
Coronaviruses (CoVs) are a group of single stranded RNA viruses, of which some of them such as SARS-CoV, MERS-CoV, and SARS-CoV-2 are associated with deadly worldwide human diseases. Coronavirus disease-2019 (COVID-19), a condition caused by SARS-CoV-2, results in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) associated with high mortality in the elderly and in people with underlying comorbidities. Results from several studies suggest that CoVs localize in mitochondria and interact with mitochondrial protein translocation machinery to target their encoded products to mitochondria. Coronaviruses encode a number of proteins; this process is essential for viral replication through inhibiting degradation of viral proteins and host misfolded proteins including those in mitochondria. These viruses seem to maintain their replication by altering mitochondrial dynamics and targeting mitochondrial-associated antiviral signaling (MAVS), allowing them to evade host innate immunity. Coronaviruses infections such as COVID-19 are more severe in aging patients. Since endogenous melatonin levels are often dramatically reduced in the aged and because it is a potent anti-inflammatory agent, melatonin has been proposed to be useful in CoVs infections by altering proteasomal and mitochondrial activities. Melatonin inhibits mitochondrial fission due to its antioxidant and inhibitory effects on cytosolic calcium overload. The collective data suggests that melatonin may mediate mitochondrial adaptations through regulating both mitochondrial dynamics and biogenesis. We propose that melatonin may inhibit SARS-CoV-2-induced cell damage by regulating mitochondrial physiology.
Mitochondria are double membrane-bound organelles, which are closely integrated with cellular metabolism and ultimately with cellular survival (Norberg, Orrenius, & Zhivotovsky, 2010; Rambold & Lippincott-Schwartz, 2011). A major function attributed to the mitochondria is the generation of cellular energy in the form of adenosine triphosphate (ATP) through respiration. Furthermore, mitochondria play important roles in cell signaling pathways including signaling for calcium homeostasis, inflammation, proliferation, autophagy and apoptosis (Schapira, 2006; Smith, Hartley, Cocheme, & Murphy, 2012). The mitochondrial mass of cells varies depending on cellular energy demands; cells with high metabolic activity have the largest amount of mitochondria, e.g., cardiomyocytes. Cellular mitochondrial content is adjusted through the competing processes of organelle biogenesis and degradation by complex intracellular and extracellular signaling pathways (Scarpulla, Vega, & Kelly, 2012). The outer mitochondrial membrane contains proteins that form voltage-dependent anion-selective channels, allowing the free translocation of uncharged molecules up to ~5,000 Daltons into the intermembrane space in the open configuration (Mokranjac & Neupert, 2009). The inner mitochondrial membrane has restricted permeability and is loaded with protein complexes including reduced nicotinamide adenine dinucleotide (NADH) dehydrogenase-ubiquinone oxidoreductase (complex I), succinate dehydrogenase-ubiquinone oxidoreductase (complex II), ubiquinone-cytochrome c oxidoreductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase (complex V); these proteins are involved in the ATP synthesis through oxidative phosphorylation (Green & Kroemer, 2004; Green & Reed, 1998). During electron transport through the respiratory chain, protein complexes (I, II, IV) use donated electrons to pump protons into the intermembrane space, creating a proton gradient which drives formation of ATP by complex V (Green & Kroemer, 2004; Green & Reed, 1998).
Mitochondria contain several circularDNA complexes (mitochondrial DNA, mtDNA), each molecule of which contains 37 genes encoding 13 key subunits of respiratory-chain complexes and the necessary RNA machinery including two ribosomal RNAs (16S rRNA and 12S rRNA) and 22 transfer RNAs, which are required for translation of mtDNA transcripts (Wallace, 2005). The mitochondrial DNA genome encodes components of the respiratory-chain complexes I, III, IV, and V. Complex II subunits and remaining mitochondrial proteins are encoded by the nuclear genome, synthesized as precursors on cytosolic ribosomes and subsequently imported into the correct mitochondrial compartment through specific protein translocation machineries. These protein translocation processes are part of a dynamic network which perform functions related to mitochondrial biogenesis, dynamics, and quality control under physiological and pathophysiological conditions (Wallace, 2005; Wiedemann & Pfanner, 2017). The majority of mitochondrial proteins are synthesized as the unfolded form and then transported into the matrix where folding ensues; failure of this complex series of events results in the accumulation of unfolded or misfolded proteins within mitochondria triggering disruption of mitochondrial functions (Tatsuta, 2009). Mitochondrial protein quality control is essential to maintain cellular homeostasis under normal and stressful conditions. Several surveillance mechanisms monitor mitochondrial integrity and protein-folding homeostasis at molecular, organellar, and cellular levels.
The first line of quality control is monitored by an elaborate network of molecular chaperones and proteases, which prevents the accumulation of misfolded proteins in the various mitochondrial compartments (Baker, Tatsuta, & Langer, 2011). The second line of quality control occurs at the organellar level; because of the dynamic nature of this organelle, mitochondria constantly undergo fission and fusion events through dynamin-related GTPases in the outer and inner mitochondrial membrane (Hoppins, Lackner, & Nunnari, 2007). This dynamic process regulates mitochondrial function by recruiting healthy mitochondria to subcellular compartments, content exchange within a mitochondrial network, changes in mitochondrial morphology and clearance of defective organelles via mitophagy (Twig et al., 2008 ). Finally, at the cellular level, the quality control is maintained by programmed cell death (apoptosis); when molecular damage becomes extreme and the majority of organelles become irreparable, cell death occurs through apoptosis (Q. Cai & Tammineni, 2016). Disruptions in any of these processes results in the impairment of mitochondrial function and cellular metabolism, leading to disturbed homeostasis and disease (H. Chen & Chan, 2009). Herein, we briefly review the potential effect of coronaviruses (CoVs) on the mechanisms involved in the mitochondrial protein quality control and, thereafter, describe the potential modulatory effect of melatonin on these molecular pathways.
Coronaviruses (CoVs) are a group of enveloped, positive-sense, and single stranded RNA viruses of ~30 kb, which share a common protein structure composed of spike (S), envelope (E), membrane (M) and nucleocapsid (N). Coronaviruses are classified into four (α, β, γ, and δ) genera; to date, seven members of α- and β-CoVs are identified as human pathogens. Four viruses (229E, NL63, HKU1, and OC43) express low pathogenicity and generally induce mild respiratory symptoms. The other three (severe acute respiratory syndrome coronavirus (SARS-CoV), middle-east respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2) are associated with severe respiratory comorbidities, and caused large-scaled pandemics in 2003, 2012 and late 2019, respectively (J. F.-W. Chan et al., 2020; de Wilde, Snijder, Kikkert, & van Hemert, 2017; R. Lu et al., 2020; Weiss & Leibowitz, 2011). During their emergence, SARS-CoV initially infected more than 8000 individuals (with 10% mortality) while MERS-CoV reportedly infected ~2500 people (with 35% mortality). As of September of 2020, outbreak of the new identified member of β-CoV genera, SARS-CoV-2, infected and estimated 26 million with a cumulative mortality rate of ~5% (J. F.-W. Chan et al., 2020; de Wilde et al., 2017; R. Lu et al., 2020; Weiss & Leibowitz, 2011). SARS-CoV-2 has a genome sequence with ~80% identity to SARS-CoV and ~50% to MERS-CoV. Coronavirus disease-2019 (COVID-19), a condition caused by SARS-CoV-2, is highly pathogenic and mainly spreads via respiratory droplets. COVID-19 patients manifest different non-specific symptoms including fever, cough, myalgia, fatigue, and diarrhea. However, severe progression of COVID-19 results in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) associated with high mortality in elderly and people with underlying diseases (Ahmed, 2020; Salehi, Abedi, Balakrishnan, & Gholamrezanezhad, 2020).
Viral respiratory infections such as CoVs promote the generation of reactive oxygen species (ROS) and inflammatory cytokines (Khomich, Kochetkov, Bartosch, & Ivanov, 2018; Yang, 2020). In peripheral blood mononuclear cells (PBMC) of SARS patients, SARS-CoV up-regulates the expression of oxidative stress-sensitive genes, heat shock proteins, transcription factors, and cytokines (Shao et al., 2006). Zinc finger protein 331 (ZNF331) and FBJ murine osteosarcoma viral oncogene homolog (FOS) are two redox-sensitive transcription factors up-regulated by SARS-CoV. The transcription factor FOS is a component of redox-sensitive transcription factor complex activator protein 1 (AP-1); AP-1 pathway is also activated by nucleocapsid (N) protein of SARS-CoV (Shao et al., 2006). Angiotensin II (Ang-II), a component of renin–angiotensin system, triggers severe acute lung injury in SARS-CoV and SARS-CoV-2; this protein is activated by mitochondrial-induced ROS and induces the activation of AP-1 by binding to the Ang-II type 1 receptor (AT1R) (Gurwitz, 2020; D. Liu, Gao, Roy, Cornish, & Zucker, 2006; Wu et al., 2005). As a result of Ang-II activation, AP-1 up-regulates the expression of AT1R leading to the exacerbation of Ang-II signaling (Haack, Mitra, & Zucker, 2013). The angiotensin converting enzyme 2 (ACE2) is used by SARS-CoV and SARS-CoV-2 to enter ACE2-expressing cells including type II alveolar epithelial cells in the lung, esophageal upper epithelial cells and absorptive enterocytes of ileum and colon (Hoffmann et al., 2020; H. Zhang et al., 2020; P. Zhou et al., 2020). Although SARS-CoV and SARS-CoV-2 use ACE2 to gain initial entry into cells, continued viral infection and replication is associated with the down-regulation of ACE2 expression resulting in the impairment of enzyme protective effects in organs (Gurwitz, 2020; Kuba et al., 2005; Y. Liu et al., 2020). Due to the role of ACE2 in the degradation of Ang-II, down-regulation of ACE2 activity leads to the Ang-II accumulation and local renin–angiotensin–aldosterone system (RAAS) activation in lungs. The elevated level of plasma Ang-II has been detected in patients with COVID-19, with the level of this protein being linearly associated with the total viral load and degree of lung injury (Y. Liu et al., 2020). Blockade of RAAS in mice exposed to SARS-CoV spike protein limits virus-induced acute lung injury (Kuba et al., 2005). Findings from these studies suggest that restoration of ACE2 and inhibition of Ang-II may be useful for treatment of COVID-19 disease.
Up-regulation of the expression of mitochondrial genes in PBMC implies that SARS-CoV markedly impacts mitochondrial function (Shao et al., 2006). SARS-CoV accessory protein 3b, as one of the interferon antagonist encoded by SARS-CoV genome, is reported to be located in the mitochondria (Q. Li et al., 2005). Furthermore, SARS-CoV non-structural protein 10 (NSP10), a critical co-factor for activation of multiple replicative enzymes, specifically interacts with the NADH-ubiquinone oxidoreductase chain 4L (ND4L) protein, a subunit of the respiratory chain complex I, and with cytochrome oxidase II, the second subunit of respiratory complex IV, in the mitochondria (Yuan et al., 2006). Master Regulator Analyses suggests that the Complex I subunit NDUFA10 is down-regulated in bronchial epithelial cells infected by MERS and SARS-CoV-2 (Guzzi, Mercatelli, Ceraolo, & Giorgi, 2020). This SARS-CoV-2-induced mitochondrial dysfunction triggers mitochondrial ROS production. Overproduction of ROS induces the expression of hypoxia-inducible factor-1α (HIF-1α), which this protein strongly induces glycolysis and IL-1β transcription in SARS-CoV-2-infected monocytes; glycolytic flux is reported to be sufficient for replication of SARS-CoV-2 in monocytes and response of SARS-CoV-2-infectd monocyte. Treatment of CoV-2-infected monocytes with antioxidants can inhibit up-regulation of HIF-1α and its target genes including ACE2 and IL-1β. Since the level of HIF-1α increases in the blood monocytes of severe COVID-19 patients, the use of antioxidant may be a therapeutic strategy to inhibit mtROS/HIF-1α/glycolysis axis in COVID-19 patients (Codo et al., 2020).
Mitochondrial morphodynamics influence the innate immune signaling through enhancing type I interferon (IFN-I) synthesis. This function is mediated by the activation of mitochondrial associated antiviral signaling (MAVS) by cytosolic pattern recognition receptors (PRRs) including RIG-I like receptors (RLRs) such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene-5 (MDA5); these genes recognize pathogen associated molecular patterns (PAMPs) in the form of viral double-stranded RNA (dsRNA) motifs (Khan, Syed, Kim, & Siddiqui, 2015); this suggests a relevant interplay between mitochondria and innate immunity. During the primary responses of host cells, RIG-I and MDA5 detect cytosolic RNA viruses, which contributes to the formation of MAVS signal complex on mitochondria. Activation of MAVS results in the activation of TANK-binding kinase 1 (TBK1) and subsequent phosphorylation of interferon regulatory factor 3 (IRF3), proteins typically associated with cytosolic chaperone Hsp90. Upon activation, IRF3 translocates into the nucleus and promotes the early production of IFN-I, establishing antiviral state by inhibiting viral replication (X.-Y. Liu, Wei, Shi, Shan, & Wang, 2010).
SARS-CoV open reading frames (ORF)-9b protein localizes in mitochondria and targets the mitochondrial MAVS signalosome; ORF-9b promotes the degradation of MAVS leading to the loss of TRAF3 and TRAF6 signaling molecules, which play essential roles in antiviral responses (Shi et al., 2014); TRAF3 functions as an adapter protein, bridging MAVS with the downstream signaling complex including TBK1, which is essential for IRF3 activation (Lui et al., 2016). SARS-CoV E, ORF3a and ORF8b proteins are recognized by PRRs such as NOD-like receptors protein 3 (NLRP3) on the cell membrane; NLRP3 is recruited to MAVS to form an NLRP3 inflammasome accompanied by adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) and pro-caspase-1 (I.-Y. Chen, Moriyama, Chang, & Ichinohe, 2019; Nieto-Torres et al., 2015; Shi, Nabar, Huang, & Kehrl, 2019). The activation of the NLRP3 inflammasome is also reported in hDPP4-transgenic mice and human macrophages infected by MERS-CoV (Y. Jiang et al., 2019). This contributes to the activation of caspase-1 which catalyzes the proteolytic activation and secretion of proinflammatory cytokines including interleukin 1 beta (IL-1β) and IL-18. The active IL-1β and IL-18 are released into the extracellular space and recruit inflammatory cells which accumulate and exaggerate the inflammatory response, thereby inducing cell pyroptosis, a highly inflammatory form of programmed cell death (Yang, 2020). Compression of the mitochondria localized ORFs encoded by SARS-CoV and SARS-CoV-2 revealed that except for ORF3b, the amino acid sequences of SARS-CoV-2 ORFs are similar to SARS-CoV ORFs (ORF7a, -8a, and -9b); these ORFs proteins are localized in the mitochondria and promote viral replication (Singh, Chaubey, Chen, & Suravajhala, 2020b). These similarities suggest that the patterns and modes of SARS-CoV-2 interaction with host antiviral defense may be similar. The storm of proinflammatory cytokines observed in the severe cases of COVID-19 indicates that SARS-CoV-2 may induce the activation of NLRP3 inflammasome, thereby causing cell pyroptosis, especially in lymphocytes (Yang, 2020).
Melatonin is a molecule synthetized by the pineal gland and locally by perhaps in all organs and cells. It has been identified in many cells including blood cells, bone marrow, cochlea, retina, lens, accessory sex glands, skin, etc. The evidence is now reasonably strong that the mitochondria of all cells probably synthesize melatonin and only 5% or less of the total melatonin produced comes from the pineal gland (Hosseinzadeh et al., 2019; Pourhanifeh, Dehdashtian, Hosseinzadeh, Sezavar, & Mehrzadi, 2020; Russel J Reiter et al., 2019)
Exogenous melatonin has been showed a number of clinical effects in various disorders including sleep disorders, chronic obstructive pulmonary disease and hypertension (Andersen, Gögenur, Rosenberg, & Reiter, 2016a). Melatonin in oral immediate-release formulations is rapidly absorbed from the small intestine by first-order kinetics. Time to maximal concentration and elimination half-life are approximately 50 and 45 min, respectively, following oral administration. In oral slow-release formulations of melatonin, time to maximal concentration and elimination half-life prolongs up to 167 and 91 min, respectively (Harpsøe, Andersen, Gögenur, & Rosenberg, 2015). Maximal serum concentration, volume of distribution and clearance show extensive variation in oral administration of melatonin; these inter-individual variations may obviously result from differences in absorption, distribution, metabolism or excretion of the drug. The average bioavailability of orally administered melatonin is approximately 15 %; this low degree of bioavailability may result from poor oral absorption, extensive first-pass metabolism in the liver or a combination of both (Harpsøe et al., 2015). Data from animal and human studies indicates that melatonin has high biological safety, even in extreme doses. Randomized clinical studies demonstrate mild adverse effects including dizziness, headache, nausea and sleepiness following the long-term administration of melatonin (Andersen, Gögenur, Rosenberg, & Reiter, 2016b). A randomized trial has reported that orally administrated melatonin in a dose of 25 mg/day for 6 months could significantly reduce serum concentrations of inflammatory cytokines including IL-6 and IL-1β (Sánchez-López et al., 2018). Furthermore, severely affected ALS patients treated with 300 mg/day melatonin for 2 years did not show any adverse effects (Weishaupt et al., 2006). Due to the destructive inflammation in severe COVID-19 patients, the sufficient amounts of melatonin are required to overcome SARS-CoV-2-induced pathological alterations; this goal is not achieved with physiological levels of melatonin (Kleszczyński, Slominski, Steinbrink, & Reiter, 2020). Administration of melatonin in mice infected with H1N1 virus could reduce the mortality in a dose of 100 mg/kg/day (200 mg/kg/48 h) (Huang et al., 2019). Conversion of murine dose to the human dose results in a human equivalent dose for melatonin of 8.1 mg/kg/day, a daily dose of 600 mg for a 75-kg human (Kleszczyński et al., 2020). Similar dose of melatonin (8.1 mg/kg/day and 8.2 mg/kg/day) has been used in a prospective nonrandomized nonblind case–control study on neonates with neonatal sepsis (El-Gendy, El-Hawy, & Hassan, 2018). Since no adverse effects have been reported by this clinical study, it is estimated that a daily dose of 600 mg of melatonin may be useful for treatment of deadly viral infectious diseases, including COVID-19 (Kleszczyński et al., 2020). A case series study performed by Castillo et al. evaluated the effect of melatonin (36–72 mg/day) as an adjuvant therapy in 10 patients admitted with COVID19 pneumonia. In 7 patients, who COVID-19 was diagnosed by positive SARS-CoV-2 reverse transcription polymerase test, the hospitalization lasted an average of 8.6 days after initiation of melatonin treatment. In 3 patients, test was negative, but it was deemed to be false because of the history, symptoms, chest imaging findings and the level of inflammatory parameters in patients; the hospitalization in these patients lasted an average of 7.3 days after initiation of melatonin treatment. The average of hospitalization was 13 days for patients who did not receive melatonin. Sleepiness was the only side-effect reported in patient treated with melatonin. These findings suggest that melatonin may be a promising agent as an adjuvant therapy against COVID-19; however, further studies are needed to evaluate the effect of different doses of melatonin in patients with severe COVID-19 (Castillo et al., 2020).
As a potent anti-oxidant, melatonin protects cells from oxidative stress-induced damage through detoxifying ROS and elevating cellular anti-oxidant enzymes (Dehdashtian, Pourhanifeh, Hemati, Mehrzadi, & Hosseinzadeh, 2020; Shahriari et al., 2020). Melatonin regulates various signaling pathways involved in inflammation, apoptosis, autophagy, angiogenesis, etc. (M. H. Mehrzadi, Hosseinzadeh, Juybari, & Mehrzadi, 2020). Melatonin influences cell physiology through both receptor-dependent and receptor-independent actions; the receptor-independent actions of melatonin are mediated by its ability to scavenge free radicals and its receptor-dependent actions are related to an interaction with membrane (MT1 and MT2) or nuclear RAR-related orphan receptors (RORs) and the retinoid Z receptors (RZRs)) receptors (Russell J Reiter et al., 2007). The MT1 and MT2 receptors are membrane-bound G protein-coupled receptors (GPCRs), which are suggested to express in various tissues, perhaps, in almost all nucleated cells (Ruediger Hardeland et al., 2011). The MT2 receptors are reported to express in the nucleus, in addition to the cytoplasmic membrane (Ahluwalia, Brzozowska, Hoa, Jones, & Tarnawski, 2018a). Furthermore, MT1 and MT2 receptors are present in mitochondrial membranes, indicating the role of melatonin in the mitochondrial signaling (Ahluwalia, Brzozowska, Hoa, Jones, & Tarnawski, 2018b). However, the expression pattern of MT1 and MT2 receptors varies depending on the cell-type and species; for example, mouse cerebellar granule cells exclusively express MT1 receptor, while the same cells in human express both MT1 and MT2 receptors (Imbesi, Uz, Dzitoyeva, Giusti, & Manev, 2008a). Furthermore, the expression pattern of MT1 and MT2 receptors changes during development, aging and various pathological conditions (Zlotos, Jockers, Cecon, Rivara, & Witt-Enderby, 2014). These melatonin membrane receptors have the ability to form homo-dimers or hetero-dimers, which these dimerizations may alter receptor pharmacology, signaling and regulation (Imbesi, Uz, Dzitoyeva, Giusti, & Manev, 2008b; Jockers, Maurice, Boutin, & Delagrange, 2008a). The MT1 and MT2 receptors couple to pertussis toxin–sensitive Gi and –insensitive Gq proteins. Binding of melatonin to these receptors leads to the inhibition of forskolin-stimulated cyclic adenosine monophosphate (cAMP) formation, protein kinase A signaling and phosphorylation of the cAMP-responsive element binding (CREB) protein through Gi protein activation (Hemati et al., 2020; Jockers, Maurice, Boutin, & Delagrange, 2008b). Both MT1 and MT2 receptors activate phospholipase C pathway leading to the up-regulation of inositol triphosphate (IP3) and 1, 2-diacylglycerol (DAG) levels through activation of Gq protein (Ng, Leong, Liang, & Paxinos, 2017). The MT1 and MT2 receptors mediate phosphorylation of c-Jun N-terminal kinase (JNK) through activation of Gi and Gq proteins (A. S. L. Chan et al., 2002). The MT1 receptor has been reported to activate mitogen-activated protein kinase kinases 1/2/ extracellular signal-regulated kinases 1/2 pathway by Gi protein activation, while MT2 receptor activates this pathway through cooperative activation of Gi and Gq proteins (M. Chen et al., 2020). Furthermore, MT2 receptors inhibit cyclic guanosine monophosphate/protein kinase G pathway upon Gi activation (Jockers et al., 2008a). Since dimerization of MT1 and MT2 receptors alters signaling of these receptors, the responsiveness of cells to melatonin depends on the presence or absence of MT1/MT2 heteromers. The effect of melatonin in the reduction of forskolin-simulated cAMP production occurs only in cells expressing both MT1 and MT2 receptors (Cecon, Oishi, & Jockers, 2018). In cells expressing both MT1 and MT2 receptors, melatonin at a low nM concentration inhibits extracellular signal-regulated kinase signaling rather than its stimulation, while silence of either receptor results in the stimulatory response of melatonin (Cecon et al., 2018).
Melatonin is synthesized from the amino acid L-tryptophan in a four-step process; (I) formation of 5-hydroxytryptophan from tryptophan by tryptophan hydroxylase, (II) decarboxylation of 5-hydroxytryptophan by l-aromatic amino acid decarboxylase to form 5-hydroxytryptamine (5-HT, also called serotonin), (III) formation of N-acetyl-5-hydroxytryptamine (N-acetyl serotonin) from serotonin by arylalkylamine N-acetyltransferase/ serotonin N-acetyltransferase (AANAT/SNAT) and (IV) conversion of N-acetyl serotonin to melatonin (N-acetyl-5-methoxytryptamine) by N-acetylserotonin-O-methyltransferase (ASMT) (Bahrami et al., 2018). The localization of AANAT/SNAT enzyme in mitochondrial matrix and intermembrane space suggests that mitochondria have the capacity to synthesize melatonin (Kerenyi, Balogh, Somogyi, & Sotonyi, 1979; Kerenyi, Sotonyi, & Somogyi, 1975). N-acetyl-coenzyme A (acetyl CoA), which is the necessary co-factor for rate limiting enzyme AANAT/SNAT, is mainly synthesized in the mitochondria (Russel J Reiter et al., 2019). The concentration of acetyl CoA in the mitochondria is much higher than other cellular compartments such as cytosol; the mitochondrial concentration of acetyl CoA can satisfy the Km of the AANAT (D.-X. Tan, Manchester, Qin, & Reiter, 2016). Furthermore, oligopeptide transporters (PEPT) 1/2 exist in the mitochondrial membrane, where they presumably facilitate the rapid transport of melatonin into the mitochondria against a gradient (Anderson & Reiter, 2019); the accumulation of melatonin in the mitochondria would be expected to prevent mitochondrial impairment in normal cells and to kill tumor cells. In the mitochondria, melatonin also promotes pyruvate conversion to acetyl-CoA through inhibiting mitochondrial enzyme pyruvate dehydrogenase kinase (PDK); an enzyme that inhibits pyruvate dehydrogenase complex (PDC) controlling the synthesis of acetyl-CoA from pyruvate. In addition to being a co-factor for the AANAT/SNAT enzyme, acetyl-CoA improves oxidative phosphorylation in the mitochondria (Russel J Reiter et al., 2019).
The existence of N(1)-acetyl-N(2)-formyl-5-methoxykynuramine (AFMK), the major metabolite of melatonin, in the mitochondria proves that mitochondria also is a site of melatonin metabolism; AFMK is produced in the mitochondria through a pseudo-enzymatic process by interaction of melatonin with cytochrome c, an iron-containing hemoprotein. In the mitochondria, cytochrome c interacts with H2O2 and is converted to oxoferryl cytochrome c with pseudoperoxidase activity. Oxoferryl derivative of cytochrome c oxidizes organic molecules such as melatonin. The interaction between oxoferryl cytochrome c and melatonin results in the restoration of normal redox cycle of cytochrome c, essential for the maintenance of mitochondrial bioenergetics under physiological and pathological conditions (Semak et al., 2005; D.-X. Tan, Manchester, Esteban-Zubero, Zhou, & Reiter, 2015). Furthermore, melatonin can directly interact with ROS produced by mitochondria to generate a variety of active metabolites with anti-oxidant activity (Hosseinzadeh, Javad-Moosavi, Reiter, Yarahmadi, et al., 2018).
Cytokine storm is believed to be one of the major causes of ARDS and multiple-organ failure in viral infections, a situation which have been reported in some cases of COVID-19 (Chousterman, Swirski, & Weber, 2017; L. Xu et al., 2019). Excessive release of pro-inflammatory cytokines amplifies immune responses, but also causes serious cell, organ and tissue damage (Huo et al., 2018; Oldstone & Rosen, 2014). Released components from injured cells (mitochondrial DNA, cardiolipin, cytochrome c and segments of nuclear DNA) along with pathogen remnants are recognized by toll-like receptors (TLR4,7, and 9) as DAMPs and PAMPs. Furthermore, Cyclic GMP/AMP synthase triggers a further large-scale pro-inflammatory cytokine release known as the “secondary cytokine storm”. Repetition of this vicious cycle results in extensive apoptosis, pyroptosis, and necrosis even of healthy cells (Huo et al., 2018). In addition to these events, macrophages/monocytes switch their metabolism from mitochondrial oxidative phosphorylation to aerobic glycolysis upon activation by PAMP or DAMP (Russel J Reiter, Ma, & Sharma, 2020). This change allows these cells to provide metabolic precursors required for hyper cellular proliferation and cytokine synthesis and release. PAMP and DAMP exaggerated innate immune responses can effectively be regulated by melatonin (Srinivasan, Mohamed, & Kato, 2012; Sung et al., 2018). In addition, reduced tissue and organ damage associated with higher survival rates have been observed in septic animal models (L. Xu et al., 2019) and in human subjects (Gitto et al., 2001) after treatment by melatonin.
Clinical characteristics of COVID-19 include elevated levels of pro-inflammatory cytokines (IL-10, IL-6, and TNF-α) accompanied by lymphopenia in severe cases. The severe progression of the disease may lead to ALI/ARDS associated with high mortality in elderly and people with underlying disease (Ahmed, 2020; Salehi et al., 2020). Advanced age COVID-19 patients are more vulnerable to the disease due to the weakened immune responses (Varricchi et al., 2020). Moreover, a number of studies have documented a significant decrease in physiological levels of melatonin in the elderly (Scholtens, van Munster, van Kempen, & de Rooij, 2016; Z.-Y. Zhao, Xie, Fu, Bogdan, & Touitou, 2002) and it is suggested that higher endogenous levels of melatonin may play a major positive role in healthy ageing and longevity (Rüdiger Hardeland, 2013; Poeggeler, Reiter, Tan, Chen, & Manchester, 1993). Thus, we hypothesize that lower concentrations of melatonin and lack of well-functioning immune system in advanced age patients, as poor prognostic factors, make them more vulnerable to COVID-19.
Melatonin influences immune-inflammatory processes through regulating immune cell function where it acts as both an activator and an inhibitor of inflammatory and immune processes (Carrillo-Vico, Guerrero, Lardone, & Reiter, 2005). Immune cells produce melatonin via enzymatic pathways independent of the pineal gland (P. Lardone, Carrillo-Vico, Molinero, Rubio, & Guerrero, 2009). Human lymphoid cells are an important physiological source of melatonin; both resting and phytohemagglutinin-stimulated human lymphocytes have the necessary machinery to synthesize and release large amounts of melatonin, several times greater than the nocturnal physiological levels in human serum. The endogenous synthesis of melatonin by lymphocytes is involved in the regulation of IL-2 production acting as an intracrine, autocrine, and/or paracrine substance (Carrillo-Vico et al., 2004; P. J. Lardone et al., 2006). The regulatory effect of melatonin on the function of immune cells including T-lymphocytes, natural killer (NK) cells, eosinophils, macrophages, and mast cells may be due to its direct action on melatonin receptors (Calvo, Gonzalez-Yanes, & Maldonado, 2013).
Melatonin exhibits exaggerated anti-inflammatory effects by inhibition of the production of pro-inflammatory cytokines, prostaglandins, adhesion molecules and cyclooxygenase-2 (Favero, Franceschetti, Bonomini, Rodella, & Rezzani, 2017; Pourhanifeh, Hosseinzadeh, Dehdashtian, Hemati, & Mehrzadi, 2020). Scavenging free radicals and enhancing the activity of antioxidant enzymes could be mechanisms by which melatonin inhibits ROS-mediated activation of NF-κB, leading to the inhibition of NF-κB nuclear translocation and subsequent inflammatory cytokine expressions (Maldonado, García-Moreno, González-Yanes, & Calvo, 2016). Furthermore, melatonin directly inhibits the NLRP3 inflammasome assembly and blunts NF-κB/NLRP3 inflammasome signaling activation through stimulation of nuclear (ROR-α) receptors (García et al., 2015), a possible mechanism by which melatonin exerts its mitochondrial protective effects (Ortiz et al., 2015). Inhibition of NLRP3 inflammasome assembly by melatonin results in the reduction of caspase-1 and IL-1β activation (Fernández-Gil et al., 2017; Y. Zhang et al., 2016). Based on anti-inflammatory, anti-oxidative and immunoregulatory features, melatonin is suggested to be protective against coronavirus-induced lung inflammation (Juybari, Pourhanifeh, Hosseinzadeh, Hemati, & Mehrzadi, 2020; Russel J Reiter et al., 2020).
Chaperone proteins and ATP-dependent proteases form an interconnected functional network that monitors and maintains protein homeostasis in mitochondria (Fig 1 ) (Voos, 2013). Only 13 of the human mitochondrial proteins are encoded by mtDNA and synthesized within this organelle. The vast majority of the mitochondrial proteins are encoded by the nuclear genome and synthesized by cytoplasmic ribosomes and subsequently imported into the organelle (Chacinska, Koehler, Milenkovic, Lithgow, & Pfanner, 2009). Nuclear-encoded proteins are presented in an unfolded state for efficient translocation across the mitochondrial membranes. This translocation proceeds through narrow pores formed by tightly-gated translocons including TOM (translocase of the outer membrane) complex and TIM (translocase of the inner membrane) complex called TIM23. TOM complex consists of central pore forming Tom40 and receptor proteins Tom20, Tom22 and Tom70 as well as the three small Tom proteins Tom5, Tom6 and Tom7; small Tom proteins are not essential for TOM functions but are required to promote assembly and stability of the TOM complex. Subunits of the TIM23 complex are divided into two groups of proteins, one of which forms the membrane sector of the complex (Tim17, Tim 21, Tim23, Tim50 and Mgr2), called as the TIM23 core channel, and the other which pairs with the presequenced translocase-associated motor (PAM), referred to as an import motor, that drives the translocation into the matrix space (Tim14/Pam18, Tim16/Pam16, Pam 17, Tim44, mtHsp70 and Mge1) (Craig, 2018; Horvath et al., 2015; van der Laan, Hutu, & Rehling, 2010).

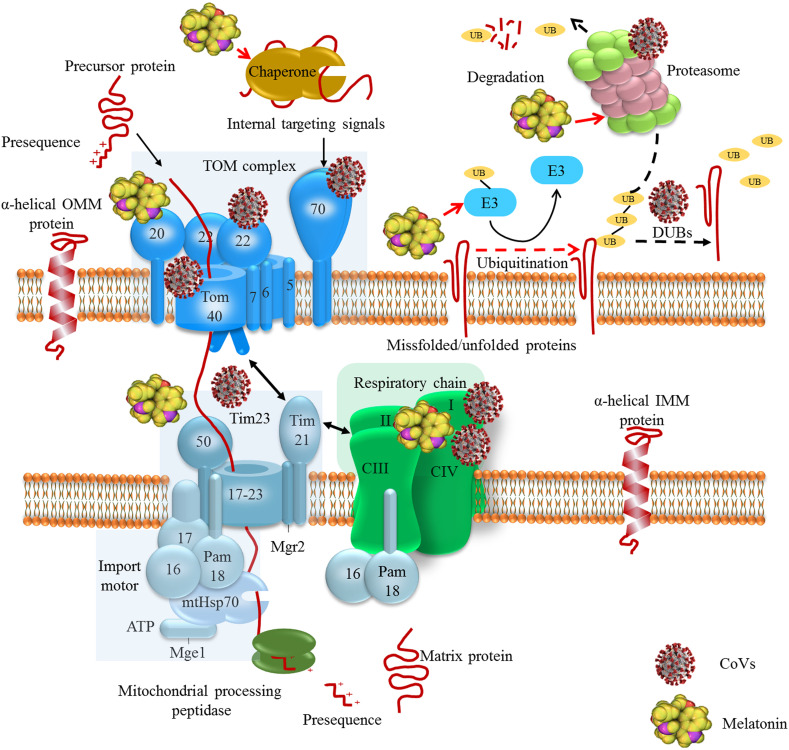
The employment of chaperone–protease networks by CoVs and the regulatory effects of melatonin. Coronaviruses such as SARS-CoV and SARS-CoV-2 employ mitochondrial protein translocation machinery to inhibit antiviral cellular responses and maintain virus replication. SARS-CoV-2 interacts with Tom complex components. Interaction with may inhibit the interaction of Tom70 with MAVS and induction of antiviral cellular responses (X.-Y. Liu et al., 2010). Polyproteins of SARS-CoV and MERS-CoV have deubiquitinating activity, which influence infected cell ubiquitination. SARS-CoV interacts with proteasome through its nucleocapsid protein to suppress proteolytic activity of proteasomes; this contributes to the inhibition of proteolysis of viral proteins and also suppression of host misfolded protein degradation (J. Lei et al., 2014; Lindner et al., 2005; Q. Wang et al., 2010). Melatonin affects chaperone–protease networks through regulating the expression of HSPs, Tom20 and Tim23, and interacting with UPS machinery, this effects result in the inhibition of aberrant protein accumulation in cells (Bonior et al., 2005; Corpas et al., 2018; Kireev et al., 2014; Martins Branco et al., 2010; S. Mehrzadi, Hemati, et al., 2020; Vriend & Reiter, 2014).
Translocation of mitochondrial proteins through the TIM23 is driven by the membrane potential across the inner membrane and the hydrolysis of ATP in the matrix (Banerjee, Gladkova, Mapa, Witte, & Mokranjac, 2015). Mitochondrial precursor proteins have a strong tendency to expose hydrophobic regions leading to the unwanted protein–protein interactions and protein aggregation. Therefore, these precursor proteins need to bind a cytosolic chaperone for maintenance in an unfolded import-competent conformation before interaction with TOM translocase (Jensen & Johnson, 1999). Molecular chaperones that belong to the Hsp70, Hsp90 and mitochondrial import stimulation factor (MSF) families facilitate translocation and folding reactions of mitochondrial proteins (Voos, 2013). These proteins deliver precursors to the Tom70 receptor recognizing signals embedded in their internal sequence of membrane-targeted precursors (Fan & Young, 2011). Mitochondrial precursor proteins bind to Tom20 and Tom70 (Hsp70-dependent binding) or Tom20 alone (Hsp70-independent binding) and pass through Tom40 channel after interaction with Tom22 and Tom5 (Endo & Yamano, 2010). Cytosolic chaperone MSF binds to Tom70 and delivers precursor proteins to Tom20 and Tom22 upon ATP hydrolysis (Endo & Yamano, 2010). Upon translocation through Tom, presequenced-containing proteins are directed into the TIM23 complex. The channel-forming Tim23 protein, Tim17 and Tim50 mediate membrane potential-dependent insertion of precursor proteins into the inner membrane (Mokranjac & Neupert, 2010). The Tim21 and Mgr2 subunits of the TIM23 complex are suggested to be involved in the interaction of the TIM23 complex with TOM complex as well as with respiratory chain complexes; TIM23-respiratory chain coupling may be important for the insertion of precursor proteins into the inner membrane when the energetic activity of mitochondria is reduced (Ieva et al., 2013; Mokranjac & Neupert, 2010).
The import motor PAM is crucial for the completion of precursor protein import to the matrix in an ATP-dependent manner; Tim44 is a coordinating subunit at the matrix side which binds to Tim17–Tim23 core of the complex. Furthermore, Tim44 interacts with mtHsp70 and recruits the chaperone and its cochaperones (Pam18, Pam16, Pam17 and Mge1) to the translocation channel (Banerjee et al., 2015). The sub-complex Pam17 is proposed to be involved in the organization and recruitment of Pam18–Pam16 to the Tim17–Tim23 core of the TIM23 complex (Van Der Laan et al., 2005). Sub-complexes Pam18 and Pam16 form a stable heterodimer stimulating the ATPase activity of mtHsp70 coupled to multiple import reaction cycles (Y. Li et al., 2004). Once proteins enter mitochondria, mtHSP70 (mitochondrial heat shock 70 kDa protein) mediates the ATP-dependent import of mitochondrial proteins into the mitochondrial matrix; mtHsp70 cooperates with the mitochondrial chaperonin system consisting of Hsp60 and Hsp10, allowing the polypeptide chain to fold into the correct tertiary conformation (Baker et al., 2011). The nucleotide exchange factor Mge1 is a soluble matrix protein which mediates the release of ADP from mtHsp70 enabling binding of ATP; this is followed by the release of bound precursor proteins so that freed mtHsp70 can be engaged in a new cycle (Mokranjac & Neupert, 2010).
Ubiquitin-proteasome system (UPS) is also involved in the monitoring of protein quality control in the outer mitochondrial membrane. When the molecular pathways are not able to refold misfolded proteins to their native state, these proteins are targeted to the UPS pathway (Baker, Palmer, & Stojanovski, 2014). The UPS machinery is important in the removal of individual mitochondrial proteins, while the autophagy pathway is responsible for the degradation of entire organelles by lysosomes (Pan, Kondo, Le, & Jankovic, 2008). The UPS is a dynamic multi-protein complex that eliminates impaired and/or nonfunctional proteins. Ubiquitin is a 76 amino acid protein, which is covalently attached to substrates through the formation of an isopeptide bond between the C-terminal glycine residue of ubiquitin and a lysine residue in the substrate. Protein ubiquitination occurs through a cascade of three enzymes including ubiquitin-activating E1 enzyme, ubiquitin-conjugating E2 enzyme, and ubiquitin E3 ligase (Bragoszewski, Turek, & Chacinska, 2017); Ubiquitin is activated by E1 enzymes and transferred to E2 enzymes. Ubiquitin is then conjugated to a carrier by E2 enzymes and ligated to the substrate proteins by E3 enzymes. The attachment of one ubiquitin molecule to one substrate protein residue, referred to as monoubiquitination, is a sufficient signal for proteasome-mediated degradation of small proteins. Polyubiquitin chains are built through the attachment of each subsequent ubiquitin molecule to one lysine residue that is present in the preceding ubiquitin (Schrader, Harstad, & Matouschek, 2009). Conversely, the ubiquitination process is reversed by deubiquitinating enzymes (DUBs), counteracting E3 ligase activity by removing or editing the ubiquitin chains (Nijman et al., 2005). The impairment of proteasome activity leads to mitochondrial dysfunction. Furthermore, various UPS components, as well as DUBs, are localized in mitochondria. These reveal the association between mitochondria and the UPS; since mitochondria are the main source of ROS and therefore are subject to oxidative damage, UPS in collaboration with other quality control mechanisms removes damaged mitochondrial proteins. In addition to mitochondrial proteins, excessive level of ROS may oxidize and damage the proteasomal subunits resulting in reduced catalytic activities (Ross, Olson, & Coppotelli, 2015). Therefore, mitochondrial dysfunction or proteasome system impairment leads to the initiation of a vicious cycle, resulting in the progressive failure of both systems.
Viruses activate host cell machinery for the production of their own components and virion assembly. Viruses keep evolving strategies to interfere with cellular processes and create a favorable environment for replication and survival (Williamson, DeBiasi, & Colberg-Poley, 2012). The mitochondrial function are directly or indirectly disrupted by viral infections; this results when viral proteins interfere with mitochondrial function or physiological alterations associated with infection including oxidative stress, hypoxia, impairment of calcium homeostasis and endoplasmic reticulum (ER) stress (Khan et al., 2015; Kim, Ahn, Syed, & Siddiqui, 2018). Viruses have evolved strategies to suppress the host antiviral responses. Viruses can inflict mitochondrial damage that results in the alteration in the mitochondrial morphodynamics to favor their propagation (Kim et al., 2018).
Viruses may employ mitochondrial protein translocation machinery to target their encoded products to mitochondria; a large number of viral proteins localize in the mitochondria during replication where they interact with mitochondrial proteins. Therefore, viruses manipulate mitochondrial functions including maintenance of oxidative balance, alteration of mitochondrial permeability transition pore, Δψm, electron transport and ATP production (Williamson et al., 2012). Since cellular chaperones involve protein folding processes, these are frequently recruited by viruses during their life cycle. The level of Hsp70 increases following viral infection of cells; this virus-induced elevation of Hsp70 levels is essential for virus proliferation (Mayer, 2005). The SARS-CoV-2 ORF9b protein is reported to interact with Tom70 (H.-w. Jiang et al., 2020). Upon RNA virus infection, Tom70 interacts with MAVS and induces antiviral cellular responses through potentiating IRF3-mediated gene expression (X.-Y. Liu et al., 2010). Therefore, association of SARS-CoV-2 Orf9b with Tom70 suppresses IFN-I response by disturbing MAVS signalosome. Interaction of ORF9b with Tom70 may influence IFN-I response through two possible mechanisms including competing Orf9b with HSP90 for binding to Tom70 or impairment of Tom70 function in the regulation of mitochondrial energy metabolism leading to the lactic acid production and subsequent inhibition of IFN-I responses (H.-w. Jiang et al., 2020). The RNA-Seq and matched ribosome profiling experiments show that the SARS-CoV-2 Nsp1 protein shuts down host translation machinery; Nsp1 is reported to inhibit translation of Tom22 and Tom40, which this can impair mitochondrial import of precursor proteins (Rao et al., 2020). The SARS-CoV-2 ER-resident protein Nsp4 is suggested to interact with components of TIM complex and therefore affects IMM protein import (Gordon et al., 2020). The SARS-CoV-2 ORF9b protein impedes the nuclear trans-localization of NF-κB/p65 through disrupting K63-linked polyubiquitination of NEMO; this results in the supression of IFN-I response through inhibiting RIG-I-MAVS antiviral signaling (Wu et al., 2021). The SARS-CoV-2 ORF9c and Nsp13 proteins target IFN pathway through interacting with negative regulator of MAVS signaling (NLRX1, NDFIP2) and MAVS effector TBK1, respectively (Gordon et al., 2020). SARS-CoV-2 membrane glycoprotein M also interacts with MAVS, contributing to the impairment of MAVS aggregation and its recruitment of downstream signaling components including TRAF3, TBK1, and IRF3, which this leads to the inhibition of downstream antiviral effector genes such as IFN-I. The SARS-CoV M protein similarly suppresses the stimulatory effect of RIG-I, MDA5, MAVS and TBK1 on IFN-I promoter (Siu et al., 2009; Siu, Chan, Kok, Chiu-Yat Woo, & Jin, 2014). The MERS-CoV M protein also shows IFN-I-antagonizing property through interacting with TRAF3 and disrupting TRAF3–TBK1 association leading to the inhibition of IRF3 phosphorylation (Lui et al., 2016).
Coronaviruses, as with many other viruses, subverts or manipulates UPS for their own advantage. In addition to the role of UPS in eliminating cellular misfolded proteins, UPS acts as a host defense mechanism to eliminate viral components. Viruses can encode specific proteins with E3-like or DUB-like activities or employ host UPS machinery to degrade cellular proteins that restrict viral propagation and to evade the host immune response (Luo, 2016; Raaben et al., 2010). Depending on the coronavirus replication, a functional ubiquitin proteasome pathway is suggested. In the case of wild-type SARS-CoV (strain Frankfurt-1) and recombinant CoVs feline infectious peritonitis virus (FIPV)Δ3abcFL, MHV-EFLM, MHV-nsp2EGFP and SARS-CoV-GFP, the replication of viruses is impaired upon proteasomal inhibition (Raaben, Posthuma, et al., 2010; Schneider et al., 2012). However, treatment of C57BL/6 mice infected by MHV-A59 and A/J mice infected by MHV strain 1 (MHV-1) with a proteasome inhibitor resulted in increased viral titers and pathology (Ma et al., 2010; Raaben, Grinwis, Rottier, & de Haan, 2010). Coronaviruses encode a number of proteins, enzymes, and undefined domains which are essential for viral replication. The replicase genes are encoded in two large open reading frames (ORF 1a and 1b) situated in the 5′-proximal three quarters of the genome. This mechanism produces two large viral polyproteins, pp1a and pp1ab, which are further cleaved by viral proteases to produce functional nonstructural proteins (Báez-Santos, John, & Mesecar, 2015). The coronaviral proteases, papain-like protease (PLpro) and 3C-like protease (3CLpro) process the viral polyprotein in a coordinated manner; 3CLpr process the C-terminal regions of the pp1a and pp1ab polyproteins, while PLpro process the N-terminal regions of pp1a/pp1ab. In addition to the processing of the viral polyproteins, it is established that SARS-CoV PLpro and MERS-CoV PLpro have deubiquitinating activity that influence host cell ubiquitination (J. Lei et al., 2014; Lindner et al., 2005). Therefore, deubiquitinating activity of SARS-CoV PLpro and MERS-CoV PLpro inhibits degradation of viral proteins and host misfolded proteins including mitochondrial proteins. Since the structure of CoV PLpro is different from homologous host enzymes such as USP14, CoV PLpro could be an important target for antivirals agents. Furthermore, SARS-CoV nucleocapsid protein (SARS-CoV NP) interacts with proteasome subunit p42; NP is postulated to be the central organizer of virus assembly. The interaction of SARS-CoV NP with proteasome subunit p42 leads to the suppression of the proteolytic activity of proteasomes; this results in the impairment of proteolysis of viral proteins and evasion of SARS-CoV from immune effectors (Q. Wang et al., 2010). It is suggested that this inhibition of proteolytic activity of proteasomes could also affect the degradation of host misfolded proteins (Fig 1).
Melatonin regulates the expression of HSPs including HSP60, HSP70, HSP90 in various cells and after different stimuli (Bonior, Jaworek, Konturek, & Pawlik, 2005; Kireev, Vara, Viña, & Tresguerres, 2014). In oxidative stress conditions associated with the induction of HSP70 expression, melatonin restores the level of HSP70 (Ozacmak, Barut, & Ozacmak, 2009). The impairment of anti-oxidant defense results in the induction of HSP70 expression. The overexpression of HSP70 is proposed to be protective by directly impeding inflammation and cell death pathways including apoptosis and necrosis. Melatonin inhibits oxidative stress-induced induction of HSP70 expression through normalizing the impaired antioxidants status (Ozacmak et al., 2009). Melatonin also improves mitochondrial protein import in oxidative stress condition. Melatonin restores the expression of Tim23 and Tom20, which this effect results from the induction of adenosine monophosphate-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC1α) pathway (Qi & Wang, 2020).Melatonin regulates multiple cellular functions through interacting with UPS machinery. Under pathogenic conditions such as hypoxia associated with the excessive level of proteasomes, melatonin reduces the level of Nedd4-1, an E3 ligase found most abundantly in neurons. Melatonin is not demonstrated to have a direct effect on UPS similar to that of bortezomib, a proteasome inhibitor (Vriend & Reiter, 2014). This effect is mediated through affecting the otoferlin (OTOF), substrate RNA splicing factor (SF3B2), importin 5 (IPO5), Hsc70 interacting protein (ST13), fibroblast growth factor receptor 3 (FGFR3), Mx1/Mx2, which play roles in RNA splicing and trafficking, growth factor and interferon signaling (Yalcin et al., 2019). In cancer cells, melatonin is reported to affect the same targets as bortezomib. The major proteins involved in cancer susceptibility including tumor suppressor protein p53, the cell cycle regulator p27, vascular endothelial growth factor (VEGF), transcription factors NF-κB, NRF2 and beta-catenin, and apoptotic and anti-apoptotic proteins, all of which are controlled by UPS; these proteins could be a target for bortezomib. The regulatory effect of melatonin on these signal transduction proteins suggests that it may influence the activity of the major targets of bortezomib (Vriend & Reiter, 2014).
Melatonin exhibits neuroprotective and cellular health-promoting effects by enhancing proteasomal activity in animal models of Alzheimer’s disease-like pathology. Aging has a negative effect on the activity of the proteasome in a variety of tissues leading to an elevated vulnerability of cells to oxidative stress and inflammation; the loss of protein clearance mechanisms is directly linked to the aging process. Thus, melatonin by improving the activity of proteasome may inhibit accumulation of aberrant proteins in cells (Corpas et al., 2018). During aging, circadian disturbances result in a decline in melatonin production, which contributes to a reduction of both proteasomal activity and mitochondrial dysfunction (Martins Branco et al., 2010; S. Mehrzadi, Hemati, Reiter, & Hosseinzadeh, 2020). Despite the direct mitochondrial protective effect of melatonin, the methoxyindole may also indirectly affect mitochondrial quality control by regulating proteasomal activity and, likewise, regulating proteasomal activity by affecting mitochondrial function (Fig 1) (Corpas et al., 2018; Martins Branco et al., 2010; Mehrzadi, Hemati, et al., 2020). Although no study has been performed on the effect of melatonin on coronavirus-mediated disruption of chaperone–protease networks, the regulatory effect of melatonin on the activity of HSPs and proteasome may improve mitochondrial function and limit the replication of CoVs such as SARS-CoV-2. Given that COVID-19 is more severe in elderly patients, and that the amount of both melatonin and the proteasome is reduced during aging, it is possible that melatonin may be useful during a COVID-19 infection, especially in elderly patients.
Mitochondrial dynamics refers to repetitive cycles of fusion and fission of the mitochondrial network. The balance between these opposing processes regulates mitochondrial shape and influences mitochondrial physiology, quality and homeostasis (Twig, Hyde, & Shirihai, 2008). A fusion process is a route by which matrix metabolites, intact mtDNA copies, and mitochondrial membrane components are quickly exchanged and equilibrated between neighboring mitochondria; fusion is a complementory mechanism for renewing the function of damaged mitochondria (Mouli, Twig, & Shirihai, 2009). Mitochondrial fusion in mammals is mediated by mitochondrial dynamin-related GTPases including the mitofusins (Mfn1 and Mfn2) and optic atrophy 1 (OPA1). Mitofusins are responsible for the outer mitochondrial membrane (OMM) fusion. In addition to the involvement in mitochondrial fusion, Mfn2 localizes in the endo/sarcoplasmic reticulum and regulates the mitochondria-ER/sarcoplasmic contact site tethering (Filadi et al., 2015). Mitofusins include an N-terminal GTPase domain, two coiled-coil domains (also called heptad-repeat domains, HR1 and HR2) and a bipartite carboxy-terminal transmembrane domain. The transmembrane domain is responsible for anchoring mitofusins to the OMM. Both the GTPase and coiled-coil domains are exposed to the cytosol and mediate MOM fusion in a GTPase-dependent manner (Chandhok, Lazarou, & Neumann, 2018). OPA1 is responsible for the inner mitochondrial membranes (IMM) fusion (Wu, Zhang, & Ren, 2019a). Constitutive processing of OPA1 by a number of proteases results in the generation of two isoforms of OPA1 including long-OPA1 (L-OPA1) and short-OPA1 (S-OPA1) (Ishihara, Fujita, Oka, & Mihara, 2006). The L-OPA1 form is integral to the inner membrane while the S-OPA1 is located in the intermembrane space. Various proteases such as the rhomboid-related protease presenilin-associated rhomboid-like (PARL), matrix AAA proteases (m-AAA; paraplegin, AFG3L1 and AFG3L2) and intermembrane space AAA proteases (i-AAA; Yme1L) are involved in OPA1 processing in mammals (J. Zhao, Lendahl, & Nistér, 2013). Processing of OPA1 by proteases is regulated both by ATP levels and by the inner membrane potential (Δψ m), implicating mitochondrial energetic status as a regulator of OPA1 processing (X. Wang et al., 2014). The heterotypic interaction of L-OPA1 on one side of the membrane to cardiolipin on the other side of the membrane is suggested to be sufficient for mitochondrial fusion; GTP-independent membrane tethering through L-OPA1 binding with cardiolipin promotes GTP-hydrolysis-dependent membrane fusion, modulated by the presence of S-OPA1 (Ban et al., 2017). The mitochondrial deacetylase SIRT3 directly deacetylates OPA1 and activates this protein by elevating its GTPase activity (Samant et al., 2014). Mitochondrial fusion proteins are ubiquitylated by several E3-ligases including Parkin and MARCH5/MITOL to be subsequently removed by proteases (Wu, Zhang, & Ren, 2019b). Mitochondrial depolarization leads to the cleavage of L-OPA1 by inducible protease OMA1 leading to a reduced mitochondrial fusion and autophagic degradation of mitochondrial organelles (Ni, Williams, & Ding, 2015).
A mitochondrial fission process leads to the division of the organelle into small and depolarized mitochondria. This process requires force coming from the activity of the dynamin-like GTPase protein Drp 1, a master regulator of mitochondrial division in most eukaryotic organisms. The Drp 1 protein is a soluble protein containing an N-terminal GTPase domain followed by a middle domain, a variable domain and a c-terminal GTPase effector domain that is involved in self-assembly (Pagliuso, Cossart, & Stavru, 2018).
Dynamin-related protein 1 is predominantly located in the cytosol, where it associates with microtubules. In response to fission signals, Drp1 is recruited to the mitochondrial surface where it forms ring-like structures around the organelle. On mitochondrial OMM Drp1 presumably docks on mitochondrial fission factor (Mff), the mitochondrial dynamic proteins 49 and 51 (MiD49 and MiD51) and Fission 1 (Fis1), its OMM receptor (Pagliuso et al., 2018). The ER–mitochondria contacts allow formation of Drp1 complexes at the site of fission. ER-associated inverted formin 2 (Inf2) and the mitochondria-anchored formin-activating protein Spire1C induce accumulation of actin filaments around the ER-mitochondria contact points; Ca2+ mobilization from the ER triggers actin redistribution around mitochondria (Moore, Wong, Simpson, & Holzbaur, 2016; Wales et al., 2016). Actin filaments induce a pre-constriction event leading to the reduction of mitochondrial cross-sectional diameter which allows Drp1-driven secondary constriction to promote fission (Moore et al., 2016). Then, Fis1, Mff, MiD49, and MiD51 recruit GTPase DRP1 to fission sites. Although the interaction between Fis1 and Drp1 has a minor role in regulating mitochondrial fission, Fis1 may mediate mitophagy and certain types of stress-induced fission. The self-assembly of Drp1 stimulates GTP hydrolysis which is likely to drive conformational changes in these oligomeric protein complexes which promote membrane scission (Lackner & Nunnari, 2009). The mechanism by which IMM fission occurs remains elusive. This process is suggested to be mediated by S-OPA1 isoforms and the IMM protein MTP18 (MTFP1); however, it is speculated that the constriction of mitochondria by Drp1 and actin may be sufficient to drive both OMM and IMM fission simultaneously (Wai & Langer, 2016).
Mitochondrial translocation of Drp 1 depends on phosphorylation–dephosphorylation of its residues such as Ser637, Ser-656 and Ser-616. Dependent on internal parameters or on external parameters including cell type, age or status, Drp1 phosphorylation at the same residue may have opposite effects on the mitochondrial fission progress. Phosphorylation of Drp1 at Ser-637 by a kinase anchoring protein 1 (AKAP1), a scaffold protein that recruits protein kinase A (PKA) and integrates several second messenger cascades to regulate mitochondrial function, and Ca2+/calmodulin dependent protein kinase Iα induces Drp1 translocation to mitochondria; however, Drp1 phosphorylation at Ser-637 by cAMP-dependent protein kinase results in the inhibition of Drp1 activity (Hu, Huang, & Li, 2017). Furthermore, activation of AMPKinduces phosphorylation of Drp1 at Ser-637 leading to the inhibition of Drp1-mediated mitochondrial fission, thereby blocking ER stress-associated NLRP3 inflammasome activation (J. Li et al., 2015). Depolarized mitochondria produced by the fission machinery are targeted by autophagy, as a receiver of the segregation output (Fig 2 ) (Calvani et al., 2013).
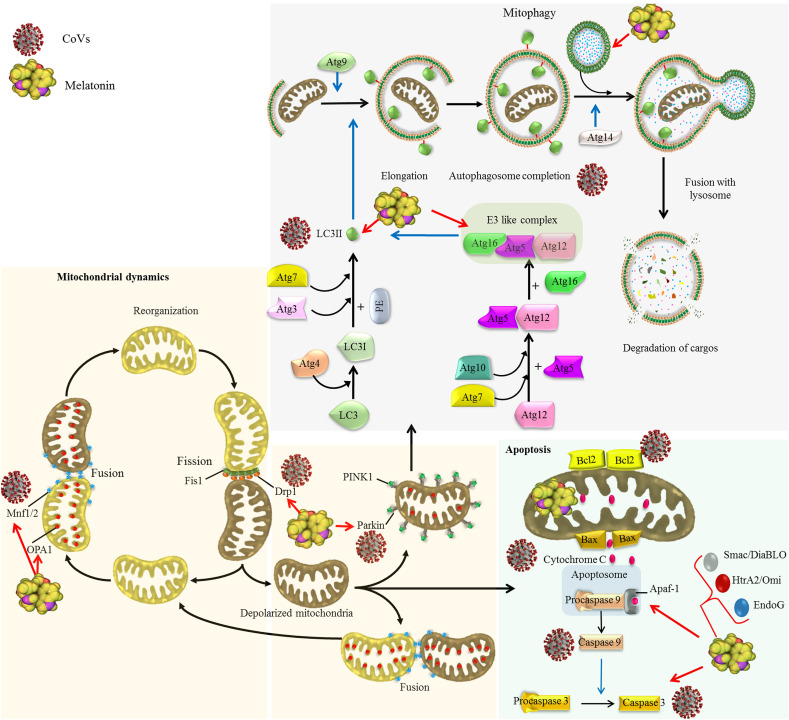
Effects of CoVs on mitochondrial dynamics and autophagy as a quality control axis and the regulatory effect of melatonin.
Coronaviruses impacts mitochondrial dynamics and mitophagy. Coronaviruses such as SARS-CoV-2 inhibits mitochondrial fusion through suppressing deubiquitination of ubiquitylated forms of Mfn1 and Mfn2 by targeting mitochondrial deubiquitinase (Singh, Chaubey, et al., 2020a). SARS-CoV promotes proteasomal degradation of Drp1 leading to induction of mitophagy (X.-Y. Liu et al., 2010). Some CoVs elevate the ratio of LC3-II/LC3-I leading to the autophagosome accumulation in infected cells (Ko et al., 2017). SARS-CoV-2 targets USP30 protein and inhibits Parkin-mediated mitophagy (Zarandi, Zinatizadeh, Zinatizadeh, Yousefi, & Rezaei, 2021). Difference in the effect of various CoVs on autophagy may result from the fact that autophagy inhibits the replication of some viruses but promotes the replication of other (Guo et al., 2016b). Melatonin regulates mitochondrial dynamics through increasing Mfn2 and OPA1 expression and diminishing Drp1 expression (Ding et al., 2018; Elesela et al., 2020; X. Zhang, Kang, et al., 2020). Melatonin also regulates autophagy/mitophagy pathway (Coto-Montes et al., 2012). In virus infected cells, melatonin shows different effect on autophagy markers including beclin-1, Atg5, Atg12 and Atg16L levels, and LC3-II/LC3-I ratio, dependent on the type of viruses and infection time (Sang et al., 2018; San-Miguel et al., 2014).
Autophagy includes several sequential steps (I-V), which are strictly regulated by more than 35 autophagy related genes (ATGs) and their corresponding proteins. (I) Induction and phagophore nucleation step is regulated by ULK1 (Unc-51-like kinase1 (mammalian homologues of Atg1), Atg13, Atg101 and RB1CC1/FIP200) and PI3 class III (class III PI3-kinase (Vps34), p150 (mammalian homolog of Vps15) complexes. The ULK1 complex involved in autophagy initiation and promotes activation of PI3 class III complex through phosphorylation of Beclin-1; PI3 class III complex induces phagophore nucleation (Glick, Barth, & Macleod, 2010). (II) The phagophore elongation and autophagosome formation steps are regulated by Atg9 protein and two ubiquitin-like (Atg12–Atg5/Atg16L and Atg8/LC3-phophatidylethanolamine (PE)) conjugation systems. (III) Autophagosome-lysosome fusion (autolysosome formation) step is regulated by Atg14 (R. Liu, Zhi, & Zhong, 2015). (IV) The degradation of sequestered cargo occurs in autophagosome by lysosomal/vacuolar hydrolases. (V) Degraded macromolecules are exported to the cytosol and recycled for the synthesis of essential components to overcome various stress conditions (Mehrzadi, Hemati, et al., 2020).
The level of autophagosome formation is negatively regulated by mammalian target of rapamycin complex 1 (mTORC1), containing GβL, a regulatory-associated protein of mTOR (raptor), PRAS40 and mTOR. The inhibition of mTORC1 results in the activation of Vps34, which is essential for the induction of phagophore nucleation. Upon energy deprivation, AMPKactivates and promotes the autophagy pathway through activation of ULK1, inactivation of mTORC1 and up-regulation of LC3 expression (Mehrzadi, Hemati, et al., 2020).
The elimination of damaged and dysfunctional mitochondria through a selective autophagic process is termed mitophagy. The delivery of damaged mitochondria to the phagophore is mediated by outer mitochondrial membrane (OMM)-spanning proteins including NIX/BNIP3L, BNIP3, FUN14 domain containing 1 (FUNDC1), p62/SQSTM1 and histone deacetylase 6 (HDAC6), which function as receptors for LC3-II (Higgins & Coughlan, 2014; Saito & Sadoshima, 2015). Serine/threonine kinase PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin are the key regulators of mitophagy by promoting mitophagic degradation of impaired mitochondria (Narendra, Tanaka, Suen, & Youle, 2008). Upon mitochondrial depolarization, PINK1 and Parkin physically associate and functionally cooperate in the identification and selection of mitochondria for degradation via mitophagy (Fig 2) (Higgins & Coughlan, 2014; L. Liu, Sakakibara, Chen, & Okamoto, 2014).
Coronaviruses alter mitochondrial dynamics and mitophagy to promote interference of cellular signaling pathways and to maintain persistent infection.
The SARS-CoV-2 ORF3a protein contains a 20-base sequence targeting USP30 protein, a mitochondrial deubiquitinase. The USP30 protein promotes mitochondrial fusion through deubiquitinating ubiquitylated forms of Mfn1 and Mfn2. Therefore, SARS-CoV-2 ORF3a protein may inhibit mitochondrial fusion by targeting USP30 (Singh, Chaubey, et al., 2020b). Furthermore, SARS-CoV-2 infected cells show reduction in the expression of mitochondrial ribosomal, complex I and mitochondrial fission promoting SOCS6 and MTFP1 genes. Decreased expression of MTFP1 and SOCS6 results in the disruption of mitochondrial fission leading to hyperfused mitochondria, which is observed in SARS-CoV-2 infected cells (Singh, Chen, et al., 2020).
Results from various studies show that different CoVs have different effects on various autophagy components in different cells. It seems that autophagic components are hijacked by CoVs to supply double-membrane vesicles needed for virus replication. Porcine epidemic diarrhea virus (PEDV) activates autophagy in infected Vero cells and inhibits NF-κB activation and subsequent production of proinflammatory cytokines (X. Guo et al., 2017). In porcine intestinal epithelial cells, however, autophagy induction could be an early effective strategy for the prevention of PEDV infection (Ko et al., 2017). Furthermore, infection of porcine kidney-15 (PK-15) and swine testicular (ST) cells with transmissible gastroenteritis coronavirus (TGEV) causes an elevation of the ratio of LC3-II to LC3-I resulting in the autophagosome accumulation in these cells. Since pharmacological inhibition of autophagy results in the elevation of TGEV replication, induction of autophagy may be a cellular strategy to prevent TGEV pathogenicity in testicular and renal cells (Guo et al., 2016a). In contrast, Zhu and colleges reported that TGEV induces oxidative stress in porcine epithelial cells resulting in the reduction of Δψm. Although the reduction of Δψm is often associated with cell apoptosis, TGEV infection does not promote the expected apoptosis. In TGEV-infected cells, reduction of total mitochondrial mass, up-regulation of p62/SQSTM1 mitochondrial level and an increase in LC3-II/LC3-I ratio and Beclin 1 expression indicates that TGEV may induce mitophagy to suppress oxidative stress and apoptosis for promoting cell survival and possibly the viral infection (Zhu, Mou, Yang, Lin, & Yang, 2016). The activity of nonlipidated LC3 is required for MHV replication, independent of intact host autophagy machinery; down-regulation of LC3 expression, but not the suppression of ATG5 and ATG7 which is required for autophagy pathway inhibits MHV replication in HeLa CEACAM1a cells(Reggiori et al., 2010; Z. Zhao et al., 2007). Furthermore, the lack of ATG5 protein and autophagy inhibition do not influence MHV replication in bone marrow derived macrophages and embryonic fibroblasts (Z. Zhao et al., 2007). However, the impairment of autophagy and the lack of ATG5 expression results in the inhibition of MHV replication in embryonic stem cells (Prentice, Jerome, Yoshimori, Mizushima, & Denison, 2004).
The NSP6 protein of avian coronavirus, infectious bronchitis virus, MHV and SARS-CoV induces autophagosome generation from ER independent of starvation (Cottam et al., 2011). Interestingly, autophagosomes induced by NSP6 have a small diameter (0.5 μm) resulting from NSP6-induced limitation in autophagosome expansion. Since NSP6 protein does not affect SQSTM1 recruitment to autophagosomes, small diameter autophagosomes induced by NSP6 can accumulate cargo and deliver autophagic cargo to lysosomes (Cottam, Whelband, & Wileman, 2014). MERS-CoV is reported to inhibit Beclin1 expression and suppress autophagosomes-lysosome fusion. This effect may be mediated by the poly-ubiquitination of Beclin1 through activating S-phase kinase-associated protein 2 (SKP2) as E3 ligase (Gassen et al., 2019). However, inhibition of the PI3K/AKT/mTOR signaling pathway is reported to inhibit MERS-CoV replication. These finding suggests that regulation of autophagy may be an important target for therapeutic intervention strategies for MERS-CoV (Kindrachuk et al., 2015).
SARS-CoV and SARS-CoV-2 also result in perturbations specific aspects of autophagy and mitochondrial processes in infected cells (Fig 2). The ORF-9b protein of SARS-CoV localizes in mitochondria, where it promotes proteasomal degradation of Drp1 leading to the mitochondrial elongation. This effect of ORF-9b in promoting mitochondrial elongation is in contrast to the elimination of unhealthy mitochondria by mitochondrial fission which occurs following viral infection. This suggests that ORF-9b promotes cell survival during viral replication through inhibition of mitochondrial fragmentation induced by other cellular stresses. Therefore, SARS-CoV may use ORF-9b to maintain viral replication by altering mitochondrial dynamics and targeting MAVS to evade host innate immunity. Furthermore, permanent transfection of cells with ORF-9b protein has no significant effect on LC3 processing or induction of autophagy, but, acute ORF-9b expression results in the significant induction of Atg5 expression and autophagosome formation (X.-Y. Liu et al., 2010).
The USP30 protein also plays a role in mitochondria homeostasis through cooperating with Parkin-mediated mitophagy. Thus, SARS-CoV-2 by using ORF3a protein may alter ubiquitination in mitochondria through interacting with USP30; this leads to the mitochondrial collapse and premature death of infected cells (Zarandi, Zinatizadeh, Zinatizadeh, Yousefi, & Rezaei, 2021) In ACE2-expressing cells, infection with SARS-CoV-2 has been reported to up-regulate the expression of PINK1; this suggests that SARS-CoV-2 may affect mitochondrial dynamics depending on the type of cells (Miller et al., 2020). The expression of SKP2 reduces in SARS-CoV-2 infected cells, which this results in the inhibition of Beclin1 degradation leading to the induction of autophagic flux (Singh, Chen, et al., 2020). Since the amino acid sequences of ORF-9b in SARS-CoV-2 are similar to SARS-CoV (Singh, Chaubey, et al., 2020b), it is suggested that SARS-CoV-2 ORF-9b inhibits mitochondrial fragmentation and induces autophagosome formation to promote cell survival for increasing viral replication. However, coexpression network analysis of the RNA seq data from SARS-CoV-2 infected cells demonstrates that SARS-CoV-2 impairs autophagic flux through up-regulation of glycogen synthase kinase 3 beta (GSK3B), or down-regulation of autophagy genes including synaptosome associated protein 29 (SNAP29) and SQSTM1 as well as lysosome acidification genes (Singh, Chen, et al., 2020). Based on these findings, it is proposed that CoVs activate autophagy during infection but it is unclear why CoVs would limit autophagy. More studies are needed to clarify the exact role of autophagy in the coronavirus infection.
Melatonin inhibits mitochondrial fission by down-regulating the expression of Drp1. This effect of melatonin may be mediated by activation of the sirtuin 1-PGC1α pathway; PGC-1α negatively regulates the expression of Drp1 by binding to its promoter. Since activation of sirtuin 1 improves Δψm and reduces generation of ROS during viral infection, melatonin seems to improve mitochondrial function by induction of sirtuin 1 expression; sirtuin 1 increases the expression of downstream transcription factor PGC1α which, in addition to the reducing Drp1 expression, promotes mitochondrial biogenesis. These findings reveal that melatonin may mediate mitochondrial adaptations through regulating both mitochondrial dynamics and mitochondrial biogenesis (Ding et al., 2018; Elesela et al., 2020). Melatonin is reported to inhibit prion-induced neuron damage by regulating mitochondrial function and dynamics. Melatonin corrects the imbalance of mitochondrial dynamics occurring in prion-induced neural cell damage by overexpressing OPA1 and diminishing Drp1 expression (X. Zhang et al., 2020). In addition to the reduction in Drp1 expression, melatonin reduces the co-localization of Drp1 and Tom20 proteins, which indicates that melatonin reduces mitochondrial translocation of Drp1 (Hongyan Zhou et al., 2018). Suppression of Drp1-mediated mitochondrial fission by melatonin results in the inhibition of mitophagy-mediated cellular death by preventing mitochondrial permeability transition pore (mPTP) opening and PINK1/Parkin activation. Melatonin activates AMPKα which subsequently inhibits voltage-dependent anion channel 1 and hexokinase 2 disassociation-involved mPTP opening and excessive mitophagy. Furthermore, AMPKα represses mitochondrial fission via inactivation of Drp1 protein (Hao Zhou et al., 2017). Activation and mitochondrial translocation of Drp1 is dependent on a variety of factors including ROS overproduction and calcium overload; the inhibitory effect of melatonin on Drp1 activation and mitochondrial translocation may also result from the antioxidant actions of melatonin and its inhibitory effect on cytosolic calcium overload (Cui et al., 2018; S. Xu et al., 2016). Melatonin also induces mitochondrial fusion in oxidative stress condition, which this effect is mediated by the activation of AMPK/PGC1α pathway; melatonin restores the expression of Mfn2 and OPA1, which are reduced in oxidative stress condition (Qi & Wang, 2020).
The modulatory effect of melatonin on autophagy/mitophagy has been reported in several investigations; however the exact mechanisms are unknown (Coto-Montes et al., 2012). In normal cells, melatonin promotes autophagy through activating AMPK, inducing the expression of Beclin 1 and ATGs, down-regulating the mTOR-signaling pathway and increasing the ratio of LC3-II/LC3-I. In cancer cells, melatonin inhibits autophagy to increase apoptotic death of cancer cells (Mehrzadi, Hemati, et al., 2020). Melatonin reduces acute liver failure induced by rabbit hemorrhagic disease virus (RHDV). Infection of rabbits with RHDV is associated with the induction of autophagy characterized by up-regulation of beclin-1, Atg5, Atg12, Atg16L and p62/SQSTM1 levels, and LC3-II/LC3-I ratio; this autophagic response is inhibited by melatonin administration (San-Miguel et al., 2014). Myocarditis induced by coxsackievirus B3 is also associated with the alteration in autophagy. The autophagy markers including LC3-II/LC3-I ratio and Beclin-1 increase on day 7 and 14 after viral infection; following melatonin treatment, these markers are diminished on day 7 and then trend upward to day 14 (Sang et al., 2018). As already mentioned, SARS-CoV-2 induces oxidative stress in infected cells, similar to other viruses, which this results in the disruption of mitochondrial function. Since melatonin is a powerful antioxidant with the ability to regulate mitochondrial dynamics and autophagy/mitophagy, this indoleamine may inhibit SARS-CoV-2 replication and protect cells from coronavirus-induced damage through inhibiting oxidative stress and improving SARS-CoV-2-induced perturbation in mitochondrial fusion, fission and autophagy (Fig 2).
The mixture of healthy mitochondria with defective mitochondria during the fusion process can repair dysfunctional mitochondria; however, when mitochondrial damage is extensive, dysfunctional mitochondria are segregated by fission, with these organelles ultimately being eliminated by mitophagy. If the quality control at molecular and organelle levels are impaired or the mitochondrial damage is greater than the capacity of fission/fusion and mitophagy pathways, defective mitochondria can rupture (Q. Cai & Tammineni, 2016). Following mitochondrial rupture, apoptotic factors escape from the mitochondrion to the cytosol and trigger apoptotic cell death through induction of caspase activation as well as chromosome condensation and fragmentation. The release of these apoptotic factors occurs when the integrity of OMM is compromised by the pro-apoptotic Bcl-2 family members. Once activated, pro-apoptotic members of Bcl2 family proteins including Bax and Bak form mPTP leading to permeabilization of the mitochondrial outer membrane (Westphal, Kluck, & Dewson, 2014); this process leads to the release of apoptotic proteins including Smac/DiaBLO, HtrA2/Omi, apoptosis inducing factor (AIF), endonuclease G (EndoG) and cytochrome c from the IMM space into the cytosol (Q. Cai & Tammineni, 2016; Martinou & Youle, 2011). In the cytoplasm, cytochrome c interacts with apoptosis protease-activating factor 1 (Apaf-1) and procaspase-9 to create the apoptosome complex. The apoptosome catalyzes the autocatalytic activation of caspase-9, which subsequently activates caspase 3. Once activated, caspase 3 cleaves vital cellular proteins and executes cell death (Fig 2) (Montero & Letai, 2018). In addition to caspase, cell death factors released from mitochondria may activate caspase-independent apoptosis. SMAC/Diablo and HtrA2/Omi promotes caspase activity by neutralizing the inhibitory effect of inhibitor of apoptosis proteins (IAPs) on caspases. Furthermore, AIF is translocated into the nucleus, where it binds chromosomal DNA and causes chromatin condensation, thereby, directly or indirectly allowing DNA fragmentation by Endo G (Lartigue et al., 2009). Furthermore, Bax/Bak promotes sumoylation of Drp1 leading to the induction of a stable membrane-associated form of Drp1 during apoptosis. Detection of the active Bax in discrete foci at mitochondrial fission sites reveals a link between mitochondrial fission and Bax activation. This indicates a link between Bcl-2 family members and mitochondrial dynamics; however, the connection between the mitochondrial fragmentation during apoptosis and Bcl-2 family proteins effects on mitochondrial dynamics is unknown (Martinou & Youle, 2011).
Two defensive arms which are used by multicellular organisms to overcome viral infections include activation of innate immune responses and induction of programmed cell death. During viral infections, activation of apoptotic pathways in host cells may be an effective strategy to prevent viral infections by inhibiting viral replication and limiting virus spreading (Cuconati & White, 2002). Thus, inhibition of apoptotic pathways may be a strategy by which viruses maintain their replicative ability. However, results from experimental studies indicate that CoVs encode either pro-apoptotic or anti-apoptotic proteins in infected cells; this suggests that apoptosis may be regulated differently depending on particular virus–host interactions (Y.-J. Tan, Lim, & Hong, 2007).
During an MHV infection, the expression of pro-apoptotic gene BNip3 is reduced in astrocytes. This repression in the BNip3 expression has been observed for live and ultraviolet light-inactivated viruses; this indicates that viruses down-regulate BNip3 expression during cell entry showing that it is not required for virus replication (Y. Cai, Liu, Yu, & Zhang, 2003). Caspase recruitment domains (CARDs) are homotypic interaction motifs found in various proteins which are involved in both apoptotic and inflammatory signaling cascades. Caspases, Apaf-1 and some members of the NLR (nucleotide-binding domain and leucine-rich repeat containing) protein family are among the proteins that contain the domain. As already mentioned, Apaf-1 interacts with cytochrome c to form the apoptosome which functions to activate caspase-9, thereby stimulating mitochondria-dependent apoptotic signaling. The CARD-containing NLR such as Nod1, Nod2 and Nlrc4 (Ipaf) have been linked to both apoptotic and inflammatory signaling (Y. Lei et al., 2009). The MAVS protein also contains a N-terminal CARD-like domain, which suggests that mitochondrial localization of MAVS protein is involved in the induction of apoptosis, in addition to the innate immunity response. Therefore, inhibition of MAVS function may be a successful strategy for some viruses to inhibit cell death responses (Y. Lei et al., 2009; X.-D. Li, Sun, Seth, Pineda, & Chen, 2005). SARS-CoV has also been shown to inhibit MAVS-mediated apoptotic responses, in addition to inhibition of IFN-I responses; SARS-CoV NSP15 protein, an important protein for SARS-CoV replication, completely abrogates MAVS-induced apoptosis. The exact mechanism by which SARS-CoV NSP15 inhibits MAVS-induced cell death is not fully understood (Y. Lei et al., 2009). The murine coronavirus NSP15 prevents detection of viral dsRNA by RIG-I and MDA5 sensors of macrophages, which are equipped with multiple dsRNA sensors; this results in the delay in the activation of IFN and inhibition of macrophage apoptosis (Deng et al., 2017).
Various studies also indicate the existence of pro-apoptotic proteins including ORF-3a, ORF-3b, ORF4, ORF-6, ORF7a and ORF7b in SARS-CoV genome, which result in the induction of apoptosis in specific cell types (Ye, Wong, Li, & Xie, 2008; Yuan, Shan, Zhao, Chen, & Cong, 2005). The ORF-3a protein, a viral protein localized to the ER, induces chromatin condensation and DNA fragmentation leading to apoptosis in SARS-CoV-susceptible cells (Law et al., 2005); this protein is reported to promote mitochondrial apoptosis through activation of p38 MAP kinase (MAPK) (Padhan, Minakshi, Towheed, & Jameel, 2008). The 3b protein (ORF4) is found in both the nucleus and mitochondria and its overexpression leads to the induction of cell cycle arrest at the G0/G1 phase (Yuan et al., 2005). Overexpression ORF-6 induces apoptosis by promoting ER stress resulting in the activation of caspase-3 and JNK MAPK pathway (Ye et al., 2008). The ORF7a protein may interact with anti-apoptotic protein Bcl-XL in the ER, leading to the sequestration of Bcl-XL in the Golgi apparatus; this subsequently tips the balance towards apoptosis, with this being associated with the activation of p38 MAPK (Schaecher, Touchette, Schriewer, Buller, & Pekosz, 2007). The ORF7a protein also interacts with human Ap4A-hydrolase (asymmetrical diadenosine tetraphosphate hydrolase, EC 3.6.1.17), a member of a superfamily of enzymes involved in the elimination of cells from toxic nucleotide metabolites; this results in the activation of cascade of mechanisms leading to cell cycle arrest and apoptosis (Vasilenko, Moshynskyy, & Zakhartchouk, 2010). Furthermore, ORF7a protein induces apoptosis through interfering with Bcl-XL and other pro-survival proteins (Bcl-2, Bcl-w, Mcl-1, and A1) (Tan et al., 2007). The SARS-CoV membrane (M) structural protein induces apoptosis through the regulation of Akt pro-survival pathway and induction of mitochondrial cytochrome c release (C.-M. Chan, Ma, Chan, & Chan, 2007). The expression of ORF8a has also been reported to be increased in SARS-CoV infected patients. The ORF8a protein induces apoptosis in addition to the viral replication indicating that this protein plays an important role in the SARS-CoV pathogenesis. The ORF8a protein is localized in mitochondria, where it induces mitochondrial hyperpolarization, overproduction of ROS, and subsequent caspase 3-dependent apoptosis (Fig 2) (C.-Y. Chen et al., 2007).
The SARS-CoV-2 ORF3a protein also induces apoptotic cell death by promoting the activation of caspase-3, truncation of Bid (tBid), cleavage of caspase-9, and release of cytochrome c (Ren et al., 2020). SARS-CoV-2 induces apoptosis in lymphocytes (CD3, CD4, and CD8 T cells) leading to a reduction of lymphocyte count; this results in the cytokine storm syndrome associated with the uncontrolled production of pro-inflammatory cytokines, leading to the hyper-inflammation as well as cellular and, eventually, organ destruction (Gao et al., 2020; Juybari et al., 2020). The non-clonal population of T cells found in acutely infected COVID-19 patients is characterized by up-regulating the expression of voltage dependent anion channel, mitochondrial membrane protein involved in outer mitochondrial membrane permeabilization and induction of cell death. The increased expression of voltage dependent anion channel in these T cells is associated with the mitochondrial dysfunction leading to the release of cytochrome c and activation of caspases; this caspases-induced T cell apoptosis may contribute to the lymphopenia, observed in COVID19 patients (Thompson et al., 2020). Since the inhibition of SARS-CoV-induced apoptosis does not alter susceptibility of cells to infection or viral replication and release, apoptosis seems may not be involved in facilitating production and dissemination of CoVs (Bordi et al., 2006). These findings suggest that apoptosis may be activated due to alterations in host-infected cells; however further studies are needed to determine that SARS-CoV-2-induced apoptosis results from virus replication; alternatively, it may be a response of the infected cell to virus-induced stress.
The influence of melatonin on the processes of apoptosis has been evaluated in various cells and tissues (Fig 2). The actions of melatonin in healthy and tumor cells are clearly different (K. H. Lu et al., 2018; R. J. Reiter et al., 2017; Su et al., 2017). Intracellular interactions of melatonin contributes to the stimulation of apoptosis in cancer cells (Bizzarri, Proietti, Cucina, & Reiter, 2013; Pourhanifeh, Mehrzadi, Kamali, & Hosseinzadeh, 2020), while it prevents apoptotic cell death in healthy cells (Pourhanifeh, Mehrzadi, & Hosseinzadeh, 2020); the specific explanation for this differential response is not known but could relate to any of a number of cellular processes including intercellular signaling, metabolism, gene regulation, or stress responses (Sainz et al., 2003; H. M. Zhang & Zhang, 2014). In viral infected cells, melatonin may modulate apoptosis and differentially regulate the expression of pro-apoptotic and anti-apoptotic mediators to limit the extent of viral spread. Since viruses are dependent on the host for each stage of replication, these intracellular parasites cause cell stress and alter expression of genes regulating cell survival (Boga, Coto-Montes, Rosales-Corral, Tan, & Reiter, 2012).
The anti-apoptotic properties of melatonin are related to its regulatory actions on mitochondrial function as well as on its antioxidant and free-radical scavenging activities. Melatonin maintains more optimal mitochondrial physiology by inhibiting the mPTP opening as well as suppressing oxidative stress-induced Ca2+ overload, which subsequently inhibits mitochondrial membrane depolarization, Ca2+-mediated mPTP opening and Ca2+-induced Ca2+ release (Andrabi, Sayeed, Siemen, Wolf, & Horn, 2004; Suwanjang, Abramov, Charngkaew, Govitrapong, & Chetsawang, 2016). Furthermore, melatonin stabilizes the level of parvalbumin and hippocalcin, as Ca2+-buffering proteins, resulting in the prevention of large rises intracellular Ca2+ levels and induction of apoptosis (Koh, 2012). Melatonin is also capable of both donating and accepting electrons to mitochondrial ETC omplexes; this contributes to the enhancement of the activities of respiratory complexes I, III and IV and in the maintenance of efficient oxidative phosphorylation and ATP synthesis (López et al., 2009; Martín, Macías, Escames, León, & Acuña-Castroviejo, 2000). By influencing these processes, melatonin improves mitochondrial respiration and energy production, thereby limiting mitochondrial depolarization, mPTP opening, and the release of apoptotic proteins such as Smac/DiaBLO, HtrA2/Omi, AIF, EndoG and cytochrome c from IMM space into the cytosol (Acuna-Castroviejo, Escames, Rodriguez, & Lopez, 2007). Melatonin inhibits Bax activation/dimerization within mitochondria by stimulating Bcl-2 localization in OMM; this effect of melatonin is mediated by the activation of plasma membrane MT1/MT2 receptors. Inhibition of Bax activation/dimerization prevents the formation of mPTP and release of pro-apoptotic factors from mitochondria (Radogna et al., 2007; Radogna et al., 2008). In a mechanism related to the activation of membrane MT1 and MT2 receptors, melatonin inhibits phosphorylation of p53 by down-regulating phosphorylation of JNK and p-38 MAPKs; this leads to the reduction of Bax/Bcl-2 ratio, inhibition of cytochrome c release, and prevention of Bid proteolytic cleavage and activation of caspases (Das et al., 2010). Furthermore, melatonin binds to MT1 receptors localized in OMM leading to the activation of MT1/Gαi signaling in the mitochondrial intermembrane space; this event suppresses adenylate cyclase activity and inhibits Ca2+-mediated cytochrome c release, thereby preventing caspase activation (Suofu et al., 2017).
The anti-apoptotic effect of melatonin has been reported in some virus-infected cells, e.g., during a venezuelan equine encephalitis (VEE) virus (VEEE) infection. Pre-treatment with melatonin clearly modified the VEE infection since it reduced apoptosis in infected mice and in VEEV-infected neuroblastoma cells (Montiel et al., 2015). Melatonin also inhibits apoptosis of hepatocytes induced by RHDV; this effect is associated with the up-regulation of Bcl-2 and Bcl-xL expressions and down-regulation of Bax expression, cytochrome c release and caspase-9 activation (Tuñón et al., 2011). Melatonin restores mitochondrial function in cardiomyocyte infected with coxsackievirus B3, which subsequently leads to the inhibition of apoptotic cell death (Ouyang et al., 2019; Sang et al., 2018). Since SARS-CoV-2 induces the release of cytochrome c and subsequently activates caspase-3 in infected cells, mitochondrial dysfunction is suggested to be a mechanism by which SARS-CoV-2 induces apoptosis. The findings summarized above suggest that a reduction of melatonin levels in the mitochondria of SARS-CoV-2 infected cells may contribute to the dysregulation of cellular and mitochondrial metabolisms (Anderson & Reiter, 2020). Therefore, treatment with melatonin may inhibit apoptosis induced by SARS-CoV-2 in infected cells by regulating mitochondrial physiology.
This review summarizes the consequences of a CoVs infection on the molecular pathways involved in the mitochondrial protein quality control. Impairment of mitochondrial function may be an important mechanism by which CoVs influence innate immune signaling and survival of infected cells. The protective actions of melatonin on mitochondrial function suggest that this molecule likely would protect coronavirus-infected cells against virus-induced mitochondrial dysfunction. In addition to the direct mitochondrial protective actions, melatonin could indirectly impact mitochondrial quality control by regulating proteasome activity, mitochondrial dynamics, autophagy and apoptosis. However, further studies are needed to clarify the effect of CoVs such as SARS-CoV-2 on mitochondrial quality control and melatonin effects on signaling pathways dysregulated by these viruses. This review will hopefully stimulate additional research related to the mechanisms of action of melatonin against CoVs-induced cell damage.
This research did not receive any specific grant from funding agencies in the public, commercial, or not for-profit sectors.
The authors declare that there are no conflicts of interest.
 SARS-CoV-2 and other coronaviruses negatively influence mitochondrial quality control: beneficial effects of melatonin
SARS-CoV-2 and other coronaviruses negatively influence mitochondrial quality control: beneficial effects of melatonin
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
