl AceQ Universal SYBR qPCR Master Mix, 0.4μl forward primer, 0.4μl reverse primer, 4.2μl ultrapure water and 5μl template DNA with the following conditions: pre-incubation at 95 °C for 5min, followed by 40 cycles of denaturation at 95 °C for 10s, annealing and extension at 60 °C for 30s. The primers of mtDNA fragment (117bp) were: forward 5’-CCCAGCTACTACCATCATTCAAGT-3’, reverse 5’-GATGGTTTGGGAGATTGGTTGATG-3’; nuclear encoded β-actin was used as genomic DNA control, the primers were: forward 5’- GCCAGCCTGACCCATAGCCATAATAT-3’, reverse 5’-GAGAGATTTTATGGGTGTAATGCGG-3’. The level of mtDNA was calculated by the comparative Ct methods from triplicate detections.
Small pieces from the lung tissues of C57BL/6J mice were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (PH 7.4) for 2h at room temperature. After rinsed in 0.1 M phosphate buffer, specimens were post-fixed with 1% osmium tetroxide for 2h, dehydrated in a graded series of ethanol, and then embedded in acetone and sectioned approximately at 70nm. Ultrathin sections were stained with 3% uranyl acetate and lead citrate and viewed under a JEM-1230 transmission electron microscope at 80 KV. Similarly, the methods of cultured NR8383 cells subjected to different reagents were treated the same as above mentioned [29]. The transmission electron microscopy images of mitochondria from at least three randomly selected fields per mice of each group were assessed by a blinded pathologist.
Lung tissues were fixed in 4% paraformaldehyde for 48h, embedded in paraffin, and sliced into 5 μm sections based on routine protocols. Then, the sections were dewaxed in xylene, rehydrated with graded ethanol, and antigen retrieval by PH 9.0 EDTA. After rinsing in TBS three times for each 5min, slices were blocked with 10% donkey serum at 37 °C for 30min, and incubated with hydrated, and then incubated with rabbit polyclonal anti-HO-1/HMOX1 antibody (Proteintech, 10701-1-AP, 1:100 dilution) or anti- HIF-1a antibody (Abcam, ab2185, 1:100 dilution) at 4 °C overnight. Following washing triple in TBST, the sections were incubated with diluted fluorescent-labeled secondary antibodies at room temperature for 30min, and counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) to indicate the nuclei for 10min in the dark. The images were captured using a fluorescent microscope (Olympus U-25ND25, Tokyo, Japan).
The frozen lung samples were homogenized by lysis buffer on ice. The cell lysates were centrifuged 9000g for 10 min at 4 °C, and the supernatant was collected. Protein concentrations were quantified by the bicinchoninic acid assay (Sigma, USA). The protein extracts (40μg) were heated, denatured, and loaded on 10% SDS-PAGE for electrophoresis followed by transferring to a PVDF membrane. The membrane was blocked with 5% skimmed milk in TBST for 1h at 37 °C, and then probed with primary antibodies for HIF-1a (1:1000, ab2185), HO-1 (1:2000, PTG10701), Mfn1 (1:1000, ab57602), Mfn2 (1:1000, PTG12186), OPA1 (1:1000, ab157457), Drp1 (1:1000, ab184247), Fis1 (1:1000, ab71498) and β-actin (1:8000, KM9001) at 4 °C overnight. After being washed four times with TBST, the blots were incubated with appropriate horseradish peroxidase–conjugated secondary antibody at room temperature for 1h. The loading control was the constitutively expressed protein β-actin, and the blots were visualized by enhanced chemiluminescence system (Bio-Rad) and analyzed using Image J.
Total RNA in lung tissues or NR8383 cells were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) in accordance with the manufacturer’s manuals. 1μg total RNA was converted to cDNA using the RevertAid reverse transcriptase (Thermos Fisher, EP0442, USA) under the following conditions: 25 °C for 10min, 42 °C for 60 min and 70 °C for 10min. RT-PCR analysis was performed using the Applied Biosystems 7300 real-time PCR system and software (Applied Biosystems, USA) with the specific mRNA primers of HIF-1a, HO-1, Mfn1, Mfn2, OPA1, Drp1, Fis1 and β-actin listed in Table 2. The pre-degeneration of the PCR mix was performed at 95 °C for 5min, followed by 40 thermal cycles consisting of denaturing for 10 s at 95 °C, annealing and extension for 30 s at 60 °C. The copy number of each transcript was calculated as the relative copy number normalized to the housekeeping gene β-actin. The 2−ΔΔCt method was used to determine the relative mRNA expression of target genes [30].
| Genes | Forward primer | Reverse primer | Length(bp) | Access Number |
|---|---|---|---|---|
| Mfn1 | GCAGAAGGATTTCAAGCAAGAT | CACTGCTGACTGCGAGATACAC | 81 | NM_024200.4 |
| Mfn2 | GGATTCCCTGCACATCGC | GCCTTTCCACTTCCTCCGTAA | 158 | NM_001285922.1 |
| OPA1 | CTGCTTTTGGAAAATGGTTCG | CCCAGTGCTTTGGCGTGA | 200 | NM_001199177.1 |
| Drp1 | GTAGGCGATCAGCCCAAGG | CAGCAGTGACGGCGAGGA | 97 | NM_001276340.1 |
| Fis1 | CTGTCTCCAAGTCCAAATCCTG | GAAAAGGGAAGGCGATGGT | 118 | NM_025562.3 |
| HIF-1a | ATCTCGGCGAAGCAAAGAGT | TCCATCTGTGCCTTCATCTCA | 186 | NM_001313919.1 |
| HO-1 | GCCACCAAGGAGGTACACAT | GGGGCATAGACTGGGTTCTG | 170 | NM_010442.2 |
| β-actin | CTGAGAGGGAAATCGTGCGT | CCACAGGATTCCATACCCAAGA | 208 | NM_007393.5 |
Abbreviations: Mfn1, mitofusin 1; Mfn2, mitofusin 2; OPA1, optic atrophy 1; Drp1, dynamin-related protein 1; Fis1, fission 1; HIF-1a, hypoxia inducible factor-1α; HO-1, heme oxygenase-1.
All values are expressed as means ± standard deviation (S.D.) of three individual experiments performed in duplicate or triplicate. Paired T-test was used for comparisons between two groups. For comparisons among multiple groups, One-way analysis of variance followed by Bonferroni’s post hoc test or Mann-Whitney U test was used as appropriate by a Graph Prism 6.7 software (GraphPad Software, La Jolla, CA). P values of less than 0.05 were considered statistically significant (*P<0.05; **P<0.01; ***P<0.001).
All mice survived until euthanized. A primary glance of lung specimens collected from LPS group indicated visibly pathological lesion including severe leukocyte infiltration, thickened alveolar wall, pulmonary edema and hemorrhage, which were markedly alleviated in the DEX-pretreated mice (Fig. 1A). As illustrated in Fig. 1B, applying a semiquantitative system for lung injury scores, consistent results were found as LPS (13.3 [11 to 16]) significantly decreased lung injury scores which were increased by DEX pretreatment (7.17 [5 to 9]). In addition, compared with sham group, serum levels of TAS, as an indicator to ensure protecting lung tissue from oxidative damage by scavenging ROS, were decreased in LPS group. Meanwhile, serum levels of TOS as a sensitive index of oxidative stress, and the TOS/TAS ratio namely OSI, which stand for the redox balance between antioxidant and oxidant status, were both increased in LPS mice [31]. DEX administration suppressed the increase of TOS and OSI levels, while enhanced the release of TAS levels (Fig.1C–E). Of note, the DEX alone group was proven to have no effect compared with sham group (P>0.05).

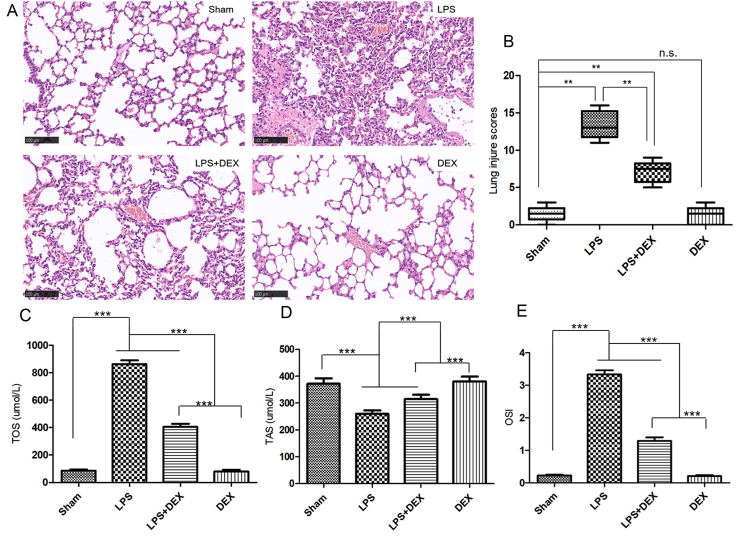
Pretreatment with DEX protected the lung against LPS-driven oxidative injury in mice. A, Photomicrographs of histopathologic changes of lung sections stained with hematoxylin and eosin at 12h after LPS challenge (original magnification, ×200). Scale bar: 100μm. B, Semiquantitative analysis of lung tissues by lung injury scores. Values are expressed as medians (range) using the Mann-Whitney U test (n = 6 mice/group). C-E, Serum levels of total oxidant status (TOS), total antioxidant status (TAS) and oxidative stress index (OSI) calculated as TOS/TAS ratio. Data were represented as mean ± SD in bar graph C, D and E. Analysis of variance (ANOVA), Bonferroni multiple comparison test, n = 6 per group. Significant differences were indicated with an asterisk: *P < 0.05, **P < 0.01, ***P < 0.001. DEX, dexmedetomidine; LPS, lipopolysaccharide; N·S., not significant; TOS, total oxidant status; TAS, total antioxidant status; OSI, oxidative stress index.
The imbalance of ROS production led to upsurge of oxidative stress, and further caused mtDNA damage under septic lung injury conditions, which result in the abnormal mitochondrial ultrastructure [32]. As shown in Fig. 2A-B, exposure mice to LPS showed a striking increase in the levels of mitochondrial ROS and mtDNA contents (P<0.05), which were significantly suppressed by pretreatment with 50μg/kg DEX but no effects in DEX alone mice. Accordingly, the normal architecture of mitochondria as shown in sham group was deformed with isolated, small, rounded mitochondria and lamellar-body vaculation with loss of clearly defined cristae after LPS challenge (Fig. 2C). However, these changes were less evident in the DEX-pretreated mice (P<0.05).
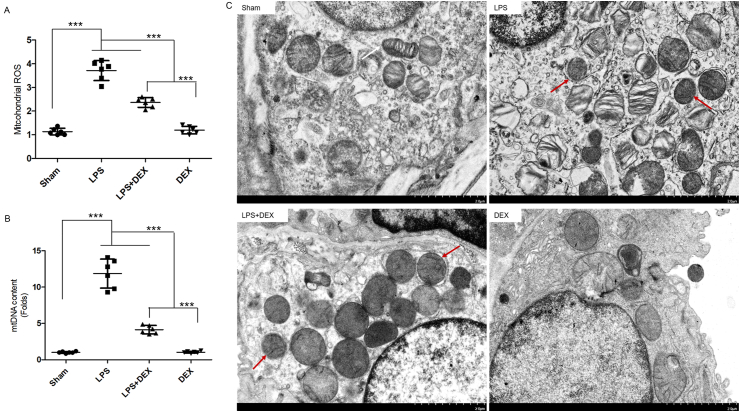
Pretreatment with DEX mitigated LPS-induced mitochondrial oxidative damage in the lung tissue of mice. A, Mitochondrial ROS production detected spectro-fluorometrically. using DCFH-DA as a fluorescent dye. B, Mitochondrial DNA (mtDNA) content using real time PCR. C, The morphological alterations of mitochondria by transmission lectron microscopy (original magnification, ×5000). Scale bar: 2μm. Red arrows denoted deformed mitochondria manifested as isolated, small, rounded mitochondria with loss of clearly defined cristae. White arrows relatively marked normal shape of mitochondria in the DEX alone group. Data were. representative of mean ± SD in bar graph A, B (n = 6), *P < 0.05, **P < 0.01, ***P < 0.001. one-way ANOVA corrected with Bonferroni coefficient. DEX, dexmedetomidine; ROS, reactive oxygen species; LPS, lipopolysaccharide; DCFH-DA, 2’,7’-dichlorofluorescein diacetate. . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To ascertain whether DEX conferred defense against sepsis-related lung injury through preserving mitochondrial dynamics, we tested the expressions of the crucial mitochondrial fusion/
Fission proteins and mRNA in lung tissues by Western Blotting and Real-time PCR (Fig.3A–C). Compared with sham group, exposure to LPS shifted the balance towards a fission phenotype, manifested as reduced mitochondrial fusion protein and mRNA levels of Mfn1, Mfn2, OPA1 while enhanced mitochondrial fission protein and mRNA levels of Drp1 and Fis1 (P<0.001). Notably, pretreatment with DEX markedly upregulated the expressions of Mfn1, Mfn2, OPA1 proteins and mRNA, yet downregulated the expressions of Drp1, Fis1 proteins and mRNA, which restored the imbalance of mitochondrial dynamics induced by LPS (P<0.001). No significant differences of above-mentioned proteins and mRNA levels between DEX alone and sham group (P>0.05).
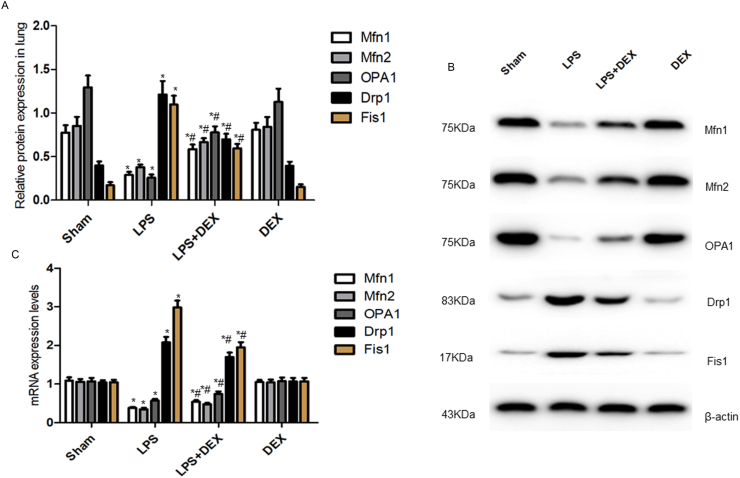
Pretreatment with DEX reinforced mitochondrial fusion while restrained mitochondrial fission in the context of septic lung injury in mice. A and B, Representative western blots of mitochondrial fusion (Mfn1, Mfn2, OPA1) and fission (Drp1 and Fis1) markers shown on right and quantitation shown on left. Analyses of band intensity on the western blotting images were presented as their relative ratios compared with β-actin. C, Expressions of the mRNA levels of mitochondrial fusion/fission markers detected by real-time PCR. Values were presented as mean ± SD of six separate experiments using one-way ANOVA and the Bonferroni test for multiple comparisons. *Significant difference from sham group (P< 0.05). #Significant difference from LPS-exposed mice (P < 0.05). DEX, dexmedetomidine; LPS, lipopolysaccharide; Mfn1, mitofusin 1; Mfn2, mitofusin 2; OPA1, optic atrophy 1; Drp1, dynamin-related protein 1; Fis1, fission 1.
As shown in Fig. 4A-C, Immunofluorescence study revealed increased fluorescence intensity of FITC labeled HO-1 and TRITC labeled HIF-1a was found in lung tissue of mice subjected to LPS. Besides, green fluorescence of FITC-HO-1 and red fluorescence of TRITC-HIF-1a were co-localized in the nucleus, the localization in the nucleus was significantly higher than that in the cytoplasm. Furthermore, DEX pretreatment obviously increased the fluorescence intensity of HO-1 and HIF-1a in the nucleus, which uncovered the temporal and spatial consistency of HO-1 and HIF-1a expression after DEX precondition. As a parallel, pretreatment with DEX 30min ahead of LPS induced a dramatic increase of protein and mRNA levels of HO-1 and HIF-1a (HO-1 protein: LPS + DEX vs. LPS, 1.092 ± 0.103 vs. 0.637 ± 0.081; HO-1 mRNA: LPS + DEX vs. LPS, 5.971 ± 0.569 vs. 3.042 ± 0.299; HIF-1a protein: LPS + DEX vs. LPS, 1.162 ± 0.11 vs. 0.776 ± 0.094; HIF-1a mRNA: LPS + DEX vs. LPS, 8.257 ± 0.841 vs. 6.285 ± 0.594; n = 6; mean ± SD, P<0.001). However, DEX alone was proven to have no effects on the above variables compared with sham group (P>0.05).
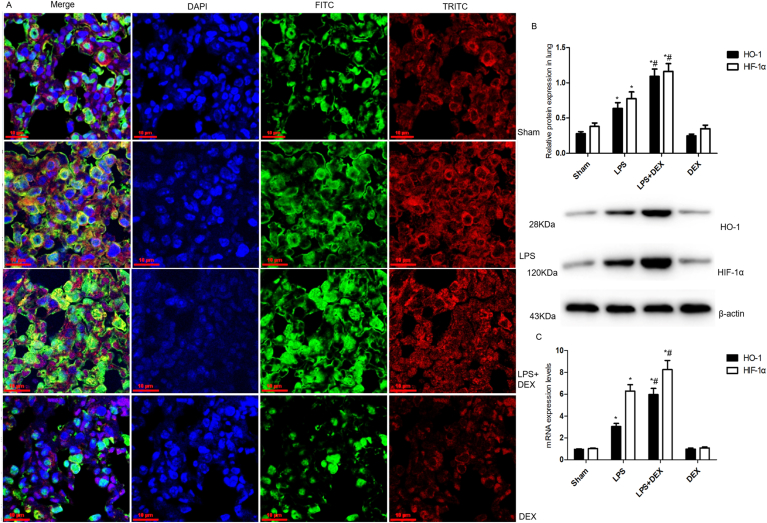
HIF-1a/HO-1 pathway mediated the regulation of DEX on mitochondrial dynamics following LPS challenge. A, Immunofluorescence assays of HO-1 and HIF-1a protein by fluorescence microscope (original magnification, ×400). Green stood for FITC-HO-1 stained sections, red stood for TRITC-HIF-1a stained sections, while blue stood for images of DAPI- stained nuclei. HO-1 was found to colocalize with HIF-1a in the nucleus of the lung tissue section. Scale bar: 10μm. B, Quantification and example bands from western blotting to assess the expressions of HO-1 and HIF-1a protein in the lung tissue of mice. β-actin served as a standard for protein loading. C, The expressions of mRNA of HO-1 and HIF-1a detected by real-time PCR in in the lung tissue of mice. Values were expressed as mean ± SD of six individual samples using one-way ANOVA corrected with Bonferroni coefficient for multiple comparisons. *Significant difference from sham group (P< 0.05). #Significant difference from LPS-treated mice (P < 0.05). DEX, dexmedetomidine; LPS, lipopolysaccharide; HO-1, heme oxygenase-1; HIF-1α, hypoxia inducible factor-1α. . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Lung histology and morphology revealed aggravated lung injury in HO-1 knockout (HO-1 KO) mice compared with WT mice, as characterized by thickened alveolar wall, increased inflammatory cells infiltration, edema formation, and alveolar hemorrhage (Supplementary Fig. 1A). And, this was concurrent with elevated lung injury scores in LPS challenged HO-1 KO mice compared to wild type mice (Supplementary Fig. 1B). With respect to oxidative stress, the alterations of oxidative stress markers were analyzed in the experiments. As shown in Supplementary Fig. 1C–I, serum TOS, OSI were increased, while TAS levels were decreased in LPS-treated mice with HO-1 knockout. Besides, reduced levels of lung GSH, GSH/GSSG ratio, whereas enhanced oxidative products GSSG, H2O2 were found in HO-1 KO mice in contrast to WT mice subjected to LPS, indicating the functional importance of HO-1 in mitigating LPS-evoked oxidative lung damages. Interestingly, it was found that HO-1 knockout significantly prevented LPS-induced increase in protein expression of HO-1 and HIF-1a (Supplementary Fig. 1J and K), validating HIF-1a/HO-1 pathway role as a protective regulator against excessed oxidative stress in answer to LPS.
To gain insight into the effects of DEX on overall changes in the general antioxidant system, the glutathione redox cycle which involved levels of lung GSH, GSSG, and the GSH/GSSG ratio were evaluated (Supplementary Fig. 2A–C). Not surprisingly, DEX precondition reduced excessed ROS production during septic lung injury via the synergistic 28.7% vs 49.1% increase in the levels of lung GSH, GSH/GSSG ratio, while 28.1% decrease in GSSG levels in LPS-stimulated mice. Besides, H2O2 production was measured by Amplex Red assay and a distinct reduction in formation of H2O2 was found in 50ug/kg DEX treated mice after LPS challenge (Supplementary Fig. 2D). However, HO-1 deficiency significantly reversed the sustained antioxidant capacity of DEX in response to LPS, as indicated by decreased GSH, and GSH/GSSG ratio, whereas increased GSSG, and H2O2 levels.
To ascertain whether DEX sustained mitochondrial dynamics following HO-1 or HIF-1a silencing, the HO-1, HIF-1a siRNA and negative control siRNA were incubated in the NR8383 alveolar macrophages. As shown in Supplementary Fig. 3A–I, preincubation of NR8383 cells with DEX evidently elevated cell viability, enhanced levels of TAS, while reduced levels of TOS and OSI (P<0.001). Besides, DEX treatment decreased excessive production of mitochondrial ROS and mtDNA contents induced by LPS, which indicative of mitochondrial dysfunction. As expected, DEX incubation ahead of LPS induced a dramatic increase of HIF-1a, HO-1, Mfn1, Mfn2 and OPA1 mRNA and proteins, while a distinct decrease of Drp1 and Fis1 mRNA and proteins (P<0.05), which was in line with the established lung protective role of DEX in mice model of septic lung injury. Conversely, HO-1 or HIF-1a knockdown blocked DEX-mediated preservation of mitochondrial dynamics, characterized by remarkable reduced levels of HIF-1a, HO-1, Mfn1, Mfn2 and OPA1 mRNA and proteins, while elevated levels of Drp1 and Fis1 mRNA and proteins (P<0.05). Negative control siRNA did not affect the above variables compared with DEX-treated NR8383 cells exposed to LPS (P>0.05).
To further elucidate the role of HIF-1a/HO-1 pathway in the protection of DEX against sepsis-related lung injury, we performed identical experiments as above using HO-1 KO mice subjected to DEX and DMOG (HIF-1a stabilizer) pretreatment. The thickened alveolar septal wall, aggravated neutrophil infiltration and alveolar hemorrhage induced by LPS were reduced in those DEX and DMOG-treated WT mice. While, the pathologic changes were reinforced in HO-1-/- mice exposure to DEX and DMOG (Fig. 5A). As shown in Fig. 5B, semiquantitative evaluation showed that the degree of lung injury in the DEX and DMOG-treated WT mice (8 [6 to 11]) was distinctly lower than that in HO-1-/- mice (14.17 [12 to 16]). Besides, the serum levels of TAS decreased by 46.36%, yet the serum levels of TOS increased by 50.7%, as well as OSI, the TOS/TAS ratio, elevated by 73.7% in HO-1-/- + LPS + DEX + DMOG group compared with WT + LPS + DEX + DMOG group (Fig.5C–E). As sepsis-induced lung injury damaged mitochondrial structure and resulted in a deficiency in mitochondrial function, thus we use transmission electron microscopy to visually compare mitochondrial morphology in the lung tissue from HO-1-/- and WT mice following LPS. As shown in Fig. 5F-H, more swollen, fragmented mitochondria with disorganized cristae were accumulated in DEX and DMOG-treated HO-1-/- mice, along with increased levels of mitochondrial ROS and mtDNA contents, which were less evident in DEX and DMOG-treated WT mice (P<0.001).
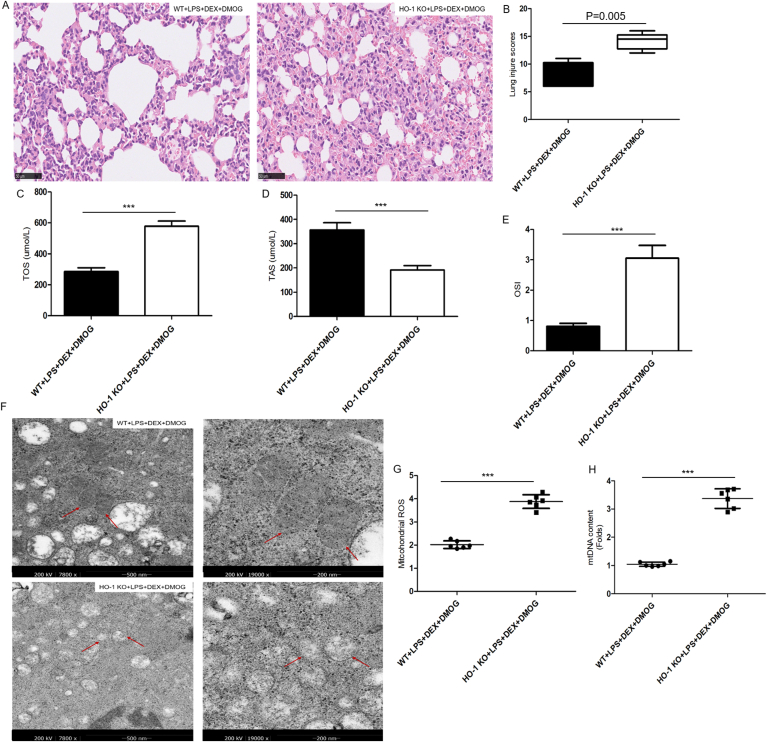
HO-1 knockout counteracted the effects of DEX-mediated lung protection in mice subjected to LPS. A, The histopathologic changes of lung sections stained with hematoxylin and eosin following 50mg/kg DMOG intraperitoneally exposed 0.5h before DEX pretreatment for 0.5h prior to LPS challenge (original magnification, ×200). Scale bar: 50μm. B, Semiquantitative analysis of lung tissues by lung injury scores. Values are expressed as medians (range) using the Mann–Whitney U test (n = 6 mice/group). C-E, Serum levels of TOS, TAS and OSI. F, The morphological alterations of mitochondria by transmission electron microscopy (original magnification, ×7800). Scale bar: 500nm. The right panels (original magnification, ×19000) were enlarged transmission electron microscopy images of mitochondria in the left panels. Scale bar: 200nm. Red arrows marked as isolated, small, rounded mitochondria with loss of clearly defined crista. G, Mitochondrial ROS generation. H, Mitochondrial DNA contents. Data were represented as mean ± SD in bar graph C, D, E, G, H, and analyzed by paired T-test (n = 6 per group). Significant differences were indicated with an asterisk: *P < 0.05, **P < 0.01, ***P < 0.001. DEX, dexmedetomidine; LPS, lipopolysaccharide; HO-1, heme oxygenase-1; DMOG, dimethyloxalylglycine; TOS, total oxidant status; TAS, total antioxidant status; OSI, oxidative stress index; ROS, reactive oxygen species; mtDNA, Mitochondrial DNA. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Additionally, the protein and mRNA expressions of specific mitochondrial fusion/fission markers (Mfn1/2, OPA1, Drp1, Fis1), HO-1 and HIF-1a were presented as Fig.6A–C. The deletion of HO-1 led to 74.02% versus 93.1% decreased in the levels of HO-1 protein and mRNA in response to DEX and DMOG pretreatment. Similar to HO-1 levels, the down-regulated expression of HIF-1a, Mfn1, Mfn2 and OPA1 proteins and mRNA were detected in DEX and DMOG-treated HO-1-/- mice. However, the levels of Drp1 and Fis1 proteins and mRNA were significantly up-regulated in HO-1-/- mice of priming with LPS and DEX, DMOG pretreatment(P<0.001). These results implicated that HO-1deletion counteracted the lung protective effects of DEX and HIF-1a stabilizer pretreatment in the settings of sepsis-related lung injury. Given these findings, it was reliable that DEX acted via HIF-1a/HO-1 pathway to preserve the balance of mitochondrial dynamics to mitigate septic lung injury in vivo.
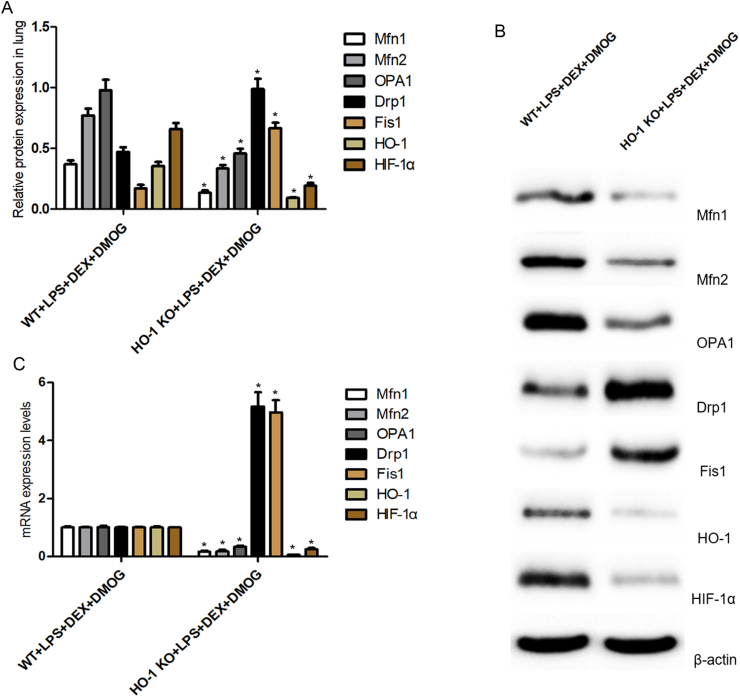
HO-1 knockout reversed the effects of DEX-afforded preservation of mitochondrial dynamics in mice septic lung injury. A-B, Quantification and example bands from Western blot analysis to determine the expression levels of Mfn1, Mfn2, OPA1, Drp1, Fis1 protein required for mitochondrial fusion/fission, as well as HO-1 and HIF-1a protein in the lung tissue of mice subjected to DEX and DMOG pretreatment. Band density was quantified relatively to β-actin. C, Representative mRNA levels of Mfn1, Mfn2, OPA1, Drp1, Fis1, HO-1 and HIF-1a expression assessed by real-time PCR in the lung tissue of DEX and DMOG-pretreated mice. Values representative of mean ± SD obtained from six independent experiments using paired T-test. *P < 0.05 versus DEX + DMOG + LPS-treated wild type mice. DEX, dexmedetomidine; LPS, lipopolysaccharide; HO-1, heme oxygenase-1; DMOG, dimethyloxalylglycine; HIF-1α, hypoxia inducible factor-1α; Mfn1, mitofusin 1; Mfn2, mitofusin 2; OPA1, optic atrophy 1; Drp1, dynamin-related protein 1; Fis1, fission 1.
To assess the effects of DEX pretreatment in different concentrations on the viability of LPS-treated NR8383 cells, a CCK-8 assay was conducted (Fig. 7A). NR8383 alveolar macrophages were stimulated with 10μg/ml LPS for 24h with pre-incubated of 0.01, 0.1, 1, 10, or 50 μmol/L DEX for 2h. Gradual increase of cell viability was observed with the concentrations of 0.01, 0.1, 1, 10μmol/L DEX pre-incubation and reached the highest value as 77.2 ± 7.244% at dose of 10μmol/L DEX in LPS-treated cells (P<0.05). In the settings, 10μmol/L DEX was used for the subsequent experiments. Having established that HIF-1a/HO-1 pathway mediated the protective efficacy of DEX against septic lung injury through improving mitochondrial dynamics in vivo, we then validate this finding using the in vitro model of LPS-primed NR8383 alveolar macrophages subjected to siRNA-mediated silencing of HO-1. As shown in Fig. 7B-D, we detected TOS, TAS and OSI of the cells to reflect the oxidative status of LPS primed NR8383 cells co-treated with 10μmol/L DEX and 100μmol/L DMOG. Compared with NR8383 cells transfected NC siRNA, the macrophages transfected with HO-1siRNA or elicited the apparently reduced levels of TAS, while enhanced levels of TOS and OSI (P<0.001). In the meantime, excessive production of mitochondrial ROS and mtDNA contents were observed in HO-1siRNA-transfected cells co-incubated with DEX and DMOG prior to LPS (P<0.001), which indicated that the knockdown of HO-1 offset the improved mitochondrial function reflected by lower mitochondrial ROS and mtDNA levels afforded by DEX and HIF-1a stabilizer (Fig. 7E–F).
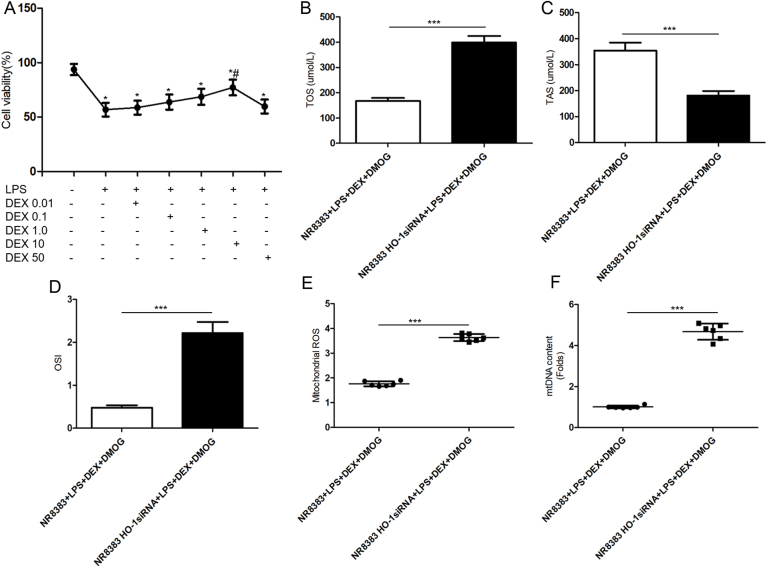
HO-1 knockdown offset the effects of DEX-mitigated LPS-induced oxidative injury in NR8383 alveolar macrophages. A, The viability of NR8383 cells preincubated with different concentrations of DEX prior to LPS conducted by CCK8 assay. Data were analyzed using the Mann-Whitney U test followed by Bonferroni correction for six individual experiments. *Significant difference from sham group (P<0.05). #Significant difference from LPS-incubated cells (P<0.05). B-D, The levels of TOS, TAS and OSI to reflect the oxidative status of LPS-primed NR8383 cells pretreated with 100μmol/L DMOG 1h before 10μmol/L DEX for 2h. E, Mitochondrial ROS production in cells. F, mtDNA contents in cells. Data in B, C, D, E and F were presented as mean ± SD using paired T-test (n = 6 replicates for each group). Significant differences were indicated with an asterisk: *P < 0.05, **P < 0.01, ***P < 0.001. DEX, dexmedetomidine; LPS, lipopolysaccharide; HO-1, heme oxygenase-1; DMOG, dimethyloxalylglycine; TOS, total oxidant status; TAS, total antioxidant status; OSI, oxidative stress index; ROS, reactive oxygen species; mtDNA, Mitochondrial DNA.
Immunofluorescence analysis of HO-1 and HIF-1a was performed to confirm that pretreatment with DEX and DMOG in NC siRNA-transfected NR8383 cells subjected to LPS induced HO-1 and HIF-1a expression, as indicated by increased number of the cells reflecting green fluorescence (Fig.8A–C). In contrast, NR8383 cells transfected with HO-1siRNA under above conditions distinctly decreased the fluorescence intensity of HO-1 and HIF-1a in the nucleus, suggesting the successful knockdown of HO-1 by siRNA, and the role of HIF-1a in the transcriptional activation of HO-1. Furthermore, as shown in Fig. 8D-F, western blotting and RT-PCR results demonstrated that HO-1siRNA substantially reduced HO-1 protein and mRNA expression (approximately 81.2% versus 52.5%) in NR8383 cells, certifying an effective knockdown of HO-1 by siRNA. In support, lower levels of HIF-1a, Mfn1, Mfn2, OPA1 protein and mRNA, while higher levels of Drp1, Fis1 protein and mRNA were confirmed in HO-1 siRNA-transfected cells upon pre-incubation with DEX and DMOG prior to LPS (P<0.05), which collectively supported a key role of HIF-1a/HO-1 pathway on DEX-afforded the preservation of mitochondrial dynamic equilibrium.
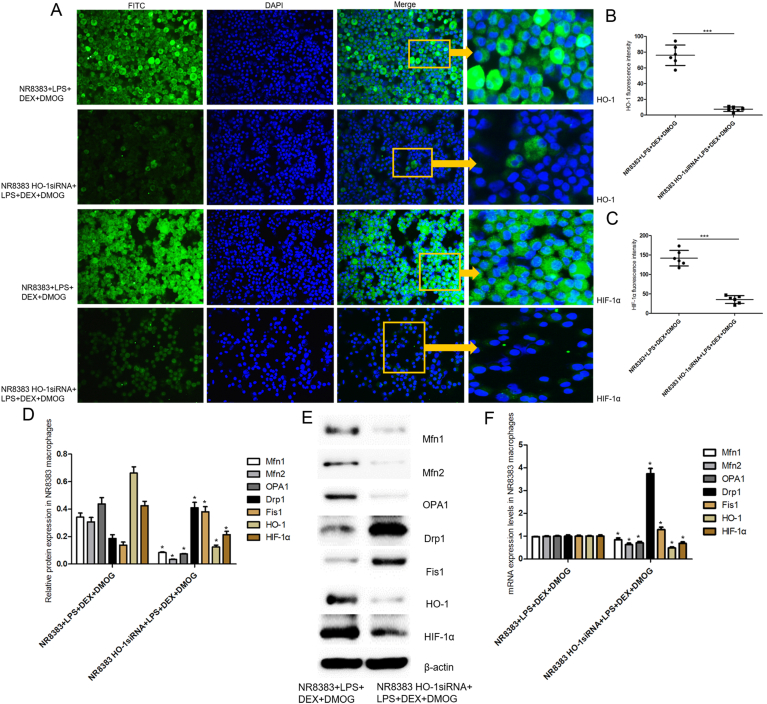
HO-1 knockdown abrogated the effects of DEX-regulated mitochondrial dynamics in in NR8383 alveolar macrophages exposed to LPS. A, Expression of HO-1 and HIF-1a in LPS-cultured NR8383 cells preincubated with DMOG and DEX detected by immunofluorescence assay. Green stood for HO-1 or HIF-1a-FITC stained sections, and nuclear was counter stained with DAPI. The overlay color of blue staining in nucleus, along with green staining both in cytoplasm or nucleus was considered to be positive. The right panels were enlarged images of the orange boxed areas in the panels of merge. Six representative fields of each slice were randomly selected from one of three individual experiments. B–C, Fluorescence intensity of HO-1 and HIF-1a quantified by Image J version1.49r software. The proportion of positive cells were calculated as the number of positive cells relative to the number of DAPI-positive cells. D-E, Representative western blots of mitochondrial fusion/fission markers including Mfn1, Mfn2, OPA1, Drp1 and Fis1 as well as HO-1 and HIF-1a protein shown on right and quantitation shown on left. β-actin was served as a loading control. F, Representative mRNA levels of Mfn1, Mfn2, OPA1, Drp1, Fis1, HO-1 and HIF-1a expression assessed by real-time PCR in LPS-primed NR8383 cells pretreated with DMOG and DEX. Values were expressed as mean ± SD in bar graph B, C, E, D and F. Paired T-test was used to detect statistically significant changes, n = 6/per group. Significant differences from DEX + DMOG + LPS-treated NC siRNA-transfected NR8383 cells were shown by (*) (**) and (***) which respectively indicated the P<0.05, P<0.01 and P<0.001 levels. DEX, dexmedetomidine; LPS, lipopolysaccharide; HO-1, heme oxygenase-1; DMOG, dimethyloxalylglycine; HIF-1α, hypoxia inducible factor-1α; Mfn1, mitofusin 1; Mfn2, mitofusin 2; OPA1, optic atrophy 1; Drp1, dynamin-related protein 1; Fis1, fission 1. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Our in vivo model of intravenous administration of LPS recapitulated major of the features of human systemic septic lung injury [33], with a markedly rise in pulmonary histopathological damages and lung injury scores followed by increased levels of TOS, OSI, GSSG, and H2O2 while decreased levels of TAS, GSH, and the GSH/GSSG ratio. For the first time, the current study explored whether dexmedetomidine (DEX), a potent α2 receptor agonist, attenuated sepsis-related lung injury by regulating mitochondrial fusion/fission through HIF-1a/HO-1 pathway in vivo and in vitro. Our findings delineated that (1) DEX conferred lung protection against LPS-driven oxidative stress injury, and reduced levels of mitochondrial ROS and mtDNA contents as well as improved mitochondrial morphology; (2) DEX sustained the balance of mitochondrial fusion and fission to mitigate LPS induced lung injury; (3) By utilizing HO-1 KO mice, HO-1siRNA and HIF-1a siRNA NR8383 macrophages to validate the pivotal role of HIF-1a/HO-1 pathway in DEX afforded the regulation of mitochondrial dynamic equilibrium (Fig. 9).
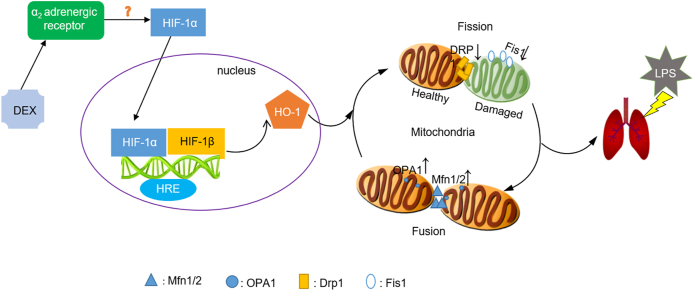
Graphical illustration of proposed DEX-mediated preservation of mitochondrial fusion and fission involved in regulating HIF-1a/HO-1 pathway. When LPS was administrated to C57BL/6J mice or NR8383 alveolar macrophages, DEX activated HIF-1a/HO-1 pathway, led to HIF-1a translocated into nuclear, heterodimerization with HIF-1β, which binds to hypoxia-response elements (HREs) to induce target gene of HO-1 expression. Meanwhile, pretreatment with DEX upregulated the expressions of mitochondrial fusion proteins Mfn1, Mfn2, OPA1, yet downregulated the expressions of mitochondrial fusion proteins Drp1, Fis1, accompanied with improved mitochondrial morphology and dysfunction in HIF-1a/HO-1 dependent pathway. Of note, DEX as a specific agonist of α2-adrenoceptor exerted efficacy of decreased sympathetic tone while increased vagal tone, thus, further studies were needed to validate the upstream mechanism of HIF-1a/HO-1pathway that DEX aimed at, and the relationship between HIF-1a/HO-1pathway and sympathetic or vagal tone in this area of research.
Acute lung injury/acute respiratory distress syndromes, as a hallmark of sepsis, is one of leading causes of mortality with extremely limited therapeutic options despite significant improvement in ICU [34]. To date, abnormal balance of mitochondrial fusion and fission probably evoked by aberrantly activated ROS has been extensively proposed in pathogenesis of sepsis-related lung injury [35]. As known, mitofusin (Mfn) 1/2 situated in mitochondrial outer membrane, and optic atrophy 1 (OPA1) situated in mitochondrial inner membrane mediated mitochondrial fusion, while mitochondrial fission is primarily regulated by dynamin related protein 1 (Drp1) and its adaptor fission 1(Fis1). Indeed, oxidative stress originated from excessive generation of mitochondrial ROS and mtDNA release induced by endotoxin caused mitochondrial dysfunction and ultrastructural damages [36,37]. And, serum levels of TOS, TAS and OSI, namely the ratio of TOS to TAS, as well as lung GSH, GSSG, GSH/GSSG ratio and H2O2 were applied in current study to dynamically reflect the oxidant/antioxidant balance. In this context, our in vivo study showed that 15mg/kg LPS administration for 12h was sufficient to cause mitochondrial oxidative damage as supported by an increase in mitochondrial ROS and mtDNA levels, lower levels of TAS, GSH and GSH/GSSG ratio while higher levels of TOS, OSI, GSSG and H2O2 correlated with status of enhanced oxidative stress. Consistently, more deformed mitochondria with disintegrated or absent cristaes were observed, and the upset balance of mitochondrial fusion/fission as indicated by reduced protein and mRNA levels of Mfn1, Mfn2, OPA1 whereas elevated protein and mRNA levels of Drp1 and Fis1 in C57BL/6J mice subjected to LPS, which was in agreement with the studies of ours and others [5,20,38]. Thus, to explore the sustain of mitochondrial dynamics in septic lung injury could benefit potential treatment strategy development.
Dexmedetomidine (DEX) as a highly selective α2-AR has been widely used during perioperative period attributed to its sedative, analgesic, sympatholytic and hemodynamic effects [39]. A growing body of studies reported that DEX has antioxidative, anti-inflammatory and antiapoptotic effects to attenuate sepsis-related lung injury in experimental models [[40], [41], [42]]. Recently, the published reports by Cui J et al. and Fu C et al. proved that DEX conferred lung protective effects against oxidative stress induced mitochondrial dysfunction and mitochondrial-dependent apoptosis [43,44]. Accordantly, we certified herein that pretreatment with DEX before LPS challenge not only elevate the viability of NR8383 alveolar macrophages but also alleviate LPS-driven pulmonary oxidative injury of C57BL/6J mice as evidenced by decreased lung injury scores, higher levels of TAS, GSH, and GSH/GSSG ratio while lower levels of TOS, OSI, GSSG and H2O2 correlated with status of inhibited oxidative stress. Moreover, DEX pretreatment upregulated the expressions of Mfn1, Mfn2, OPA1 proteins and mRNA, yet downregulated the expressions of Drp1, Fis1 proteins and mRNA, accompanied by improved mitochondrial morphology, depressed levels of mitochondrial ROS and mtDNA contents. This finding was inconsistent with the study of Zhang JR et al. [12], which probably due to higher dosage of DEX (0.5mg/kg) exerted an inhibitory effect on mitochondrial fusion during sepsis-induced lung injury. Interestingly, the consistency of HIF-1a and HO-1 proteins and mRNA expressions with the levels of mitochondrial fusion Mfn1, Mfn2, OPA1 proteins and mRNA in DEX pretreatment group has caught our attention, indicating that DEX might act on HIF-1a/HO-1pathway to regulate mitochondrial dynamics so as to mitigate septic lung injury.
HIF-1a-induced HO-1 upregulation represented one of pivotal cytoprotective mechanism against various pathophysiological process such as cerebral or hepatic ischemia/reperfusion injury, septic myocardial injury, sepsis-related lung injury and so on [14,[45], [46], [47], [48], [49]]. As such, a significant decrease in protein expression of HIF-1a along with HO-1 knockout was observed in mice with LPS injection, indicating that HIF-1a directly targeted HO-1. And, HO-1 deletion broken the impaired compensatory response induced by LPS, accompanied by aggravated lung injury, higher levels of TOS, OSI, GSSG and H2O2, while lower levels of TAS, GSH, and GSH/GSSG ratio correlated with massive oxidative stress, which proposed that HIF-1a acted at HO-1 served as a pivotal antioxidant defense system in the settings of septic lung injury. The findings of the same change trend of HIF-1a with HO-1 protein were in line with the researches of Sigala F, et al. [50]. Herein, immunofluorescence analysis showed that DEX pretreatment obviously increased the co-expression of HO-1 and HIF-1a in the nucleus following LPS challenge, suggesting HIF-1a-dependent HO-1 expression play a dominant role in DEX conferred lung protection. Moreover, HO-1 and HIF-1a were found to be colocalized in alveolar macrophages in the lung tissues from DEX-pretreated mice subjected to LPS. Thus, we chose alveolar macrophage cell line NR8383 in the current study to further explore the underlying mechanism of DEX. To further ascertain the effect of HIF-1a/HO-1pathway on DEX-mediated mitochondrial dynamics during sepsis-induced acute lung injury, we pretreated LPS-exposed HO-1-/- mice or LPS-stimulated HO-1siRNA transfected NR8383 cells with DEX and DMOG, as a competitive inhibitor of prolyl hydroxylase (PHD) to activate HIF-1a. As expected, HO-1 knockout reversed the protective efficacy afforded by DEX and DMOG pretreatment, manifested by destructed architecture of lung tissue, lower levels of TAS, higher levels of TOS and OSI, increased mitochondrial ROS production and mtDNA contents. Furthermore, pretreatment with DEX and DMOG significantly relieved mitochondrial morphology damage and improved balance of mitochondrial fusion/fission indicated by elevated Mfn1, Mfn2, OPA1 proteins and mRNA levels while reduced Drp1, Fis1 proteins and mRNA levels, which were demolished by the genetic deletion of HO-1. Additionally, the expression of HIF-1a proteins and mRNA was markedly downregulated in parallel to reduced Mfn1, Mfn2, OPA1 proteins and mRNA levels in LPS-exposed HO-1-/- mice subjected to DEX and DMOG, implying that upregulation of HIF-1a in turn boost HO-1 expression was involved in DEX-mediated modulating the balance of mitochondrial dynamics. In particular, knocking down HO-1 or HIF-1a with siRNA abrogated the cytoprotective effects that DEX had on LPS-induced oxidative injury has been underscored in vitro, which was in line with studies reported by Wang Y et al. showed that DEX alleviated LPS-induced apoptosis in macrophages by eliminating dysfunctional mitochondria [10]. Considering the plasticity of HIF-1a behaved as either protective or detrimental during inflammation or oxidative stress, loss of HIF-1a by siRNA in LPS-primed NR8383 macrophages in this research significantly blocked DEX-mediated maintenance of mitochondrial dynamics, and aggravated mitochondrial oxidative damages, as reflected by decreased viability of cells, lower levels of TAS, higher levels of TOS and OSI, enhanced levels of mitochondrial ROS and mtDNA contents. In particular, HIF-1a silencing shifted the balance of mitochondria dynamic towards fission phenotype, accompanied by downregulated expression of HO-1mRNA and protein, which declared that HO-1 acted as a direct downstream regulator of HIF-1a, and the endogenous protective role of HIF-1a/HO-1 pathway against septic lung injury. The findings were in accord with studies by Eckle T, et al. and He C, et al. [14,47]. Collectively, our results provided the first piece of evidences showing that DEX pretreatment acted via HIF-1a/HO-1 pathway to preserve the balance of mitochondrial dynamics to mitigate LPS induced lung injury in mice and LPS induced oxidative injury in NR8383 alveolar macrophages.
Of note, DMOG inhibited the activity of prolyl hydroxylase domain enzymes (PHD) and lead to stabilization and accumulation of HIF-1α protein in the nucleus [24], herein DMOG was applied in the current study to activate HIF-1α. As a substitute, it might more explicit to utilize adenovirus-induced constitutively active HIF-1α, which stabilize HIF-1-mediated pathway by self-govern regardless of endogenous HIF-1α degradation [51]. Besides, DEX as a specific agonist of α2-adrenoceptor exerted efficacy of decreased sympathetic tone while increased vagal tone, thus, further lager animal study or clinical trials should be desired for validating the upstream mechanism of HIF-1a/HO-1pathway that DEX aimed at, and the relationship between HIF-1a/HO-1pathway and sympathetic or vagal tone in this area of research. Furthermore, as the endogenous cyto-protection of HO-1 induction in bronchial epithelial cells or vascular endothelial cells have been proved [52,53], therein, using markers for labeling distinct cell types may be more strength to fully illuminate the protection mechanisms of DEX on septic lung injury. In addition, it was notice that DEX was clinically administered by a continuous intravenous infusion, however, prolonged infusion by vein did not be done instead of intraperitoneal injection of DEX in this experiment [11]. Although the translational value of DEX might be questionable, it was a preclinical study which lay the basis for using DEX as the potential drug candidate for attenuating sepsis related lung injury.
In summary, we demonstrated that DEX ameliorated endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/HO-1 signaling pathway. Given that DEX has been clinically used for its sedation and analgesia, the advancing knowledge we have provided here on the regulation of mitochondrial dynamics of DEX via activation of HIF-1α/HO-1 pathway in septic lung injury may support a novel use of DEX as a treatment for sepsis patients.
This study was supported by a grant from the Youth Project of
The authors have declared that they have no conflict of interest.
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
We express our gratitude to Dr. Daqing Ma from Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Chelsea and Westminster Hospital for his critical comment during manuscript preparation.
 Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/HO-1 signaling pathway
Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/HO-1 signaling pathway
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
