
The recent emergence of coronavirus disease-2019 (COVID-19) as a global pandemic has prompted scientists to address an urgent need for defining mechanisms of disease pathology and treatment. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent for COVID-19, employs angiotensin converting enzyme 2 (ACE2) as its primary target for cell surface attachment and likely entry into the host cell. Thus, understanding factors that may regulate the expression and function of ACE2 in the healthy and diseased body is critical for clinical intervention. Over 66% of all adults in the United States are currently using a prescription drug and while earlier findings have focused on possible upregulation of ACE2 expression through the use of renin angiotensin system (RAS) inhibitors, mounting evidence suggests that various other widely administered drugs used in the treatment of hypertension, heart failure, diabetes mellitus, hyperlipidemias, coagulation disorders, and pulmonary disease may also present a varied risk for COVID-19. Specifically, we summarize mechanisms on how heparin, statins, steroids and phytochemicals, besides their established therapeutic effects, may also interfere with SARS-CoV-2 viral entry into cells. We also describe evidence on the effect of several vitamins, phytochemicals, and naturally occurring compounds on ACE2 expression and activity in various tissues and disease models. This comprehensive review aims to provide a timely compendium on the potential impact of commonly prescribed drugs and pharmacologically active compounds on COVID-19 pathology and risk through regulation of ACE2 and RAS signaling.
The recent emergence of coronavirus disease-2019 (COVID-19) as a pandemic affecting millions of individuals has raised great concern throughout the world and has spurred an urgent need for treatments. In order to enter the host cell, the causative agent “severe acute respiratory syndrome coronavirus-2” (SARS-CoV-2) binds to angiotensin converting enzyme 2 (ACE2), which is widely expressed throughout the body, including lung alveolar epithelial cells, nasal and oral mucosal cells, vascular endothelium, and enterocytes of the small intestine (Hamming et al., 2004). Higher ACE2 expression is correlated with higher pseudotype SARS-CoV-2 and SARS-CoV viral infectivity (Hofmann et al., 2004; H. P. Jia et al., 2005; W. Li et al., 2007, Ou et al., 2020), suggesting that increased ACE2 levels may predispose individuals to SARS-CoV-2 transmission. Since ACE inhibitors (ACE-Inhs.) and Angiotensin 1-Receptor (AT1-R) blockers (ARBs) have been reported to increase ACE2 expression, concerns have been raised regarding the safety of these drugs in patients exposed to SARS-CoV-2. Thus, these concerns are mainly based on the hypothesis that such medications may raise the expression of ACE2 and increase the susceptibility of patients to SARS-CoV-2 (Peron & Nakaya, 2020). Because both an ACE inhibitor (lisinopril) and an AT1-R blocker (losartan) are among the 10 most commonly used drugs with a combined 155 million prescriptions per year in the USA alone (Zolk et al., 2020), patients receiving these drugs would represent a substantial group of people at risk. This article reviews the effects not only of ACE-Inhs. and ARBs, but also of other drugs on the expression and activity of ACE2. In addition, the pharmacological effects of these drugs and naturally occurring compounds are discussed in the context of COVID-19. Thus, the focus of this review is neither the treatment of COVID-19 nor the regulation of ACE2 in specific disease conditions, but the regulation of ACE2 by the many drugs, pharmacologically active compounds and naturally occurring substances used in society today.
ACE2, a homologue of ACE, was discovered two decades ago by two independently working research groups, Donoghue et al. (2000) and Tipnis et al. (2000). Like ACE, ACE2 is an integral membrane protein and zinc metallopeptidase with an amino acid sequence that is 42% identical to ACE. While ACE contains two catalytic domains, ACE2 has only one. Importantly, ACE-Inhs. belonging to the classic “pril” group used in the treatment of cardiovascular diseases do not affect the enzymatic functions of ACE2 (Donoghue et al., 2000; Tipnis et al., 2000). Structural and functional features and topographical characteristics of ACE2 have been reviewed earlier (Kuba, Imai, Ohto-Nakanishi, & Penninger, 2010; Turner, 2015). The major functional difference between ACE and ACE2 is that ACE produces Angiotensin II (Ang-II), whereas ACE2 degrades this peptide. Specifically, ACE2 functions as a carboxypeptidase removing a single C-terminal amino acid from the octapeptide Ang-II, generating the heptapeptide Angiotensin-(1-7) [Ang-(1-7)] or, much less efficiently, from the decapeptide Angiotensin I (Ang-I) forming the nonapeptide Angiotensin-(1-9) [Ang-(1-9)]. In contrast, ACE acts as a carboxydipeptidase (peptidyldipeptidase) removing the C-terminal dipeptide from Ang-I to form Ang-II. Furthermore, whereas ACE metabolizes bradykinin to [des-Arg9]-bradykinin, ACE2 degrades [des-Arg9]-bradykinin to pharmacologically inactive breakdown products (Tipnis et al., 2000). Other substrates for ACE2, at least in vitro, include apelin-13/17, neurotensin (1-11), dynorphin A (1-13), amyloid-β peptides, β-casomorphin-(1-7), and ghrelin (Turner, 2015; Vickers et al., 2002).
In most tissues, ACE2 is found in its membrane-bound form, which contains an extracellular segment anchored to the plasma membrane through a transmembrane domain. This enzymatically active N-terminal ecto-domain can be cleaved by a membrane-bound protease, also called secretase (or sheddase), and released into the surrounding extracellular space. Thus, some fraction of membrane bound ACE2 is shed into the circulation as soluble ACE2 and can be detected in plasma, cerebrospinal fluid, and urine samples. In its soluble form, however, ACE2 is found in very low concentrations in the circulation (Epelman et al., 2008; Rice et al., 2006). While serum ACE levels were reported to be 7 nM (Rice et al., 2006), the ACE2 concentration was found to be 200-fold lower (33 pM) in over 500 subjects. In recent years, it has become increasingly apparent that the proteolytic shedding of cell surface ACE2 is an important mechanism regulating its expression, functions, and soluble concentrations in biological fluids (J. Xu et al., 2017). The major protease mediating ectodomain shedding of ACE2 is a type I transmembrane protein belonging to the adamalysin subfamily of Zn-dependent metalloproteases (“A Disintegrin And Metalloprotease 17”; ADAM17). Since this protease also mediates extracellular domain shedding and activation of the proinflammatory cytokine TNF-α (Black et al., 1997; Moss et al., 1997), it is also known as “tumor necrosis factor-α (TNF-α) cleavage enzyme” (TACE) (Lambert et al., 2005; Patel et al., 2014). Pharmacological agents, e.g., rosiglitazone (Chodavarapu et al., 2013) and the vitamin D analog paricalcitol (Riera et al., 2016), as well as endogenous peptides, e.g., Ang-II (Patel et al., 2014) and insulin (Salem, Grobe, & Elased, 2014), regulate the activity of ADAM17. Several pathologies, such as hypertension, diabetes mellitus, renal failure (Chodavarapu et al., 2016; Salem, Grobe, & Elased, 2014; Somineni, Boivin, & Elased, 2014 J. Xu et al., 2016), and SARS-CoV infections (Haga et al., 2008) are associated with significant alterations in ADAM17 activity. Importantly, soluble ACE2 levels in circulation and biological fluids are the result of a dynamic process determined not only by cell surface expression, but also by ACE2 shedding. Notably, soluble circulating ACE2 appears to serve as a biomarker in renal and cardiovascular diseases, such as hypertension, heart failure, and diabetes mellitus (Anguiano, Riera, Pascual, & Soler, 2017).
ACE2, by converting Ang-I into Ang-(1-9), and Ang-II into Ang-(1-7), degrades Ang-I and Ang-II, thereby negatively regulating the renin-angiotensin system (RAS) and mitigating the deleterious effects of these peptides (Fig. 1 ). Thus, the enzymatic functions of ACE2 are of particular significance in pathological conditions where the RAS is overstimulated by Ang-I and Ang-II (Arendse et al., 2019; Chappell, 2016; Paz Ocaranza et al., 2020). Biological actions of Ang-(1-7) are mediated mainly by the Mas receptor (MasR), and a vast array of its effects are opposite to those attributed to Ang-II activation of the Ang-II type 1 receptor (AT1-R) (Arendse et al., 2019; Santos et al., 2018). Ang-(1-7) can be degraded to Ang-(1-5) by ACE (Chappell, 2016). In addition, Ang-(1-7) inhibits the enzymatic activity of ACE (Raffai, Khang, & Vanhoutte, 2014; Tom, de Vries, Saxena, & Danser, 2001). Thus, Ang-(1-7) acts both as substrate and as inhibitor of ACE. Ang-(1-9) has also shown beneficial biological effects via the AT2-R that result in cardioprotection, vasodilation, and decreased platelet aggregation; however, expression of AT2-Rs is low in adults (Arendse et al., 2019; Paz Ocaranza et al., 2020). Nevertheless, Ang-II binds with equal affinity (in the nM range) to AT1-Rs and AT2-Rs, and AT2-R density in tissues increases significantly in some physiological conditions, e.g., fetal development, pregnancy, and parturition, as well as in pathological conditions, e.g., inflammation, ischemia, diabetes, hypertension, and pulmonary fibrosis (Karnik et al., 2015; Kaschina, Namsolleck, & Unger, 2017; Sumners, Peluso, Haugaard, Bertelsen, & Steckelings, 2019). Activation of AT2-Rs usually counterbalances the effects of AT1-Rs and induces antihypertensive, antioxidative, anti-inflammatory, and anti-fibrotic effects (de Kloet, Steckelings, & Sumners, 2017; Karnik et al., 2015; Sumners, Peluso, Haugaard, Bertelsen, & Steckelings, 2019). Thus, the ACE2/Ang-(1-7)/MasR axis, along with AT2-Rs, has emerged as a physiological antagonist that counter-regulates the activity of the classical RAS pathway (Arendse et al., 2019; Chappell, 2016; Santos et al., 2018).

![The renin angiotensin system (RAS). Classical RAS consists of angiotensin converting enzyme (ACE) breaking down Angiotensin (Ang)-I into Ang-II, both of which can bind to either the AT1 (angiotensin type 1) or the AT2 (angiotensin type 2) receptor. Non-classical RAS consists of ACE2 converting Ang-I into Ang-(1-9) and Ang-II into Ang-(1-7). Ang-(1-7) stimulates the Mas receptor. Bradykinin and [des-Arg9]-bradykinin are degraded by ACE and ACE2, respectively, into pharmacologically inactive peptides.](/dataresources/secured/content-1765835139716-9cbb036c-5927-4e06-a179-c7600ef0cbc3/assets/gr1_lrg.jpg)
The renin angiotensin system (RAS). Classical RAS consists of angiotensin converting enzyme (ACE) breaking down Angiotensin (Ang)-I into Ang-II, both of which can bind to either the AT1 (angiotensin type 1) or the AT2 (angiotensin type 2) receptor. Non-classical RAS consists of ACE2 converting Ang-I into Ang-(1-9) and Ang-II into Ang-(1-7). Ang-(1-7) stimulates the Mas receptor. Bradykinin and [des-Arg9]-bradykinin are degraded by ACE and ACE2, respectively, into pharmacologically inactive peptides.
As a major driver of the ACE/Ang-II/AT1-R axis, Ang-II downregulates ACE2 expression by activating AT1-R-mediated upregulation of “extracellular signal-regulated kinase“ (ERK)1/2 and p38 mitogen-activated protein (MAP) kinase in human tubular kidney cells (Koka et al., 2008), rat aortic vascular smooth muscle cells (Gallagher, Ferrario, & Tallant, 2008a), cardiomyocytes (Gallagher, Ferrario, & Tallant, 2008b), and catecholaminergic neurons (L. Xiao, Haack, & Zucker, 2013). In Neuro-2A cells transfected with ACE2, AT1-R activation by Ang-II leads to internalization and subsequent destruction of ACE2 in lysosomes (Deshotels, Xia, Sriramula, Lazartigues, & Filipeanu, 2014). In addition, Ang-II activation of the AT1-R promotes ADAM17-mediated proteolytic cleavage of ACE2 in COS7 cells (Mifune et al., 2005), cardiomyocytes (Patel et al., 2014) and hypothalamic neurons (Xia, Sriramula, Chhabra, & Lazartigues, 2013; J. Xu, Sriramula, et al., 2017). Furthermore, Ang-II stimulates phosphorylation of three MAP kinases, i.e., p38 kinase, ERK 1/2, and c-Jun N-terminal kinase (JNK) to mediate its actions. It also increases the production of transforming growth factor β 1 (TGF-β1), which further suppresses ACE2 expression (Chou, Chuang, Lu, & Guh, 2013; Su, Zimpelmann, & Burns, 2006) and promotes ADAM17 activation (Ohtsu et al., 2006). Thus, Ang-II-induced down regulation of ACE2 expression, eventually leads to impaired conversion of Ang-II to Ang-(1-7) and causes further accumulation of Ang-II and RAS-mediated detrimental effects in a positive feedback cycle. Similar to Ang-II, another vasoconstrictive peptide, Endothelin-1, also downregulates ACE2 transcription by activating p38 MAP kinase and ERK1/2 pathways in human bronchial epithelial cells (H. Zhang, Li, Zeng, Wu, & Ou, 2013) and rat cardiomyocytes (Gallagher, Ferrario, & Tallant, 2008b).
As opposed to Ang-II, Ang-(1-7) and atrial natriuretic peptide (ANP) do not affect ACE2 expression (Deshotels, Xia, Sriramula, Lazartigues, & Filipeanu, 2014). However, both peptides counteract Ang-II-AT1-R-mediated down regulation of ACE2 by activating MAP kinase phosphatase in rat aortic vascular smooth muscle cells (Gallagher, Ferrario, & Tallant, 2008a), astrocytes (Gallagher, Chappell, Ferrario, & Tallant, 2006), and cardiomyocytes (Gallagher et al., 2008b). In addition, Ang-(1-7) and ANP inhibit ADAM17 activity (X. Ma et al., 2016; Zhai et al., 2018). In summary, while Ang-II downregulates ACE2 expression initiating a positive feedback mechanism leading to further elevation of Ang-II, Ang-(1-7) activates the Mas receptor and counteracts these cellular actions of Ang-II.
In addition to the RAS, ACE2 is involved in the regulation of the kinin-kallikrein system (KKS). Effector peptides of the KKS, mainly bradykinin (BK) and its active metabolite [des-Arg9]-BK (DABK), recognize two pharmacologically distinct G protein-coupled receptors: the B1 receptor, whose main agonist is DABK, and the B2 receptor, whose ligand is BK (Rhaleb, Yang, & Carretero, 2011). The RAS enzymes ACE and ACE2 degrade BK and DABK, respectively (Fig. 1) (Donoghue et al., 2000; Vickers et al., 2002). In addition, Ang-(1-7) produced by ACE2 downregulates ACE activity (Tom, de Vries, Saxena, & Danser, 2001) and potentiates BK-induced vasodilatations (Raffai, Khang, & Vanhoutte, 2014). Thus, downregulation of ACE2 activity by disease conditions, such as lung injury and SARS-CoV infections, may increase DABK levels. Over-activation of B1 receptors can contribute to the pathogenesis of these diseases. In an endotoxin-induced lung inflammation model, the loss of ACE2 function leads to an accumulation of DABK, an activation of B1 receptors, and the release of proinflammatory chemokines from airway epithelia. Examples of released chemokines are C-X-C motif chemokine 1 and 5, macrophage inflammatory protein-2, and cytokines, e.g., TNF-α. In this model, neutrophil infiltration as well as lung inflammation and injury have been increased (C. P. Sodhi et al., 2018).
In clinical studies, commonly used antihypertensive medications, such as β-adrenergic receptor blockers (βARBs) and calcium channel blockers (CCBs), are not associated with changes in plasma or urine ACE2 levels (Furuhashi et al., 2015). Interestingly, the A1075 allele of the ACE2 gene has been associated with increased mortality in male patients with acute coronary syndrome in the absence of, but not in the presence of βARB treatment, suggesting a pharmacogenetic effect linking ACE2 and βARBs (Palmer et al., 2008).
βARBs, mainly through β1 receptor-mediated inhibition of the sympathetic nervous system, negatively regulate the release of renin from juxtaglomerular cells in the kidney and are thereby involved in RAS regulation. The contribution of renal sympathetic activity on the ACE2/Ang-(1-7)/Mas receptor pathway of the RAS has been investigated in disease models. Renal denervation decreases blood pressure, cardiac and renal fibrosis, cardiomyopathy, and oxidative stress. It upregulates cardiac and renal ACE2 protein expression in isoproterenol-induced cardiomyopathy (Q. Liu et al., 2015), myocardial infarction (Feng et al., 2017) and hypertension models (W. Han et al., 2020; M. Wang et al., 2018). In a recent study, renal denervation improved cardiac function, decreased fibrosis, and upregulated hypothalamic ACE2 mRNA and protein expression in a heart failure model (W. J. Chen et al., 2019). In a rat model of type 2 diabetes mellitus with insulin resistance, renal denervation decreased plasma and renal tissue norepinephrine levels, improved vascular endothelial functions, and increased mRNA and protein expression of ACE2 in aortic endothelial cells. This effect was due to induction of autophagy via the “AMP-activated protein kinase” (AMPK) and “mammalian target of rapamycin” (mTOR) signaling pathways (Y. Wang, B. Rijal, et al., 2020), suggesting that ACE2 expression in different regions can be regulated by sympathetic activity. Following treatment with the non-selective β adrenoreceptor agonist isoproterenol, both increased (Nadu, Ferreira, Reudelhuber, Bader, & Santos, 2008) and decreased (Syed et al., 2016) cardiac ACE2 expression have been reported in hypertrophy models. Similarly, while, isoproterenol downregulated cardiac ACE2 expression in a myopathy model (Q. Liu et al., 2015), it upregulated cardiac ACE2 levels in a myocardial infarction model (Badae, El Naggar, & El Sayed, 2019). A recent study in rat salivary glands reported isoproterenol-induced downregulation of ACE2 mRNA expression in the parotid, but not in the sublingual and submandibular glands (Cano et al., 2019), suggesting that the effect of isoproterenol may vary between different tissues and disease models.
In spontaneously hypertensive (SH) rats, the βARB atenolol reduced the blood pressure to a similar extent as compared to hydralazine, a direct vasodilator, and olmesartan, an AT1-R blocker. However, atenolol and hydralazine showed no effect on ACE2 expression in both tissues (Igase, Strawn, Gallagher, Geary, & Ferrario, 2005), whereas olmesartan caused marked upregulation of ACE2 in aortic tissue, but had no effect on the carotid artery (Igase, Strawn, Gallagher, Geary, & Ferrario, 2005). Nebivolol, a βARB, did not decrease the blood pressure but reduced plasma renin concentration, cardiac Ang-II levels, oxidative stress, and fibrosis; cardiac ACE2 activity and mRNA levels remained essentially unchanged (Varagic et al., 2012). However, in another study on SH rats, the βARB propranolol alone or in combination with the AT1-R blocker losartan or the ACE-Inh. captopril decreased the blood pressure and markedly reduced ACE2 mRNA expression in the aorta (Lezama-Martinez et al., 2018). Of note, the βARB labetalol increased the maximal reaction rate and decreased the substrate specificity of ACE2 (Kulemina & Ostrov, 2011), suggesting that at least some βARBs can interact directly with ACE2. βARBs have also been shown to decrease proinflammatory cytokines, including IL-1β, IL-6, TNFα, IFNγ (Deten, Volz, Holzl, Briest, & Zimmer, 2003; Doo et al., 2001; Hajighasemi & Mirshafiey, 2016; Matsumura et al., 2002). Moreover, they reduce pulmonary edema (Rassler, 2012), inhibit NLRP3 inflammasome (Wong et al., 2018), and reduce the mortality and disease severity of acute respiratory distress syndrome (ARDS) (Al-Qadi & Kashyap, 2015; Noveanu et al., 2010) and chronic obstructive lung disease (COPD) (Nielsen, Pedersen, Sode, & Dahl, 2019), suggesting that βARBs may have beneficial effects on COVID-19. In addition to βARBs, ⍺1-AR antagonists have recently been shown to prevent cytokine responses and to increase the survival after inflammatory stimuli in mouse models (Staedtke et al., 2018). They also reduce morbidity and mortality in patients at risk for developing a cytokine storm syndrome (Vogelstein et al., 2020). Briefly, the sympathetic nervous system activates the RAS through βARs, βARBs inhibit renin release, and renal denervation upregulates cardiac, renal and hypothalamic ACE2 expression in various disease models. In addition, decreased reactive oxygen species (ROS) production, increased endothelial nitric oxide synthase (NOS) expression and NO formation can lead to upregulation of ACE2 expression (W. Han et al., 2020; Varagic et al., 2012). However, both increased and decreased ACE2 expression have been reported after application of the non-selective β adrenoreceptor agonist isoproterenol, and there is no consistent evidence that βARBs influence ACE2 expression or activity. In 221 hypertensive patients, no association between the use of βARBs and renal ACE2 gene expression was found (X. Jiang et al., 2020). In the context of COVID-19, a recent study with 880 COVID-19 patients reported that the use of βARBs was associated with a significantly better outcome (Pinto-Sietsma et al., 2020).
There are few studies investigating the effects of calcium channel blockers (CCBs) on ACE2 regulation. Amlodipine (an L-type CCB) and cilnidipine (an L- and N-type CCB), not alone but in combination with valsartan, decreased the blood pressure but did not change aortic ACE2 mRNA expression (Takai, Jin, Aritomi, Niinuma, & Miyazaki, 2013). In a mechanical stress model resulting in elevated ACE2 mRNA expression, Ang-II decreased ACE2 surface expression of pressurized human aortic endothelial cells, and nifedipine (an L-type CCB) reversed this effect (Iizuka, Kusunoki, Machida, & Hirafuji, 2009). Felodipine (an L-type CCB) decreased blood pressure, fibrosis and TGF-β1 expression; but no changes in renal ACE2 mRNA expression were observed in ischemic or non-ischemic hypertensive rats (S. Bai, Huang, Chen, Wang, & Ding, 2013). On the other hand, nimodipine treatment attenuated the reduction in brain ACE2 mRNA expression that occurs in ischemic brain tissue (Abdel-Fattah, Messiha, & Mansour, 2018). Similarly, amlodipine increased renal ACE2 levels in hypertensive rats (Onat & ŞAhna, 2018). In summary, data suggest that CCBs reverse reduced ACE2 expression in various disease models. Recently, in an analysis of 291 COVID-19 patients, the use of CCBs was not associated with increased disease severity or mortality rates (Fosbol et al., 2020). In cell culture experiments, CCBs of the dihydropyridine class, amlodipine, felodipine and nifedipine, at high concentrations (10-500 μM), were reported to limit the growth of SARS-CoV-2 in epithelial kidney (Vero E6) and epithelial lung (Calu-3) cells (Straus, Bidon, Tang, Whittaker, & Daniel, 2020).
The effects of most diuretic drugs on ACE2 are currently unknown. Among thiazide diuretics, hydrochlorothiazide increased cardiac ACE2 gene expression in normotensive but decreased it in hypertensive rats (Jessup, Brosnihan, Gallagher, Chappell, & Ferrario, 2008). However, mineralocorticoid receptor blockers (MRBs) have been reported to regulate ACE2 activity and expression in various disease models. In macrophages from heart failure patients, the MRB spironolactone reduced oxidative stress and lipid peroxide formation, accompanied by a markedly upregulated ACE2 expression, whereas aldosterone significantly reduced it (Keidar et al., 2005). Spironolactone also upregulated decreased ACE2 expression levels in aldosterone-treated cultured cardiomyocytes (Yamamuro et al., 2008) and kidney (Fukuda et al., 2011), as well as in kidneys of rats with obstructive jaundice (Kong et al., 2019), but not in human mesangial cells (Stoll, Yokota, Sanches Aragao, & Casarini, 2019). Eplerenone, another MRB, did not consistently reverse decreased ACE2 levels in rats with experimental heart failure (Karram et al., 2005) and in the hearts of hypertensive rats (Takeda et al., 2007), but completely reversed aldosterone- and high salt-induced down regulation of renal ACE2 expression (Bernardi et al., 2015). Similarly, eplerenone reversed the aldosterone-induced, p47-mediated downregulation of ACE2 expression in mouse macrophages, heart and kidney (Keidar et al., 2005). In addition, eplerenone was reported to inhibit ADAM17 activity in human monocytes (Satoh, Ishikawa, Minami, Akatsu, & Nakamura, 2006), which should potentially promote cell surface ACE2 activity. In line with this, in a diabetic nephropathy model, improvement of kidney pathology by spironolactone was associated with decreased serum ACE2 levels (Dong et al., 2019).
In summary, the majority of studies points to an increase in ACE2 expression after MRB treatment, mainly by counteracting aldosterone-induced down regulation of ACE2. In addition, mitigation of the deleterious effects of obesity on the RAS, possibly reducing obesity-related COVID-19 complications (Feraco et al., 2013; Vecchiola et al., 2020) and direct anti-inflammatory and antiviral effects of MRBs, could be beneficial in the treatment of pulmonary COVID-19 complications (Cadegiani, Wambier, & Goren, 2020). Importantly, MRBs such as spironolactone possess a significant anti-androgenic activity, which may be beneficial in the context of SARS-CoV-2 infection, by inhibiting the androgen-dependent expression of “Transmembrane protease, serine 2” (TMPRSS2), a transmembrane protease crucial for SARS-CoV-2 entry (Liaudet & Szabo, 2020). In addition, potassium canrenoate (the active metabolite of spironolactone) results in concentration (0.1-10 μM)-dependent reductions of the binding of the SARS-CoV-2 spike protein to the ACE2 receptor (Carino et al., 2020). Increased plasma aldosterone levels associated with disease severity in COVID-19 patients (Villard et al., 2020) suggest that MRBs may have beneficial effects in COVID-19. A recent study concluded that canrenone decreased all-cause mortality and improved the clinical outcome in a small cohort of 30 COVID-19 patients with diseases ranging from moderate to severe (M. Vicenzi et al., 2020). Another diuretic, furosemide, significantly decreased lipopolysaccharide-induced proinflammatory cytokine production in cell lines and potently inhibited IL-6 and TNF-α release (Z. Wang, Y. Wang, et al., 2020), suggesting its potential use in hypercytokinemic conditions in COVID-19.
Blockers of AT1 receptors and ACE-Inhs. are the most commonly used drugs in the treatment of hypertension and cardiovascular diseases (Zolk et al., 2020). As mentioned earlier, ACE2 antagonizes the effects of Ang II. At the cellular level, Ang-II, mainly by acting on AT1 receptors, downregulates the expression of ACE2 (Ferrario, Ahmad, & Groban, 2020). Therefore, it can be expected that either the inhibition of Ang-II production by ACE-Inhs. or the blockade of AT1 receptors may lead to upregulation of ACE2 expression. In addition, activation of peroxisome proliferator-activated receptors (Harada et al., 2016; Horiuchi, Iwanami, & Mogi, 2012; Maquigussa et al., 2018; Michel, Foster, Brunner, & Liu, 2013; Z. Z. Zhang et al., 2013) and sirtuin 1 (SIRT1) (Strycharz et al., 2018) by AT1-R blockers (ARBs), such as telmisartan, losartan and irbesartan, may further contribute to the upregulation of ACE2 expression (Goltsman et al., 2019; Gupte et al., 2008; W. Zhang et al., 2014).
Detailed lists of experimental studies assessing the effects of ACE-Inhs. and ARBs on the expression or activity of ACE2 are provided in Table 1, Table 2 , respectively. Not surprisingly, the majority of experimental studies supports the assumption that RAS inhibition upregulates ACE2 activity and expression, although there appear to be some differential responses between ARBs versus ACE-Inhs., between drugs belonging to the same group of drugs, and between different tissues and species. For example, in normotensive Lewis and hypertensive mRen2.Lewis male rats, the ARB losartan markedly increased ACE2 activity in the heart (Ferrario et al., 2005; Ferrario et al., 2005); a similar increase in cardiac ACE2 activity was reported for the ARB eprosartan in rats with heart failure (Karram et al., 2005). The ACE-Inh. lisinopril, however, either failed to increase cardiac ACE2 activity (Lewis rats) or stimulated it to a lesser extent than losartan (in murine Ren2 renin transgenic rats), despite similar reductions in blood pressure (Ferrario, Jessup, Chappell, et al., 2005; Ferrario, Jessup, Gallagher, et al., 2005; Jessup et al., 2006). In the kidneys of both strains, losartan and lisinopril increased ACE2 activity (Ferrario, Jessup, Chappell, et al., 2005; Ferrario, Jessup, Gallagher, et al., 2005; Jessup et al., 2006), although to a lesser degree compared to the heart. On the other hand, it was found that the ACE-Inh. ramipril reduced cardiac ACE2 activity in a rat model of kidney injury (L. Burchill et al., 2008).

| Pharmacological agent/class | Experimental model /Tissue/Subject | Effect | Reference |
|---|---|---|---|
| Ramipril/ ACE-Inh. | Male Sprague-Dawley rats/ Streptozocin induced diabetes model | Increased renal ACE2 immunostaining and protein expression in diabetic rats. ACE2 expression decreased markedly in diabetic rats. | Tikellis et al. (2003) |
| Ramipril/ ACE-Inh. | Sprague–Dawley rats/ Myocardial ischemia induced by ligation of the left coronary artery | Cardiac ACE2 mRNA expression and ACE2 activity increased by myocardial ischemia. Ramipril did not cause any change. | Burrell et al. (2005) |
| Lisinopril/ ACE-Inh. | Lewis rats/ Heart | Decrease in plasma Ang II, increase in plasma Ang 1–7 and ACE2 mRNA, but not cardiac ACE2 Activity | Ferrario, Jessup, Chappell, et al. (2005) |
| Lisinopril/ACE-Inh. Losartan/ARB | Lewis rats/ Kidney | Lisinopril or Losartan treatment were both associated with increases in ACE2 activity but used in combination, did not produce this effect. | Ferrario, Jessup, Gallagher, et al. (2005) |
| Lisinopril/ACE-Inh. | Lewis and hypertensive mRen2.Lewis rats | Increased renal ACE2 mRNA expression in hypertensive but not in normotensive rats. | Chappel and Ferrario (2006) |
| Enalapril/ACE-Inh. | Sprague Dawley rats/ Coronary artery ligation in heart | Increased plasma and cardiac ACE2 activity, and cardiac ACE2 mRNA levels 8 weeks post-surgery | Ocaranza et al. (2006) |
| Lisinopril/ACE-Inh. | Transgenic Ren2 rats/ Heart and kidney | Decrease in plasma Ang II, increase in plasma Ang 1–7, cardiac and renal ACE2 mRNA and activity | Jessup et al. (2006) |
| Lisinopril/ ACE-Inh. | Lewis rats/ Kidney | No change in kidney ACE2 mRNA, but increased ACE2 activity | Ferrario, Jessup, Gallagher, et al. (2005) |
| Lisinopril/ ACE-Inh. | Wistar rats/ Dietary sodium restriction | Renal ACE2 activity was unchanged with lisinopril treatment in either group | Hamming et al. (2008) |
| Perindopril/ ACE-Inh. | Male C57BL/6 mice/ Streptozotocin induced diabetes model | Decreased renal ACE2 activity and mRNA expression in both control and diabetic mice | Tikellis et al. (2008) |
| Ramipril/ ACE-Inh. | Sprague Dawley rats/ Acute kidney injury model | Decreased cardiac ACE2 activity and protein expression | L. Burchill et al. (2008) |
| Ramipril/ ACE-Inh. | Sprague Dawley rats/ Kidney nephrectomy model | No change in renal ACE2 activity. Increased with nephrectomy | Velkoska, Dean, Burchill, Levidiotis, & Burrell (2010) |
| Perindopril/ ACE-Inh. | Male Wistar rats and HSC-T6 cells/ CCl4-induced liver fibrosis model | Increased ACE2 mRNA and protein expression in fibrotic liver. Perindopril alone no effect on HSC-T6 cells | M. L. Huang et al., 2010, Huang et al., 2020 |
| Ramipril/ ACE-Inh. | Sprague Dawley rats/ Kidney after subtotal nephrectomy | Ramipril had no effect on ACE2 in cardiac or renal tissue. Reduced ACE2 activity in renal cortex by nephrectomy was reversed by ramipril | Burrell et al. (2012) |
| Fosinopril/ ACE-Inh. | Male Sprague-Dawley rats/ Coronary artery ligation induced disease model | No change in cardiac ACE2 mRNA expression | Y. Wang, C. Li, et al. (2012) |
| Ramipril/ ACE-Inh. + Valsartan/ ARB | Sprague Dawley rats/ Myocardial infarction model | ACE2 expression was not altered but may have decreased in viable myocardium border or infarct zones, (unclear statistical analysis). | L. J. Burchill et al. (2012) |
| Perindopril/ ACE-Inh. +Losartan/ ARB | Akita Agt-Transgenic C57BL/6 mice/ Hypertension model | Marked increase in renal ACE2 mRNA and protein expression in hypertensive mice | Lo et al. (2012) |
| Enalapril/ ACE-Inh. | Spontaneously Hypertensive rats/ Heart | ACE2 mRNA expression was increased but ACE2 protein expression did not change with ACE-Inh. treatment | Z. Yang et al. (2013) |
| Enalapril/ ACE-Inh. | Male C57BL/6 mice/ High fat diet model | Increased pancreatic ACE2 protein expression | Frantz, Crespo-Mascarenhas, Barreto-Vianna, Aguila, and Mandarim-de-Lacerda (2013) |
| Imidapril/ ACE-Inh. | Broiler chickens/ Low temperature induced cardiac remodeling | Decreased cardiac ACE2 mRNA expression | X. Q. Hao et al. (2013) |
| Imidapril/ ACE-Inh. | Broiler chickens/ Low temperature induced pulmonary hypertension model | Decreased pulmonary ACE2 mRNA expression | X. Q. Hao et al. (2014) |
| Captopril/ ACE-Inh. | Male Wistar rats/ Coronary artery occlusion induced myocardial infarction model | Markedly decreased cardiac ACE2 mRNA and protein expression in infarcted heart | Flores-Monroy, Ferrario, Valencia-Hernandez, Hernandez-Campos, & Martinez-Aguilar (2014) |
| Enalapril/ ACE-Inh. +Losartan/ ARB | Sprague Dawley rats/ Cardiac remodeling from aortic constriction | ACE2 cardiac protein expression was increased (~3-fold) with both drugs in rats with cardiac remodeling; data were not provided for animals with sham surgery. | Y. Zhang et al. (2014) |
| Captopril/ ACE-Inh. | Mouse Lewis lung carcinoma cells/ Hypoxia model | Increased ACE2 protein expression in hypoxic cells with markedly decreased ACE2 protein expression levels | L. Fan et al. (2014) |
| Perindopril/ ACE-Inh. | Male Wistar rats/ Streptozotocin induced diabetes model | Increased cardiac ACE2 protein expression in diabetic rats | P. P. Hao et al. (2015) |
| Captopril/ ACE-Inh. | Male Sprague Dawley rats/ Lipopolysaccharide induced lung injury model | Increased pulmonary ACE2 protein expression in controls and marked increase in injured lungs | Y. Li et al. (2015) |
| Captopril/ ACE-Inh. | Landrace pigs/ porcine cardiac arrest model | No change in serum ACE2 | H. L. Xiao et al. (2016) |
| Captopril/ ACE-Inh. | Male Wistar rats/ Aortic coarctation-induced hypertension model | No effect on cardiac ACE2 mRNA expression in sham and hypertentensive group. | Ibarra-Lara et al. (2016) |
| Enalapril/ ACE-Inh. | Swine/ cardiac arrest and resuscitation model | Compared to controls, enalapril did not alter myocardial ACE2 mRNA and protein expression | G. Wang, Zhang, Yuan, Wu, & Li et al. (2016) |
| Cilazapril/ ACE-Inh. | Male Wistar rats/ Doxorubicin-induced cardiomyopathy model | No change in cardiac ACE2 protein expression in doxorubicin treated rats | H. Ma et al. (2017) |
| Ramipril/ ACE-Inh. | Male Sprague Dawley rats/ Subtotal nephrectomy induced kidney disease model | No effect on cardiac ACE2 activity in subtotal nephrectomized rats | Burrell, Gayed, Griggs, Patel, & Velkoska (2017) |
| Captopril/ ACE-Inh. | Female Wistar rats/ Ovariectomized rat model for osteoporosis | Increased bone ACE2 protein expression in osteoporotic rats. But no effect in control rats | Abuohashish, Ahmed, Sabry, Khattab, & Al-Rejaie (2017) |
| Captopril/ ACE-Inh. | Land race pigs/Pulmonary embolism model | No change in pulmonary ACE2 protein expression | H. L. Xiao et al. (2018) |
| Captopril/ ACE-Inh. | Spontaneously hypertensive and Wistar Kyoto rats/ Aortic tissue | Markedly decreased aortic ACE2 mRNA expression in hypertensive rats with significantly upregulated ACE2 levels | Lezama-Martinez et al. (2018) |
| Captopril/ ACE-Inh. | Male Sprague-Dawley rats/ Focal cerebral ischemia model | Increased brain ACE2 activity in controls and ischemic brains | Tao et al. (2018) |
| Captopril/ ACE-Inh. | Landrace pigs/ Acute pulmonary embolism model | No change in cardiac ACE2 immunostaining and protein expression | H. L. Xiao et al. (2019) |
| Enalapril/ ACE-Inh. | Male Wistar albino rats/ Isoproterenol induced myocardial infarct model | No change in cardiac ACE2 concentration in infarcted cardiac tissue | Badae, El Naggar, and El Sayed (2019) |
| Captopril/ ACE-Inh. | Male Wistar rats/ SiO2 inhalation induced lung injury model | Increased ACE2 protein expression in lung and pulmonary fibroblasts, also increased serum ACE2 level | B. N. Zhang et al. (2019) |
| Perindopril/ ACE-Inh. | Female Sprague Dawley rats/ Hyperlipidemic Alzheimer disease model | Reversed the decreased hippocampal ACE2 mRNA expression in rats with Alzheimer disease | Messiha, Ali, Khattab, & Abo-Youssef (2020) |
| Enalapril/ ACE-Inh. | Male Swiss mice/ Hyperlipidic diet-induced obesity model | Highly significant increase in hepatic ACE2 gene expression | Moraes et al. (2020) |
| Losartan/ARB Olmesartan/ARB | Lewis rats /Coronary artery ligation in heart | Increase in plasma Ang II, Ang 1–7 and ACE2 mRNA 28 days post surgery | Ishiyama et al. (2004) |
| Eprosartan/ARB | Male Wistar rats/ Aortocaval fistula induced heart failure model | Heart failure caused decreased cardiac ACE2 expression and enzyme activity was restored by eprosartan | Karram et al. (2005) |
| Losartan/ ARB | Lewis rats/ Heart | Increase in plasma Ang II, Ang 1–7 levels, ACE2 mRNA and cardiac ACE2 activity | Ferrario, Jessup, Chappell, et al. (2005) |
| Olmesartan/ ARB | Spontaneously hypertensive rats/ Aorta | Markedly increased aortic ACE2 immunostaining and mRNA expression. But no effect on carotid artery | Igase et al. (2005) |
| Losartan/ ARB | Transgenic Ren2 rats/ Heart and kidney | Increase in plasma Ang II, Ang 1–7, cardiac and renal ACE2 mRNA and activity | Jessup et al. (2006) |
| Losartan/ ARB | Lewis and hypertensive mRen2.Lewis rats | Increased renal ACE2 mRNA expression in hypertensive but not in normotensive rats. | Chappel and Ferrario (2006) |
| Olmesartan/ ARB | Spontaneously hypertensive rats and Wistar Kyoto rats/ Heart | Olmesartan significantly increased the cardiac ACE2 expression level compared to that in Wistar Kyoto rats and SHRSP treated with a vehicle | Agata et al. (2006) |
| Valsartan/ ARB | Male transgenic Ren2 and Sprague-Dawley rats/ Hypertension model | Iincreased renal ACE2 mRNA expression in Ren2 rats | Whaley-Connell et al. (2006) |
| Candesartan/ ARB | Dahl salt-sensitive hypertensive rats/ Hypertension model | Increased cardiac ACE2 mRNA and protein expression in hypertensive rats | Takeda et al. (2007) |
| Losartan/ ARB | 3T3-L1 murine adipocytes | No change on the ACE2 mRNA expression. | Gupte et al. (2008) |
| Olmesartan/ ARB | Male spontaneously hypertensive rats/ Balloon induced carotid artery injury | Increased carotid artery intima ACE2 immunostaining in injured group. But no effect in uninjured intima | Igase, Kohara, Nagai, Miki, & Ferrario (2008) |
| Losartan/ ARB | Transgenic and hypertensive C57BL/6J mice | No change in brain ACE2 protein expression, but activity increased. | Xia, Feng, Obr, Hickman, & Lazartigues (2009) |
| Telmisartan/ ARB | C57BLKS/J mice/ Kidney | Following 2 weeks administration, increased ACE2 protein levels, and ACE2 mRNA expression | Soler et al. (2009) |
| Olmesartan/ ARB | Male Wistar rats/ Pressure-overload cardiac hypertrophy model | Increased cardiac ACE2 mRNA expression in hypertrophic hearts | Kaiqiang, Minakawa, Fukui, Suzuki, & Fukuda (2009) |
| Losartan/ ARB | Male Wistar rats/ Lipopolysaccharide induced septic shock model | Increased lung ACE2 protein expression | Hagiwara et al. (2009) |
| Losartan/ ARB | Male Sprague-Dawley/ cigarette smoke induced pulmonary hypertension model | No effect on ACE2 protin expression in pulmonary smooth muscle cell cultures, but increased ACE2 expression in smoke exposed lungs and cell cultures. | S. X. Han et al. (2010) |
| Losartan/ ARB | Male FVB/NJ mice/ Nephrectomy induced kidney disease model | No effect on renal ACE2 avtivity and protein expression in nephrectomized rats | Dilauro, Zimpelmann, Robertson, Genest, & Burns (2010) |
| Losartan/ ARB | Sprague Dawley rats/ cigarette smoke-induced lung damage | ACE2 expression was unchanged in control rats by either dose of losartan. Animals exposed to cigarette smoke had reduced ACE2, which losartan treatment restored | S. X. Han et al. (2010) |
| Losartan/ ARB | Male C57BL/6 mice/ Fructose diet | Losartan alone increased renal ACE2 protein expression but no effect on ACE2 activity; also reversed the increasing effect of fructose | Senador et al. (2010) |
| Losartan/ ARB | Sprague Dawley rats/ Acute Respiratory Distress Syndrome model in the lung | Restored ACE2 activity decreased by the injury. ACE2 activity decreased in controls | Wosten-van Asperen et al. (2011) |
| L-158,809/ ARB | Fischer 344 rats/ Dorsomedial medulla of the brain | L-158,809 induced 2-fold increase in brain ACE2 mRNA expression | Gilliam-Davis et al. (2011) |
| Losartan/ ARB | Male Sprague-Dawley rats/ Lipopolysaccharide and mechanical ventilation induced lung injury models | Decreased pulmonary ACE2 activity in only ventilated rats, increased activity in lung injured rats | Wosten-van Asperen et al. (2011) |
| Candesartan/ ARB | Male Lewis rats/ Fischer-to-Lewis renal transplantation model | Decreased serum ACE2 activity | Rusai et al. (2011) |
| Telmisartan/ ARB | Male Lewis rats/ Experimental autoimmune myocarditis model | Decrease of ACE2 protein expression and immunoreactivity caused by myocarditis was partially reversed by telmisartan | V. Sukumaran et al. (2011) |
| Olmesartan/ ARB | Transgenic C57BL/6J mice overexpressing renin and angiotensinogen | Markedly increased cardiac ACE2 activity and mRNA expression after NOS inhibition | Inaba et al. (2011) |
| Telmisartan/ ARB | Male spontaneously hypertensive and male Wistar–Kyoto rats/ | Decreased ACE2 mRNA in aorta of hypertensive group was upregulated by telmisartan | J. C. Zhong et al. (2011) |
| Irbesartan/ ARB | C57BL/6 mice/ Aorta | Treatment with irbesartan significantly augmented ACE2 protein levels and ACE2 mRNA expression | Jin et al. (2012) |
| Olmesartan/ ARB | C57BL/6J mice/ Vascular cuff injury model | Increased vascular ACE2 mRNA expression in injured rats. | Iwai et al. (2012) |
| Olmesartan/ ARB | Male Lewis rats/ Cardiac myosin-induced dilated cardiomyopathy model | Decrease of myocardial ACE2 mRNA and protein expression in cardiomyopathy group was partially reversed by olmesartan | V. Sukumaran et al. (2012) |
| Telmisartan/ ARB | Male Lewis rats/ Autoimmune myocarditis cardiomyopathy model | Increased cardiac ACE2 immunostaining and protein epression in cardiomyopathic rats | V. Sukumaran et al. (2012) |
| Telmisartan/ ARB | Male Sprague-Dawley rats/ Bile duct ligation induced hepatic fibrosis model | Increased liver ACE2 immunostaining, mRNA and protein expression | Yi, Liu, Wen, & Yin (2012) |
| Losartan/ ARB | Akita Agt-Transgenic C57BL/6 mice/ Hypertension model | Marked increase in renal ACE2 mRNA and protein expression in hypertensive mice | Lo et al. (2012) |
| Candesartan/ ARB | Male Lewis rats/ Myosin induced cardiotoxicity | Increased cardiac ACE2 protein expression | Arumugam et al. (2012) |
| Losartan/ ARB | Male C57BL/6 mice/ High fat diet model | No change in pancreatic ACE2 protein expression | Frantz et al. (2013) |
| Losartan/ ARB | Balb/c, FVBN wild and Mas receptor knockout mice/ Adriamycin-induced nephropathy model | Increased renal ACE2 protein expression in Adriamycin treated mice. | Silveira et al. (2013) |
| Olmesartan/ ARB | mRen2.Lewis hypertensive rats/ Kidney | Increased ACE2 mRNA and protein | Varagic et al. (2013) |
| Valsartan/ ARB | Male Wistar-Kyoto and spontaneously hypertensive rats | Increased aortic ACE2 mRNA expression | Takai, Jin, Aritomi, Niinuma, and Miyazaki (2013) |
| Losartan/ ARB | Male Wistar rats/ Aaortic coarctation induced hypertrophy model | No change in coronary ACE2 immunostaining. | Souza et al. (2013) |
| Azilsartan/ ARB | AT2 and Mas knockout mice, both on C57BL/6J Background/ Vascular injury model | Increased vascular ACE2 mRNA expression in injured tissues from wild and knockout mice | Ohshima et al. (2014) |
| Irbesartan/ ARB | C57BL/6 mice/Heart | Increase in cardiac ACE2 mRNA, Irbesartan prevented Ang II induced decrease in ACE2 protein levels | Patel et al. (2014) |
| Azilsartan, Olmesartan/ ARB | Transgenic hRN/hANG-Tg mice | Decrease of ACE2 mRNA expression in transgenic mice was attenuated by azilsartan but not olmesartan | Iwanami et al. (2014) |
| Losartan/ ARB | Mouse Lewis lung carcinoma cells/ Hypoxia model | Increased ACE2 protein expression in hypoxic cells with markedly decreased ACE2 protein expression levels | L. Fan et al. (2014) |
| Losartan/ ARB | Male Sprague–Dawley rats/ Cigarette smoke induced pulmonary hypertension | No effect on lung ACE2 protein expression, but cigarette smoke decreased ACE2 protein expression | Y. M. Yuan et al. (2015) |
| Losartan/ ARB | Male New Zealand white rabbits/ High-cholesterol diet atherosclerosis model | ACE2 activity, protein expression increased in aortic plaque. Losartan further increased these values. | Y. H. Zhang et al. (2015) |
| Losartan/ ARB | Spontaneously hypertensive rats | Increased renal, but not cardiac ACE2 mRNA expression. | Klimas et al. (2015) |
| Valsartan/ ARB | Male Wistar rats/ Balloon-injured neointimal hyperplasia model | Injury induced ACE2 mRNA and protein expression was reversed by valsartan | Y. Li et al. (2016) |
| Olmesartan, Candesartan, Telmisartan, Losartan, Valsartan and Irbesartan/ ARB | Male C57BL/6 mice/ Transverse aortic constriction induced heart failure model | Heart failure suppressed the ACE2 protein expression and all ARBs tested upregulated ACE2. | X. Wang et al. (2016) |
| Telmisartan/ ARB | Male Sprague-Dawley rats/ Angiotensin II induced hypertension model | Reversed Ang-II-induced reduction in activity and immunostaining of cardiac ACE2 | F. Bai et al. (2016) |
| Candesartan/ ARB | Male transgenic diabetic mice | Increased renal ACE2 protein expression in diabetic mice | Callera et al. (2016) |
| Olmesartan/ ARB | Transgenic and C57BL/6N mice/ Cardiac hypertrophy model | Reversal of cardiac ACE2 mRNA expression decreased in cardiac hypertrophy model | Tanno et al. (2016) |
| Losartan/ ARB | Male C57BL/6 mice/ Unilateral ureteral obstruction model | Increased renal ACE2 mRNA expression | de Jong et al. (2017) |
| Azilsartan/ ARB | Male db/db mice/ Diabetic mice model | Increased cardiac ACE2 protein expression in diabetic mice. No effect on non-diabetic mice | Vijayakumar Sukumaran, Tsuchimochi, Tatsumi, Shirai, & Pearson (2017) |
| Irbesartan/ ARB | Male C57BL/6J mice/ Restraint stress model | Increase of intestinal ACE2 immunostaining and mRNA expression that was suppressed by stress. | Yisireyili et al. (2018) |
| Olmesartan/ ARB | Male Golden Syrian hamsters/ Fluorouracil-induced mucositis model | ACE2 mRNA expression upregulated by fluorouracil was reduced by olmesartan | Araujo et al. (2018) |
| Losartan/ ARB | Spontaneously hypertensive and Wistar Kyoto rats/ Aortic tissue | Markedly decreased aortic ACE2 mRNA expression in hypertensive rats with significantly upregulated ACE2 levels | Lezama-Martinez et al. (2018) |
| Olmesartan/ ARB | Male renin overexpressing, Ren-TG, and C57BL/6N mice/ Hypertension model | Decreased renal ACE2 mRNA and protein expression in hypertensive mice was reversed by olmesartan | Ichikawa et al. (2018) |
| Telmisartan/ ARB | Male Wistar rats / Cerebral ischemia-reperfusion model | Increased brain ACE2 mRNA expression of down regulated ACE2 in ischemic brain tissue. | Abdel-Fattah, Messiha, and Mansour (2018) |
| Losartan, telmisartan / ARB | C57BL/6 mice / High-fat obesity model | High-fat induced decrease of ACE2 mRNA expression was reversed by losartan and telmisartan | Graus-Nunes et al. (2019) |
| Losartan / ARB | Male albino rats / High fat high sucrose induced diabetes model | Increased adipose tissue ACE2 protein expression diabetic rats | Sabry et al. (2019) |
| Valsartan/ ARB | Female spontaneously hypertensive and Wistar-Kyoto rats | Increased cardiac ACE2 mRNA and protein expression | Y. Zhao et al. (2019) |
| Azilsartan/ ARB | Male Wistar-Kyoto rats / Adenine-induced chronic renal failure model | No significant change of renal ACE2 levels in immunostaining and immunoblotting analysis compared to vehicle group | Kidoguchi et al. (2019) |
| Losartan / ARB | Male Wistar rats / Losartanl treatment of salivary gland | No effect on ACE2 mRNA expression in parotid, sublingual and submandibular glands | Cano et al. (2019) |
| Telmisartan/ ARB | Male Wistar rats / Streptozotocin induced diabetes model | No change in renal ACE2 protein level. | Malek, Sharma, Sankrityayan, & Gaikwad (2019) |
| Telmisartan/ ARB Captopril/ ACE-Inh. act | Male Sprague-Dawley rats / Pregabalin-Induced Heart Failure | Pregabalin induced suppression of cardiac ACE2 protein expression was completely reversed by telmisartan and captopril | Awwad, El-Ganainy, ElMallah, Khattab, & El-Khatib (2019) |
| Telmisartan/ ARB | Female Wistar rats / D-Galactose treated ovariectomised, Alzheimer model | Increased hippocampal ACE2 protein expression | Abdelkader, Abd El-Latif, & Khattab (2020) |
Interestingly, a recent study reported that renal ACE2 levels were decreased and pulmonary ACE2 levels remained unchanged in ACE knockout mice or in mice treated with ACE-Inhs. or ARBs (Jan Wysocki, Lores, Ye, Soler, & Batlle, 2020). In another recent study, treatments with the ACE-Inh. enalapril or the ARB losartan did not affect ACE2 mRNA expression in lung, ileum, kidney, and heart of normotensive healthy C57BL/6J mice (Congqing Wu et al., 2020). Similarly, treatment with the ACE-Inh. lisinopril (100 nM) did not alter ACE2 expression in A549 lung cancer cells (Bartova, Legartova, Krejci, & Arcidiacono, 2020). In another recent study on human alveolar adenocarcinoma (A549) and lung cancer (Calu-3) cell lines, Ang-I (10-1000 nM) and Ang-II (1-100 nM) did not alter ACE2 expression. Treatment with ARBs, such as losartan and valsartan, and ACE-Inhs., such as lisinopril and captopril, did not affect ACE2 expression in these pulmonary cells (Baba et al., 2020).
The results of clinical studies investigating the effects of therapeutic concentrations of ARBs and ACE-Inhs. on ACE2 levels in biopsy, plasma and urine samples are provided in Table 3 . The majority of these studies reports no effect of ACE-Inhs. and ARBs on samples obtained from patients with cardiovascular diseases. In a recent study on kidney biopsies of diabetic patients, the use of ARBs and ACE-Inhs. did not change ACE2 mRNA expression (R. E. Gilbert et al., 2020). However, in atrial biopsies from patients with cardiovascular diseases, treatment with ARBs and ACE-Inhs. significantly increased ACE2 mRNA expression (Lebek et al., 2020).
| Pharmacological agent/class | Experimental model /Tissue/Subject | Effect | Reference |
|---|---|---|---|
| ACE-Inh. undefined | 58 patients with renal disease | No change in immunolocalization of renal ACE2 | Lely, Hamming, van Goor, & Navis (2004) |
| ACE-Inh. and ARB undefined | Plasma ACE2 activity was assayed from 228 patients with heart failure. | No association was found between ACEI/ARB use and ACE2 levels. | Epelman et al. (2008) |
| ACE-Inh. and ARB undefined | 13 patients with diabetic nephropathy | No change in kidney ACE2 mRNA levels compared to controls. But ACE-Inh. or ARB was associated with increased renal ACE2 mRNA expression in control subjects | Reich, Oudit, Penninger, Scholey, & Herzenberg (2008) |
| ACE-Inh. and ARB undefined | 113 patients with chronic systolic heart failure | No association was found between ACE-Inh. and ARB use and ACE2 levels. | Epelman et al. (2009) |
| ACE-Inh. and ARB undefined | 859 patients with type 1 diabetes and 204 healthy control subjects. | Mild increase in serum ACE2 was increased ~10 to 20% (higher in women) In diabetics using ACEIs, No association was found between ARB usage and ACE2 levels. | Soro-Paavonen et al. (2012) |
| ACE-Inh. and ARB undefined | 113 kidney transplant patients. 45 patient using ACE-Inh. and ARB | No effect on serum ACE2 activity | Soler et al. (2012) |
| ACE-Inh. and ARB undefined | 239 patient with chronic kidney disease | No effect on plasma ACE2 activity | Roberts, Velkoska, Ierino, & Burrell (2013) |
| ACE-Inh. undefined | 95 patients with ST-elevation myocardial infarction. | No association was found between ACE-Inh. and serum ACE2 levels. | Ortiz-Perez et al. (2013) |
| ACE-Inh. and ARB undefined | 70 patients with acute decompensated heart failure | Baseline or changes in serum ACE2 activity were not associated with the use of ACE-Inh. and ARB | Shao et al. (2013) |
| ACE-Inh. and ARB undefined | 46 patients/ intestinal biopsies | Increased intestinal ACE2 mRNA levels in ACE-Inh. treatment group compared to controls. But no change in ARB group | Vuille-dit-Bille et al. (2015) |
| ACE-Inh. and ARB undefined | 2004 chronic kidney patient. | ARB, but not ACE-Inh. increased plasma ACE2 activity compared to non-treated patients. | Anguiano et al. (2015) |
| ACE-Inh. and ARB undefined | 239 hypertensive patients, and 188 patients with heart failure | No association was found between ACE-Inh. and ARB use and serum ACE2 levels | Uri et al. (2016) |
| ACE-Inh. and ARB undefined | 161 hypertensive patients.45 patients are treated with ACE-Inh. and ARB | No effect on serum ACE2 concentration | S. Li et al. (2017) |
| Captopril/ ACE-Inh. Losartan/ ARB | 71 patients with chronic kidney disease in hemodialysis | Both drugs did not change ACE2 mRNA expression in hemodialysis patients. | Trojanowicz et al. (2017) |
| Lisinopril/ ACE-Inh. | 140 patients with essential hypertension | Lower serum ACE2 levels in patients treated with Lisinopril | Hristova, Stanilova, & Miteva (2019) |
| ACE-Inh. and ARB undefined | 127 patients with aortic stenosis | No association was found between ACE-Inh. and ARB use and plasma ACE2 activity | Ramchand et al. (2020) |
| ACE-Inh. and ARB undefined | 88 patients with atrial fibrillation. | No association was found between plasma ACE2 levels and ACEI/ARB use. | Walters et al. (2017) |
| ACE-Inh. and ARB undefined | 79 patients with obstructive coronary artery disease. | Plasma ACE2 levels had no association with use of ACE-Inh. and ARB | Ramchand, Patel, Srivastava, Farouque, and Burrell (2018) |
| ACE-Inh. and ARB undefined | 50 patients with diabetic nephropathy. All patients were treated with ACE-Inh. and/or ARB | No effect on urinary ACE2 mRNA expression compared to controls | G. Wang et al. (2008) |
| ACE-Inh. and ARB undefined | 190 patients with chronic kidney disease | No significant difference in urinary ACE2 compared to controls | Mizuiri et al. (2011) |
| Olmesartan/ ARB | 31 type 2 diabetes patients with nephropathy | Increased urinary ACE2 levels independently of blood pressure | Abe, Oikawa, Okada, & Soma (2015) |
| ACE-Inh. and ARB undefined | 152 patients with chronic kidney disease | Associated with increased urine ACE2 levels | Abe, Maruyama, Oikawa, Maruyama, Okada & Soma. (2015) |
| ACE-Inh. (enalapril) | 100 hypertensive patients. | Olmesartan increased urinary ACE2. Enalapril, losartan, valsartan, candesartan, valsartan and telmisartan had no effect. | Furuhashi et al. (2015) |
| ARB (losartan, valsartan, candesartan, valsartan and telmisartan, olmesartan). | |||
| ACE-Inh. and ARB undefined | 132 Type-2 Diabetic patients, 58 patients using ACE-Inh. and ARB | Elevated urinary ACE2 levels in diabetic hypertensive patients were significantly decreased by ACE-Inh. and ARB | Y. Liang et al. (2015) |
| ACE-Inh. and ARB undefined | 75 patients with Type-2 diabetes | No effect on urinary ACE2 levels | Mariana et al. (2016) |
| ACE-Inh. and ARB undefined | 76 patients with and without chronic kidney disease | No change in urine ACE2 concentrations | J. Wysocki et al. (2017) |
Concerning plasma ACE2 levels, a recent study with 2,022 heart failure patients reported that neither the use of an ACE-Inh. nor of an ARB was associated with higher plasma ACE2 concentrations (Sama et al., 2020). In clinical studies involving patients with heart failure (Chirinos et al., 2020; Epelman et al., 2009; Sama et al., 2020), atrial fibrillation,(Walters et al., 2017), hypertension (Kuznetsova & Cauwenberghs, 2020), aortic stenosis (Ramchand et al., 2020), and coronary artery disease (Ramchand, Patel, Srivastava, Farouque, & Burrell, 2018), plasma ACE2 protein levels or ACE2 activities were not higher among patients who were taking ACE-Inhs. or ARBs than among untreated patients. In addition, in patients with genetic variants of the ACE gene, no association of genetically predicted serum ACE levels with lung ACE2 and TMPRSS2 expression or with plasma levels of ACE2 was found (Gill et al., 2020). In a recent study, serum ACE2 levels of 1,452 individuals on ACE-Inh. or ARB treatment remained unaffected compared to those not using these medications (Emilsson et al., 2020). Similarly, another recent study reported that plasma ACE2 activity remained unaltered in patients treated with ACE-Inhs. and ARBs (Kintscher et al., 2020). However, this study reported that plasma ACE2 activity was significantly increased in a small cohort of COVID-19 patients using ACE-Inhs. In line with this finding, ACE-Inh. and ARB treatment was associated with high plasma ACE2 levels in a large cohort of patients with atrial fibrillation (Wallentin et al., 2020).
In earlier human studies measuring plasma Ang-(1-7) levels as surrogate for ACE2 activity, while acute administration of ACE-Inhs. did not alter Ang-(1-7) levels (Campbell, Zeitz, Esler, & Horowitz, 2004; Luque et al., 1996), chronic use (6 months) of ACE-Inhs. increased Ang-(1-7) levels (Luque et al., 1996). Importantly, plasma ACE2 activity may not represent enzymatic activity at the tissue level, as Ang-II infusion into mice decreases myocardial ACE2 protein level and activity but increases plasma ACE2 activity (Patel et al., 2014). Interestingly, the antihypertensive effects of captopril (X. Fan et al., 2007), benazepril (Q. Chen et al., 2011; Y. Y. Chen et al., 2016), and imidapril (Y. Y. Chen et al., 2016) are reportedly associated with polymorphisms or variations in the ACE2 gene in a gender-specific manner; however serum ACE2 levels have not been reported in these studies.
In a longitudinal cohort study involving Japanese patients with hypertension, urinary ACE2 levels were higher among patients who received long-term treatment with the ARB olmesartan than among untreated control patients. However, this association was not observed with the ACE inhibitor enalapril or with other ARBs, such as losartan, candesartan, valsartan, and telmisartan (Furuhashi et al., 2015). Correlation analysis of cardiac tissue samples from 11 patients with heart failure did not show any significant relation between angiotensinase activity and prior use of ACE-Inhs. (Zisman et al., 2003). Notably, ACE2 mRNA expression remained unchanged in bronchial epithelial cells from a small cohort of patients with COPD using ACE-Inhs. (Higham & Singh, 2020). In a small cohort of 11 patients with kidney disease, a statistically significant increase in ACE2 expression with use of ACE-Inhs. or ARBs was detected in renal epithelial and endothelial cells, but the underlying diseases confounded the association (Subramanian et al., 2020). In another study, ACE2 expression slightly, but significantly decreased in nasal cilia of patients taking ACE-Inhs. and remained unchanged in patients using ARBs (Ivan T Lee et al., 2020). In 221 hypertensive patients, no association between ACE-Inhs. or ARBs and renal ACE2 gene expression was found (X. Jiang et al., 2020). In addition, in kidney biopsies from 49 diabetic patients, treatment with ARBs and ACE-Inhs. did not change ACE2 mRNA expression (R. E. Gilbert et al., 2020). A recent gene expression analysis of 1,051 lung tissue samples indicated that the use of ACE-Inhs. was associated with lower expression of ACE2 and of the SARS-Cov-2 activator TMPRSS2, while the use of ARBs was not associated with an increased expression of these genes (Milne, Yang, Timens, Bosse, & Sin, 2020). However, in 62 patients undergoing coronary artery bypass grafting, treatment with ARBs and ACE-Inhs. was independently associated with an increased myocardial ACE2 mRNA expression (Lebek et al., 2020). Importantly, in sino-nasal biopsies from patients, treatment with ACE-Inhs. or ARBs did not increase ACE2 expression in the cilia of the upper respiratory tract (I. T. Lee et al., 2020). Altogether, these clinical studies strongly suggest that treatment with ACE-Inhs. and ARBs is not associated with increased ACE2 expression.
In a propensity analysis of 12,594 patients tested for COVID-19, there was no association between any single medication class, including ACE-Inhs., ARBs, CCBs, βARBs, and thiazide diuretics, and an increased likelihood of a positive test. Moreover, none of these medications was associated with an increased risk of severe illness among patients who tested positive (Reynolds et al., 2020). In a population based retrospective study of 34,936-hypertensive adults, the use of antihypertensive drugs, including diuretics, CCBs, βARBs, ACE-Inhs. and ARBs, did not alter the risk of COVID-19 (Vila-Corcoles et al., 2020). Similarly, in a recent study analyzing 6,272 COVID-19 patients, no association between the use of ACE-Inhs. and ARBs (as well as CCBs and βARBs), and COVID-19 risk was found (Mancia, Rea, Ludergnani, Apolone, & Corrao, 2020). Equally, another propensity analysis of 18,472 patients tested for COVID-19 did not reveal any association between ACE-Inh. or ARB use and COVID-19 test positivity (Mehta et al., 2020). In a retrospective study with 4,480 COVID-19 patients, prior use of ACE-Inhs. and ARBs was not significantly associated with COVID-19 diagnosis among patients with hypertension or with severe disease conditions (Fosbol et al., 2020). In a large population study, patients using ARBs or CCBs had a lower risk of COVID-19 (J. Kim et al., 2020). Other studies also found no association between the use of ACE-Inhs., ARBs and an increased risk of testing positive for SARS-CoV-2 or a more severe outcome (Chang et al., 2020; De Spiegeleer et al., 2020; Son, Seo, & Yang, 2020). Instead, the use of ARB and ACE-Inhs. was associated with no risk (Dublin et al., 2020; Raisi-Estabragh et al., 2020) or a reduced risk of COVID-19, as determined by an 8.3 million cohort study (Hippisley-Cox et al., 2020).
In line with these findings, the clinical outcome of 136 diabetic and hypertensive COVID-19 patients using ACE-Inhs. or ARBs was not different from that of patients who do not use these drugs (Y. Chen, D. Yang, et al., 2020). In a study with 1,200 COVID-19 patients, no evidence for increased disease severity was found in hospitalized patients on chronic treatment with ACE-Inhs. or ARBs (Bean et al., 2020). Similarly, in 50 high-risk aged COVID-19 patients with cardiovascular disease, the ACE-Inh. ramipril had no impact on the incidence or the severity of the disease (Amat-Santos et al., 2020). In another recent study with 880 COVID-19 patients, no evidence for an adverse outcome was found in severely affected COVID-19 patients that had used ARBs prior to admission (Pinto-Sietsma et al., 2020). In 2,263 hypertensive COVID-19 patients, the use of ACE-Inhs. or ARBs was not associated with an altered risk of hospitalization or mortality. In analyses stratified by insurance group, the use of ACE-Inhs. lowered the risk of hospitalization by nearly 40% in the Medicare group, a phenomenon not observed in commercially insured patients (Khera et al., 2020). Similarly, in a case-population study of 1,139 COVID-19 patients, the risk of hospitalization among users of ACE-Inhs. or ARBs was not different from that of users of other antihypertensive drugs; and no increased risk of hospitalization was associated with the use of either ACE-Inhs. or ARBs (de Abajo et al., 2020). Equally, in 543 hypertensive COVID-19 patients, no association was found between disease severity and treatment with ARBs and ACE-Inhs. (Bravi et al., 2020). In a rather large multinational cohort, no clinically significantly increased risk of COVID-19 diagnosis or hospitalization was found in patients using ACE-Inhs. or ARBs (Morales et al., 2020). In addition, the use of ACE-Inhs. and ARBs did not affect mortality rates in small cohorts of COVID-19 patients (Amat-Santos et al., 2020; Iaccarino et al., 2020; Inciardi et al., 2020; Tedeschi et al., 2020). Another recent study of 5,179 COVID-19 patients in Korea concluded that prior use of ACE-Inhs. and ARBs was not independently associated with increased mortality rates (S. Y. Jung, Choi, You, & Kim, 2020). In small cohorts of hypertensive COVID-19 patients, the use of ACE-Inhs. and ARBs did not significantly change the clinical course, disease severity and mortality rates (Wang et al., 2020, Wang et al., 2020; Sardu et al., 2020; Jiuyang Xu et al., 2020). A retrograde analysis of 2,700 intensive care patients with severe sepsis and septic shock unrelated to COVID-19 indicates no difference in mortality rates between users of ACE-Inhs. or ARBs and non-users within the subgroup of patients with respiratory infections (Sunden-Cullberg, 2020). Also, previous treatment with ACE-Inhs. or ARBs had no effect on mortality, heart failure, requirement for hospitalization, or ICU admission in 210 patients with COVID-19 (López-Otero et al., 2020). In recent months, several clinical studies have reported that the use of ARBs and ACE-Inhs. does not affect disease progression and mortality rates in COVID-19 patients (Anzola et al., 2020; Bae et al., 2020; Braude et al., 2020; Cordeanu et al., 2020; H. Cui et al., 2020; Di Castelnuovo et al., 2020; Gormez et al., 2020; Hippisley-Cox et al., 2020; Kalra et al., 2020; Khan et al., 2020, Kim et al., 2020; Kocayigit et al., 2020; Lafaurie et al., 2020; J. Lee et al., 2020; Sardu et al., 2020; Soleimani et al., 2020, Taher et al., 2020; Trifirò et al., 2020, Wang et al., 2020). Altogether, these results indicate that the use of ACE-Inhs. or ARBs neither increases the COVID-19 risk, nor disease severity nor mortality rates.
In line with these findings, in 188 COVID-19 patients with hypertension, the use of ACE-Inhs. and ARBs was associated with a lower risk of all-cause mortality, compared with non-users (P. Zhang et al., 2020). In small cohorts of hypertensive COVID-19 patients, the use of ACE-Inhs. and ARBs significantly improved disease severity, immune response, laboratory findings and viral load (J. Meng et al., 2020; Pan et al., 2018; G. Yang et al., 2020). In 157 critically ill elderly COVID-19 patients, medication with ACE-Inhs. was associated with lower mortality rates (C. Jung et al., 2020). Similarly, the use of ACE-Inhs. and ARBs was associated with a reduced risk of COVID-19-related hospitalization for diabetic patients (de Abajo et al., 2020). COVID-19 patients continuing to receive ACE Inhs. or ARBs had a lower risk of mortality compared with those who discontinued at the time of hospitalization (Cannata et al., 2020; Lam et al., 2020). In 892 hypertensive COVID-19 patients, the use of ACE-Inhs. and ARBs was associated with significantly improved outcome and disease severity compared with non-use or the use of other antihypertensive drugs (H. K. Choi et al., 2020). In 249 hypertensive COVID-19 patients, medication with ACE-Inhs. significantly reduced the risk of severe disease and was associated with milder lung infiltrations, milder disease progress and shorter hospitalizations (Şenkal et al., 2020). Furthermore, recent additional studies also report that treatment with ARBs and ACE-Inhs. is associated with reduced disease severity and decreased mortality rates in COVID-19 patients (Adrish et al., 2020; C. Chen et al., 2020; R. Chen et al.; H. K. Choi et al., 2020; Genet et al., 2020; Matsuzawa et al., 2020; Megaly & Glogoza, 2020; X. Meng et al., 2020; Negreira-Caamaño et al., 2020; Palazzuoli et al., 2020; Yahyavi et al., 2020; Y. Yuan et al., 2020). Another recent study concluded that among patients with influenza or pneumonia, treatment with ARBs and ACE-Inhs. did not increase the risk of admission to the intensive care unit, but reduced the mortality (Christiansen et al., 2020). Briefly, all of the above results suggest that the use of ACE-Inhs. and ARBs does not increase disease pathology; on the contrary, these medications may have some beneficial effects on the clinical outcome of COVID-19.
However, recently some studies have appeared pointing to the opposite: in a retrospective cohort study of 268 COVID-19 patients, the long-term use of ACE-Inhs. and ARBs was independently associated with a higher risk of severe COVID-19 and a poor outcome (Liabeuf et al., 2020). In a large cohort of patients taking ACE-Inhs. or ARBs, the use of ACE-Inhs. was associated with increased rates of S. Aerus and gram-negative infections, while herpes zoster was more commonly associated with ARBs (Bidulka et al., 2020). In addition, the use of ACE-Inhs. and ARBs was associated with a higher risk of in-hospital mortality in 74 hypertensive patients with COVID-19 pneumonia (Selcuk et al., 2020). In 44 patients with severe COVID-19, the use of ACE-Inhs. and ARBs was associated with an increased risk of acute kidney injury, and an increase in urea nitrogen associated with these drugs was predictive of the development of acute respiratory failure (Oussalah et al., 2020). There is a report of four COVID-19 patients, in whom ACE-Inhs. or ARBs had to be stopped due to acute kidney injury (Chenna et al., 2020). In addition, the use of ACE-Inhs. was found to be associated with an increased incidence and higher mortality rates in 466 patients infected with human Coronavirus NL63 (Krvavac et al., 2020).
Despite initial concerns, RAS inhibition was suggested to have beneficial effects for COVID-19 patients. The role of the RAS in the pathogenesis of acute lung injury appears to center around elevated Ang-II signaling through AT1 receptors. In small cohorts of COVID-19 (Liu et al., 2020, Wu et al., 2020) and H7N9 (F. Huang et al., 2014) infected patients, as well as children with respiratory syncytial virus (Gu et al., 2016), serum Ang-II levels were significantly higher in infected individuals than in non-infected individuals and were associated with viral load and lung injury. A retrospective review of 539 patients with viral pneumonia indicates that continuing in-hospital use of ACE-Inhs. or ARBs reduces the risk of pneumonia and mortality (Henry et al., 2018). Furthermore, in a meta-analysis of 37 studies, both ACE-Inhs. and ARBs were associated with a decrease in pneumonia-related mortality (Caldeira, Alarcao, Vaz-Carneiro, & Costa, 2012). Interestingly, patient populations that may benefit most were found to be those with a history of stroke and Asian patients. A retrospective cohort study with hospitalized pneumonia patients reported that prior and inpatient use of ACE-Inhs. and ARBs was associated with decreased mortality rates (Mortensen et al., 2012). Similarly, decreased mortality and better survival rates were reported in patients with ARDS taking ACE-Inhs. and ARBs, compared to those not using these medications (J. Kim et al., 2017). Analysis of a randomized control trial in patients with acute respiratory failure suggested that treatment with ACE-Inhs. and ARBs at discharge following an episode of acute respiratory failure was associated with a significant (44%) reduction in one-year mortality (Noveanu et al., 2010). More recently, preadmission use of ACE-Inhs. or ARBs was reported to be associated with a decreased risk of total hospital mortality (Hsieh, How, Hsieh, & Chen, 2020). In addition, losartan demonstrated beneficial effects in animal models of ventilator-associated lung injury (C. Chen et al., 2014; Jerng et al., 2007; S. Yao, Feng, Wu, Li, & Wang, 2008). Similarly, blockade of AT1 receptors attenuates lung injury in mice that have been administered the spike glycoprotein of SARS-CoV (Kuba et al., 2005). ARBs delay the onset of ARDS and decrease lung injury in rats challenged by Bordetella bronchiseptica (Raiden et al., 2002) or lipopolysaccharide (Wosten-van Asperen et al., 2011). Moreover, in a recent large population study, the use of ACE-Inhs. and ARBs was associated with either no effect on the incidence of influenza or a lower incidence, depending on the duration of use (Chung, Providencia, & Sofat, 2020). In summary, clinical and preclinical studies indicate that treatment with ACE-Inhs. or ARBs has beneficial effects in patients with ARDS, irrespective whether it is COVID-19 related or not.
A major complication of SARS-CoV-2 infection is the development of severe lung disease leading to pulmonary fibrosis. In the adult lung, the major source of ACE2 are the normally quiescent alveolar epithelial type II pneumocytes that, during lung fibrosis, proliferate actively and downregulate ACE2 expression (H. P. Jia et al., 2005; Uhal et al., 2013). In these cells, ACE2 expression can be further decreased by SARS-CoV-2 induced downregulation. Thus, it is plausible that a diminished ACE2/Ang-(1-7)/MasR axis and an unbalanced increase of the ACE/ Ang-II/AT1 receptor pathway can lead to pulmonary vasoconstriction. Together with inflammation (promoting the production of proinflammatory cytokines, such as IL-6, IL-8, TGF-β, and TNF-α by macrophages), oxidative organ damage, and increased collagen production, this can promote acute lung injury and subsequent fibrosis (Delpino & Quarleri, 2020; Wigén, Löfdahl, Bjermer, Elowsson-Rendin, & Westergren-Thorsson, 2020).
ACE2 decreases Ang-II levels by generating Ang-(1-7), which acts on the MasR and exerts vasodilatory, anti-inflammatory, antioxidative, and anti-fibrotic actions (J. Guo, Huang, Lin, & Lv, 2020). In patients with ARDS, a higher ratio of Ang-(1-7) to Ang-I among survivors was observed, compared to non-survivors (Reddy et al., 2019). In addition, treatment with Ang-(1-7) decreases lung injury and attenuates ARDS in rats with low Ang-(1-7) levels (Wosten-van Asperen et al., 2011), suggesting that the counter-regulation exerted by the ACE2/Ang-(1-7)/MasR axis may benefit patients with ARDS. In mice, losartan reduced mortality by blunting Ang-II-associated increases in soluble epoxide hydrolase, a promoter of lung injury (Tao et al., 2018). Activation of the Ang-(1-7)/ACE2/MasR axis inhibits pulmonary fibrosis (Meng et al., 2014; Meng et al., 2015) and protects from thrombosis (R. A. Fraga-Silva et al., 2012). Treatment with soluble ACE2 has been shown to reduce Ang-II levels and to increase Ang-(1-7) levels in a clinical trial of patients with ARDS (A. Khan et al., 2017). In line with these findings, recombinant soluble ACE2 attenuates the inflammatory response, increases oxygenation and protects from lung injury in animal models of ARDS (Imai et al., 2005; P. Yang et al., 2014; H. Zhang & Baker, 2017; Zou et al., 2014). Of note, meta-analyses of earlier results reported that ACE insertion/deletion polymorphism might contribute to disease mortality (Matsuda, Kishi, Jacob, Aziz, & Wang, 2012) and the susceptibility for ARDS (Deng et al., 2015). On the other hand, an earlier study could not find any association between ACE2 gene polymorphism and disease severity in ARDS patients (Chiu et al., 2004).
Altogether, a recent meta-analysis of clinical studies on ACE-Inhs. and ARBs concluded that high-certainty evidence suggests that ACE-Inh. or ARB use is not associated with more severe COVID-19 disease; and moderate-certainty evidence suggests no association between the use of these medications and positive SARS-CoV-2 test results among symptomatic patients. Whether these medications increase the risk for mild or asymptomatic disease or are beneficial in COVID-19 treatment remains uncertain (Kansagara et al., 2020, Tain et al., 2016).
Renin inhibitors, such as aliskiren, inhibit the first and rate-limiting step of the RAS, namely the conversion of angiotensinogen to angiotensin I; they are used primarily for the treatment of essential hypertension. Aliskiren attenuated the blood pressure without affecting glucose metabolism, insulin resistance, and pancreatic β-cell mass, and did not alter pancreatic ACE2 protein expression in high fat-induced obese mice (Frantz, Crespo-Mascarenhas, Barreto-Vianna, Aguila, & Mandarim-de-Lacerda, 2013). In the offspring of rats maternally exposed to high fructose intake, aliskiren prevented hypertension and increased renal ACE2 expression in females, but not in males (Hsu et al., 2016). In another study, aliskiren significantly reduced gingival inflammation, excessive wound healing processes, and periodontal bone loss in diabetic rats with periodontal disease (Oliveira et al., 2019), accompanied by a marked downregulation of gingival ACE2 gene expression. In non-obese diabetic mice, aliskiren decreased blood pressure and serum renin activity, raised renal ACE2 gene but not protein expression and increased ACE2 activity (Riera et al., 2016). In a renal transplantation model, aliskiren decreased not only serum Ang-II, but also levels of the renoprotective Ang-(1-7), and decreased serum ACE2 activity (Rusai et al., 2011).
Commonly used cardiac or cardiotonic glycosides, such as digoxin and digitoxin, act mainly by inhibiting cardiac Na-K-ATPase. They are employed for the treatment of congestive heart failure and cardiac arrhythmias and have not been reported to affect ACE2 transcription or activity. Interestingly, cardiotonic glycosides, such as ouabain and the vertebrate-derived analogue bufalin, at low concentrations and independently of Na-K-ATPase inhibition, prevent the fusion and interfere with clathrin-mediated uptake of Middle East respiratory syndrome (MERS)-CoV, CoV-MHV, and CoV-FIP in cell lines through the α1-subunit of the Na-K-ATPase-mediated Src signaling pathway (Amarelle & Lecuona, 2018; Burkard et al., 2015). Similarly, cardiotonic glycosides, including digoxin, digitoxin, oleandrin, and ouabain, inhibited the replication of CoV-TEG, but not CoV-MHV, and protected from virus-induced apoptosis and cytopathic effects in ST cells (C. W. Yang et al., 2017) through the phosphoinositide 3-kinase-phosphoinositide-dependent kinase-1 (PI3K-PDK1) signaling pathway (C. W. Yang, Chang, Lee, Hsu, & Lee, 2018). Digitoxin, ouabain, and bufalin, at low μM concentrations, also reportedly inhibit the replication of the “porcine reproductive and respiratory syndrome virus”, which belongs to the order Nidovirales, remotely related to SARS-CoV (Karuppannan, Wu, Qiang, Chu, & Kwang, 2012). Recently, digitoxin (Ko, Jeon, Ryu, & Kim, 2020), digoxin and ouabain (Cho et al., 2020) were shown to have antiviral activity against SARS-CoV-2 with respective IC50 values of 43 nM and 24 nM. They were also reported to inhibit viral mRNA expression, copy number, and viral protein expression in Vero cells (Cho et al., 2020). In addition, digitoxin reportedly inhibits an influenza virus-induced cytokine storm and reduces pulmonary levels of proinflammatory cytokines in rodent models (Pollard, JC, & Pollard, 2020).
Commonly used anticoagulant and thrombolytic medications have not been reported to interact with the activity or expression of ACE2. Thrombolytic effects of ACE2 activation have been demonstrated (R. A. Fraga-Silva et al., 2012; Santos et al., 2018). Similarly, Ang-(1-7) produced by ACE2 shows antithrombotic effects in animal models (Rodrigo Araujo Fraga-Silva et al., 2011). While pharmacological activation of ACE2 by xanthene (XNT) reduces thrombus formation in the vena cava of hypertensive rats, ACE2 inhibition by DX600 promotes thrombosis (R. A. Fraga-Silva et al., 2012). In addition, XNT diminishes platelet attachment to damaged blood vessels, reduces thrombus size, and prolongs the time to complete occlusion of blood vessels in mice. Therefore, a decrease in antithrombotic ACE2 activity is associated with an increase in thromboses in hypertensive rats. Under pathological conditions, AT1 receptor activation by Ang-II has been shown to induce deleterious effects, such as vasoconstriction, oxidative stress, platelet aggregation and exacerbated thrombus formation (Celi, Cianchetti, Dell'Omo, & Pedrinelli, 2010; Santos et al., 2018). Therefore, a decreased vascular ACE2/Ang-(1-7)/Mas receptor pathway and unopposed ACE/Ang II/AT1 activity during viral invasion can promote coagulation and thrombo-embolic events. Furthermore, increased bradykinin levels, due to ACE2 deficiency, may promote thrombus formation, since knockout of the bradykinin receptor B2 can prevent thrombus formation in a murine model (Shariat-Madar et al., 2006).
Sepsis-induced coagulopathy, increased risk of thromboembolism and disseminated intravascular coagulation in COVID-19 patients (Thachil, 2020; Whyte, Morrow, Mitchell, Chowdary, & Mutch, 2020) have prompted the use of anticoagulants, mainly low-molecular weight heparins. Heparin, in addition to its anticoagulant effects, can also abrogate the adverse effects of the ACE/Ang II/AT1 axis in cardiomyocytes (Akimoto et al., 1996), mesenteric arteries (Xie-Zukauskas, Das, Short, Gutkind, & Ray, 2013), and other vascular structures (Dilley & Nataatmadja, 1998; J. S. Park, Kim, Won, Koh, & Kim, 1996) and counteract Ang-II-induced aldosterone stimulation (Azukizawa, Iwasaki, Kigoshi, Uchida, & Morimoto, 1988). In addition, heparin exhibits antiviral properties, mainly due to its structural analogy with heparan sulfate (HS), a highly negatively charged linear polysaccharide attached to membrane proteins and extracellular matrix proteoglycans. It has been reported that culture-adapted HCoV-OC43 (de Haan et al., 2008), mouse CoV (de Haan et al., 2005; Watanabe, Sawicki, & Taguchi, 2007), porcine CoV (Huan et al., 2015), and avian CoV (Madu et al., 2007) employ heparan-sulfate proteoglycans for adhesion or entry to susceptible cells. In addition, HCoV-NL63 (Milewska et al., 2014), SARS-CoV (Lang et al., 2011; E. Vicenzi et al., 2004), and SARS-CoV-2 (Clausen et al., 2020) use ACE2 as an entry receptor and utilize heparan sulfate proteoglycans as attachment receptors, and heparin acts as competitor preventing the binding of the spike protein to the host cell, thereby reducing the infection rate and mortality. Treatment with heparin lyases, which degrade cell surface heparan sulfates, drastically reduces the binding of SARS-CoV-2 spike protein to the cell surface (Clausen et al., 2020). In a recent study, it has been shown that heparin forms 1:1 complexes with the receptor-binding domain of the S1 protein and disrupts its binding to ACE2 (Y. Yang, Du, & Kaltashov, 2020). In the context of SARS-CoV-2, a growing body of evidence suggests that SARS-CoV-2 can bind the glycosaminoglycans HS and unfractionated heparin (UFH), dependent on their level of sulphation (W. Hao et al., 2020; Li et al., 2020, Mycroft-West et al., 2020; Tree et al., 2020). Initial binding to heparan sulphates was suggested to trigger conformational changes (Clausen et al., 2020; Mycroft-West et al., 2020) and to keep the spike protein within an ‘open’ conformation allowing for downstream binding and processing of ACE2 and host cell proteases, respectively (W. Hao et al., 2020). It was proposed that while the receptor-binding domain of the SARS-CoV-2 spike (S) protein confers sequence specificity for heparan sulphates expressed by target cells, an additional HS binding site in the S1/S2 proteolytic cleavage site enhances the avidity of binding to ACE2 (L. Liu et al., 2020). Recent studies suggest that multiple heparin and heparan sulfate binding sites are present on the SARS-CoV-2 spike protein; one at the S1/S2 furin cleavage site, and others at the S2 protein and within the receptor binding domain of S1 (Partridge et al., 2020). UFH and two low molecular weight heparins (dalteparin and enoxaparin) inhibited SARS-CoV-2 spike protein binding in RT4 carcinoma (Partridge, Green, & Monk, 2020), Vero (Mycroft-West et al., 2020; Tree et al., 2020) and HEK293T cell lines (Tandon et al., 2020). Importantly, the IC50 values for inhibition of S protein binding to ACE2 expressing cell lines for UFH, dalteparin and enoxaparin were 0.03 U/ml, 0.5 U/ml, and 0.07 U/ml, respectively, which are below their target prophylactic and therapeutic serum concentrations (Kwon et al., 2020; Partridge, Green, & Monk, 2020; Tree et al., 2020). Furthermore, non-anticoagulant complex sulphated polysaccharides (fucoidans), such as RPI-27 (EC50 = 83 nM) and trisulfated heparin (EC50 = 5 μM), potently inhibited SARS-CoV-2 infection in Vero cells (Kwon et al., 2020).
Recently, it was reported that the heparan sulfate mimetic pixatimod, a clinical-stage synthetic sulfated compound, binds directly to the S1 protein of SARS-CoV-2. It also inhibits its interaction with ACE2 and reduces viral infection in Vero E6 cells (Guimond et al., 2020). In addition to their interaction with the spike protein, the cell surface heparan sulfate proteoglycans (HSPGs) mediate SARS-CoV-2 endocytosis. Heparin and drugs that target this HSPG-dependent endocytosis, such as mitoxantrone and sunitinib, potently inhibit SARS-CoV-2 entry (Q. Zhang et al., 2020). Similarly, HSPGs modified by the 3-O-sulfotransferase isoform-3 preferentially increase spike glycoprotein-mediated cell-to-cell fusion. Competition with either fondaparinux, a 3-O-sulfated HS-binding oligopeptide, or a small synthetic non-sugar molecule blocked spike protein-mediated cell-to-cell fusion. Finally, the synthetic sulfated molecule inhibited (0.1-1 μM) fusion of pseudo SARS-CoV-2 with HEK-293T cells (Tiwari et al., 2020). Interestingly, HS-modifying bacteria in human microbial communities may regulate viral adhesion; and loss of these commensals may predispose individuals to infection (Martino et al., 2020).
Furthermore, heparin catalyzes the conformational change of serpins (serine protease inhibitors), such as antithrombin III, to accelerate inactivation of proteases, including factor Xa, trypsin (Huntington, 2005), and cathepsin L (Higgins, Fox, Kowalski, Nielsen, & Worrall, 2010), which are involved in the entry and replication of SARS-CoV (L. Du et al., 2007; Millet & Whittaker, 2015). In summary, antiviral, anti RAS, and anti-aldosterone effects, coupled with endothelial protective, antioxidative (Thachil, 2020) and antinflammatory (Costanzo et al., 2020) properties, are useful features of heparin, besides its anticoagulant effects, in the treatment of ARDS and prevention of COVID-19 related thromboembolic events (Whyte, Morrow, Mitchell, Chowdary, & Mutch, 2020). Potential beneficial effects of heparin in the treatment of COVID-19 are illustrated in Fig. 2 . Of note, while heparin does not affect ADAM17 expression (H. Cui et al., 2011), aspirin, although at relatively high concentrations, promotes ADAM17-mediated shedding (Aktas et al., 2005). Finally, aspirin, another anticoagulant, has been shown to activate SIRT1 (Aşcı et al., 2016; Y. R. Jung et al., 2015; Kamble, Selvarajan, Aluganti Narasimhulu, Nandave, & Parthasarathy, 2013), which is expressed next to the promotor region of the ACE2 gene; hence its activation is potentially associated with ACE2 upregulation (Clarke, Belyaev, Lambert, & Turner, 2014). In a recent study of 98 COVID-19 patients, the use of aspirin was associated with a decrease in mechanical ventilation, in intensive care unit admission, and in in-hospital mortality (Chow et al., 2020). A list of the effects of cardiovascular drugs on the activity and expression of ACE2 is provided in Table 4 .
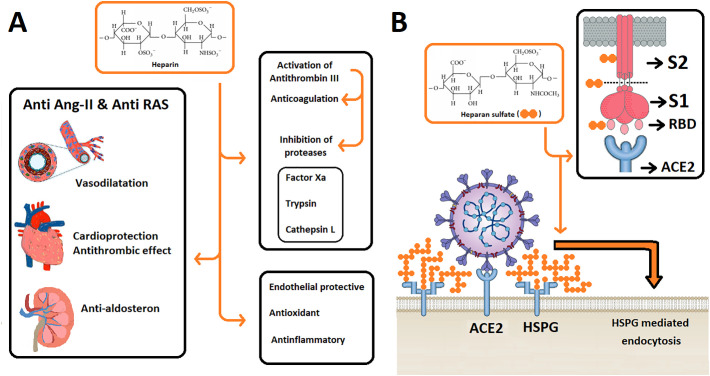
Potential beneficial effects of heparin in COVID-19 treatment. (A) Chemical structure of heparin (orange inbox) showing multiple sulphations of this compound. The anti Ang-II & Anti RAS effects of heparin also contribute to its antinflammatory and antioxidant actions. Activation of antithrombin III (serine protease inhibitor, serpin) by heparin leads to inactivation of several proteases, such as factor Xa, trypsin, and cathepsin L, which are involved in coagulation and viral replication. (B) Heparan sulfate (HS) (orange inbox; tandem filled orange circles indicate HS or heparin), a structural analog of heparin, binds to (1) S2 protein, (2) the S1/S2 region of S protein (near furin protease cut site) and (3) to the open state of the receptor binding domain (RBD) on the S1 segment of the SARS-CoV-2 spike. On the surface of the host cell, HS polymers are bound extensively to HS proteoglycans (HSPGs) and mediate HSPG-dependent endocytosis of SARS-CoV-2. Heparin competes with HS binding sites and inhibits S protein binding as well as endocytosis of the SARS-CoV-2. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
| Pharmacological agent/class | Experimental model /Tissue/Subject | Effect | Reference |
|---|---|---|---|
| Isoprenaline (isoproterenol)/ Non-selective β adrenoreceptor agonist | Male Sprague-Dawley and TGR(A1–7)3292 rats/ Isoproterenol induced cardiac hypertrophy model | Increased cardiac ACE2 mRNA expression in Sprague-Dawley rats | Nadu, Ferreira, Reudelhuber, Bader, and Santos (2008) |
| Isoprenaline (isoproterenol)/ Non-selective β adrenoreceptor agonist | Male Sprague-Dawley rats/ Isoproterenol induced cardiomyopathy model | Decreased cardiac ACE2 protein expression | Q. Liu et al. (2015) |
| Isoprenaline (isoproterenol)/ Non-selective β adrenoreceptor agonist | Male Wistar rats/ Isoprenaline induced cardiac hypertrophy model | Decreased cardiac ACE2 mRNA expression | Syed et al. (2016) |
| Isoprenaline (isoproterenol)/ Non-selective β adrenoreceptor agonist | Male Wistar albino rats/ Isoproterenol induced myocardial infarction model | Increased cardiac ACE2 protein levels | Badae et al. (2019) |
| Isoprenaline (isoproterenol)/ Non-selective β adrenoreceptor agonist | Male Wistar rats/ Isoproterenol treatment of salivary gland | Decreased ACE2 mRNA expression in parotid gland, but no effect on sublingual and submandibular glands | Cano et al. (2019) |
| Atenolol/ β1-receptor antagonist | Male spontaneously hypertensive rats/arterial tissue | No effect on ACE2 immunostaining in carotid artery and aorta. Decreased ACE2 mRNA expression carotid artery | Igase et al. (2005) |
| Nebivolol/ β1-adrenoreceptor blocker | Male spontaneously hypertensive rats/ High-salt diet model | Increased cardiac ACE2 mRNA expression. No effect of ACE2 activity. | Varagic et al. (2012) |
| Labetalol/ β1-adrenreceptor blocker | Human recombinant ACE2/ Enzyme kinetic assay | Increased maximal reaction rate of ACE2, but overall enzyme efficieny may not change | Kulemina and Ostrov (2011) |
| Propranolol/β1-adrenreceptor blocker | Spontaneously hypertensive and Wistar Kyoto rats/ Aortic tissue | Markedly decreased aortic ACE2 mRNA expression in hypertensive rats with significantly upregulated ACE2 levels | Lezama-Martinez et al. (2018) |
| Hydrochlorothiazide/ Diuretic | Spontaneously hypertensive and Wistar Kyoto rat | Cardiac ACE2 activity and mRNA expression increased, but the activity decreased in hypertensive rats | Jessup, Brosnihan, Gallagher, Chappell, and Ferrario (2008) |
| Nifedipine/ (L-type CCB) | Human aortic endothelial cells/ mechanical stress model | Increased ACE2 protein cell surface expression | Iizuka, Kusunoki, Machida, and Hirafuji (2009) |
| Cilnidipine/ (L-type CCB) | Male Wistar-Kyoto and spontaneously hypertensive rats/Hypertension model | Both of these drugs did not affect aortic ACE2 mRNA expression | Takai et al. (2013) |
| Amlodipine/ (L-type CCB) | |||
| Felodipine/ (L-type CCB) | Male Sprague Dawley/ Goldblatt hypertensive rat model | No change in renal ACE2 mRNA expression in ischemic and non-ischemic kidneys | S. Bai, Huang, Chen, Wang, and Ding (2013) |
| Nimodipine/ (L-type CCB) | Male Wistar rats/ Cerebral ischemia-reperfusion model | Increased brain ACE2 mRNA expression of down regulated ACE2 in ischemic brain tissue. | Abdel-Fattah et al. (2018) |
| Amlodipine/ (L-type CCB) | Rats/ Nitric oxide inhibition and salt induced hypertension model | Increased renal ACE2 levels | Onat and ŞAhna (2018) |
| Aliskiren/ Renin antagonist, antihypertensive | Male Lewis rats/ Fischer-to-Lewis renal transplantation model | Decreased serum ACE2 activity | Rusai et al. (2011) |
| Aliskiren/ Renin antagonist, antihypertensive | Male C57BL/6 mice/ High fat diet model | No change in pancreatic ACE2 protein expression | Frantz et al. (2013) |
| Aliskiren/ Renin antagonist, antihypertensive | Sprague-Dawley rats/ maternal high fructose induced hypertension model | Increased renal ACE2 protein expression in offspring of females exposed to high fructose intake. | Hsu et al. (2016) |
| Aliskiren/ Renin antagonist, antihypertensive | Female non obese diabetic/ ShiLtJ and NOR/LtJ mice | No effect on renal ACE2 activity, but increased ACE2 mRNA expression in kidney. | Riera et al. (2016) |
| Aliskiren/ Renin antagonist, antihypertensive | Male Balb/c mice/ Streptozotocin-induced diabetes model | Decreased gingival tissue ACE2 mRNA expression | Oliveira et al. (2019) |
| Ivabradine/ Pacemaker current inhibitor, Heart failure medication | Dogs/ heart failure model | Increased cardiac ACE2 activity | R. C. Gupta, Want, Rastogi, Zhang, & Sabbah (2012) |
| Spironolactone/ MRB | Male Wistar rats/ Aortocaval fistula induced heart failure model | Heart failure caused decreased cardiac ACE2 expression and enzyme activity was restored by eprosartan | Karram et al. (2005) |
| Spironolactone/ MRB | Heart failure patients/ Monocyte-derived macrophage | Increase in ACE2 activity and ACE2 mRNA expression one-month post therapy | Keidar et al. (2005) |
| Spironolactone/ MRB | Male Sprague-Dawley rats/ Diabetic nephropathy model | Decreased plasma ACE2 level | Dong et al. (2019) |
| Spironolactone/ MRB | Male Sprague-Dawley rats/ Obstructive jaundice model | Decreased renal ACE2 mRNA expression due to obstructive jaundice was reversed by spironolactone | Kong et al. (2019) |
| Eplerenone/ MRB | Balb/C mice/Heart and kidney | Increase in cardiac ACE2 activity and ACE2 mRNA expression, but nonsignificant increase in the kidneys | Keidar et al. (2005) |
| Eplerenone/ MRB | Wistar rats/ Heart | Prevented aldosterone induced reduction in cardiac ACE2 mRNA expression | Yamamuro et al. (2008) |
| Eplerenone/ MRB | Dahl salt-sensitive hypertensive rats/ Hypertension model | No effect on cardiac ACE2 mRNA and protein expression in hypertensive rats | Takeda et al. (2007) |
| Eplerenone/ MRB | Male Wistar rats/ Uninephrectomy and high salt induced kidney injury model | Partial reversal of aldosterone induced decrease of renal ACE2 expression in injured and high salt exposed kidneys. | Bernardi et al. (2015) |
| Hydralazine/ Directly acting vasodilator | Male spontaneously hypertensive rats/ Hydralazine treatment | No effect on ACE2 immunostaining mRNA expression in carotid artery and aorta | Igase et al. (2005) |
| Hydralazine/ Directly acting vasodilator | Transgenic C57BL/6J mice overexpressing renin and angiotensinogen | No change in cardiac ACE2 activity and mRNA expression after NOS inhibition | Inaba et al. (2011) |
| Hydralazine/ Directly acting vasodilator | Transgenic and C57BL/6N mice/ Cardiac hypertrophy model | No change in cardiac ACE2 mRNA expression | Tanno et al. (2016) |
| Hydralazine/ Directly acting vasodilator | Male renin overexpressing, Ren-TG, and C57BL/6N mice/ Hypertension model | No effect on renal ACE2 mRNA and protein expression in hypertensive mice | Ichikawa et al. (2018) |
| Sacubitril+ Valsartan/ Neprilysin inhibitor+ARB/ Treatment of heart failure | Female spontaneously hypertensive and Wistar-Kyoto rats/ Hypertension model | Reversal of decreased cardiac ACE2 mRNA and protein expression in hypertensive rats | Y. Zhao et al. (2019) |
| Sildenafil/ Vasodilator used for erectile dysfunction | Male piglets/ Myocardial ischemia induced injury model | No change in cardiac ACE2 immunostaining, protein and mRNA expression in ischemic piglets | G. Wang, Zhang, Yuan, Wu, & Li et al. (2015) |
Thiazolidinediones, such as pioglitazone and rosiglitazone, agonists of peroxisome proliferator-activated receptor gamma, are used in the treatment of type 2 diabetes mellitus (T2DM) as insulin sensitizers with anti-inflammatory and anti-atherosclerotic effects. In rats with steatohepatitis induced by high fat diet, pioglitazone increased serum ACE2 levels and ACE2 mRNA expression in the liver (W. Zhang, C. Li et al., 2013), adipose tissue and skeletal muscle (W. Zhang et al., 2014) as well as 3T3-L1 adipocytes (Gupte et al., 2008). Rosiglitazone treatment improved cardiac and renal functions; enhanced atrial natriuretic peptide responses and markedly upregulated renal ACE2 gene transcription in rats with heart failure (Goltsman et al., 2019). Similarly, rosiglitazone increased vascular ACE2 expression in hypertensive, but not in normotensive rats (Sanchez-Aguilar et al., 2019). In transgenic diabetic mice, rosiglitazone downregulated renal ADAM17 expression, decreased ACE2 shedding and reduced urinary soluble ACE2, thereby increasing ACE2 protection of the kidneys without altering renal ACE2 expression (Alawi et al., 2020; Chodavarapu et al., 2013). Notably, although rosiglitazone induced an upregulation of ACE2 mRNA and protein expression in adipocytes of wild type mice, this ACE2 increase was markedly attenuated in adipocytes of fibroblast growth factor 21 (FGF21) knockout mice (Pan et al., 2018), suggesting that endogenous FGF21 in adipocytes modulates ACE2 expression in an autocrine manner. In a small cohort of patients, pioglitazone treatment improved glucose metabolism, reduced TNF-α expression and enzymatic activity of ADAM17 in skeletal muscle of T2DM patients (Tripathy et al., 2013).
Glucagon-like peptide 1 (GLP-1), a hormone produced in the distal ileum in response to food intake, activates GLP-1 receptors, increases insulin secretion, reduces glucagon release, and regulates glucose homeostasis. Long-acting GLP-1 receptor agonists, such as liraglutide and exendin-4, are used to treat T2DM. Exendin-4, a clinically used antidiabetic drug and GLP-1 receptor activator, improved pathological changes, decreased renal Ang-II, and completely restored down-regulation of renal ACE2 expression occurring after ureter obstruction (Le et al., 2016). In diabetic rats with reduced ACE2 levels, liraglutide induced marked upregulation of pulmonary ACE2 gene transcription (Romani-Perez et al., 2015). In another study, liraglutide increased pulmonary ACE2 expression in rats with in utero growth retardation (Fandino et al., 2018). Similarly, liraglutide completely reversed reduced hepatic ACE2 mRNA expression in mice with high-fat-induced liver disease through activation of the phosphatidylinositol-3-kinase (PI3K/Akt) signaling pathway in HepG2 cells (M. Yang et al., 2020). Liraglutide and another antidiabetic, linagliptin, a dipeptidyl peptidase-4 (DPP4) inhibitor, improved Ang-II-induced cardiovascular pathology, counteracted Ang-II-induced downregulation of Smad7, reduced collagen synthesis and cardiac fibrosis, and upregulated myocardial expression and activity of ACE2 (L. H. Zhang et al., 2015). The DPP4, independently of its enzymatic activity, functions as an entry receptor for the MERS coronavirus, and its co-expression with ACE2 has been shown in bronchial epithelial cells (Radzikowska et al., 2020). DPP4 inhibitors (gliptins), in addition to their antidiabetic actions, have anti-inflammatory effects, reduce cytokine overproduction, and have been suggested as treatment of COVID-19 (Solerte, Di Sabatino, Galli, & Fiorina, 2020). However, recent studies reported that gliptins had no significant effect on disease severity, mortality and clinical outcomes in diabetic COVID-19 patients (Y. Chen, D. Yang, et al., 2020; Fadini et al., 2020). In addition, using Mendelian randomization analysis, a large genome-wide association study has reported that increased ACE2 expression is associated with both type 1 and type 2 diabetes (Rao, Lau, & So, 2020).
The commonly prescribed oral antidiabetic drug metformin activates the AMP-activated protein kinase (AMPK) in vitro and in vivo (G. Zhou et al., 2001), resulting in an attenuation of hepatic glucose production and an enhancement of peripheral glucose uptake (G. Zhou et al., 2001). Metformin also increases AMPK-mediated phosphorylation of ACE2 at Ser680 in human endothelial cell lines, thereby upregulating cell surface ACE2 activity and expression via inhibition of the ubiquitination-related degradation of ACE2 (J. Zhang et al., 2018). Notably, metformin decreased pulmonary pressure and alleviated pulmonary artery damage in wild type but not in AMPK knock out mice (J. Zhang et al., 2018). However, in an earlier study in Huh7 cells, metformin had no effect, although activation of AMPK by the AMP mimic AICAR (5-amino-4-imidazolecarboxamide riboside) markedly upregulated ACE2 expression and activity (Clarke, Belyaev, Lambert, & Turner, 2014). Metformin does not affect renal ACE2 expression or ADAM17 activity, but reduces urinary ACE2 by improving glucose levels in diabetic rats (Somineni, Boivin, & Elased, 2014). Importantly, metformin activates SIRT1 (Cuyàs et al., 2018), which is expressed next to the promotor region of the ACE2 gene. Hence increased expression or enhanced functional activation of SIRT1 is associated with an increase in expression of ACE2 (Clarke et al., 2014). Drugs that increase endosomal pH values (such as chloroquine) are known to reduce viral replication. Metformin has been reported to inhibit the Na+/H+ exchanger and the vacuolar ATPase on endosomal membranes and to subsequently increase the endosomal pH (Jeongho Kim & You, 2017), which potentially interferes with viral replication. In addition, metformin has been shown to reverse established lung fibrosis in mouse models (Rangarajan et al., 2018), a desirable pharmacological effect in the treatment of lung injury caused by viral pneumonia. Metformin also preserves alveolar capillary permeability and decreases the severity of ventilator-induced lung injury in rabbits (Tsaknis et al., 2012). It also prolongs survival and attenuates pulmonary injury by reducing pulmonary inflammation, coagulation, and fibrosis in a rat model (X. Chen et al., 2015). Analysis of one small cohort of COVID-19 patients indicates that the use of metformin is associated with a reduced mortality (Crouse et al., 2020; Hariyanto & Kurniawan, 2020), whereas another one reports an increased disease progression (Y. Gao et al., 2020). Of note, it has recently been reported that cyclic sulfonamide derivatives are potent inhibitors (IC50 = 0.9-3.1 μM) of SARS-CoV-2 in Vero cells (Y. S. Shin et al., 2020).
Statins, also known as HMG-CoA reductase inhibitors, are a class of lipid-lowering drugs used for decreasing mortality in patients at high risk of cardiovascular disease. Currently, drugs of the statin group are the most common cholesterol-lowering medications. Rosuvastatin was shown to decrease cell proliferation and intimal pathology, and to upregulate aortic ACE2 transcription and protein expression in vascular injury models (Y. H. Li et al., 2013). On the other hand, pravastatin alone did not exert any protective effect on cardiovascular pathology and did not alter cardiac ACE2 expression, but significantly potentiated cardiovascular protective actions of insulin in diabetic rats (Min et al., 2018). Similarly, fluvastatin significantly enhanced the cardio-protective effects of insulin, improved cardiac function and restored cardiac ACE2 expression in diabetic rats (Y. H. Shin et al., 2017). In vascular smooth muscle cell cultures, atorvastatin did not alter ACE2 transcription, but reversed TNF-α induced downregulation of ACE2 expression (Suski et al., 2014). In another study on statins, atorvastatin improved the lipid disturbance, decreased atherosclerotic pathology and increased cardiac and renal ACE2 protein expression; but ACE2 mRNA expression increased only in cardiac tissue of atherosclerotic rabbits maintained on a high cholesterol diet (Tikoo et al., 2015). Notably, statins (Carloni & Balduini, 2020; G. Du et al., 2014; Ota et al., 2010) and antidiabetics, such as exenatide, liraglutide (Strycharz et al., 2018), and sitagliptin (Malvandi, Loretelli, Ben Nasr, Zuccotti, & Fiorina, 2019), activate SIRT1 and can potentially increase the expression of ACE2 (Clarke et al., 2014). In addition, atorvastatin was reported to downregulate ADAM17 activity in cultured neonatal rat cardiomyocytes (Y. Liao et al., 2008). However, methyl-β-cyclodextrin, a cholesterol depleting agent, and lovastatin stimulated ADAM17 (aka TACE) activity in L428 cells (von Tresckow et al., 2004), COS-7 cells and fibroblasts (Matthews et al., 2003). Conversely, cholesterol loading of retinal epithelial cells downregulated ADAM17 expression, suggesting that lowering cholesterol levels by statins may modulate cell surface ACE2 activity (J. Wang, Ohno-Matsui, & Morita, 2012). Statins are known to improve endothelial dysfunction (Katsiki, Banach, & Mikhailidis, 2020; Katsiki et al., 2018) and to decrease elevated inflammatory markers, e.g., C-reactive protein and interleukin-6. They exert anti-inflammatory and immunomodulatory effects (Pirro et al., 2019; Zeiser, 2018). By stabilizing atherosclerotic plaques, they prevent a viral-induced acute coronary syndrome and renal injury (Katsiki, Banach, & Mikhailidis, 2020; Mohammad et al., 2019). These pharmacological features, coupled with profibrinolytic and anticoagulant effects (Biedermann et al., 2018; Bifulco & Gazzerro, 2020), make statins a desirable treatment option for COVID-19-related pathologies (Rodrigues-Diez et al., 2020). Similarly, clofibrate, another lipid-lowering drug with a different mechanism of action, decreased cardiac oxidative stress and Ang-II, improved cardiac function and upregulated cardiac ACE2 protein expression in hypertensive rats with stressed ventricles (Ibarra-Lara et al., 2016).
Coronaviruses, including SARS-CoV (G. M. Li, Li, Yamate, Li, & Ikuta, 2007; Y. Lu, Liu, & Tam, 2008) and SARS-CoV-2 (H. Wang, Yuan, Pavel, & Hansen, 2020), have been reported to require lipid rafts for cellular entry. Cholesterol was reported to be involved in binding and altering the oligomeric status of the N-terminal fusion peptide of SARS-CoV, which is essential for virus entry into the host cell (Meher, Bhattacharjya, & Chakraborty, 2019), and also to interrupt cell-cell fusion induced by the virus (K. S. Choi, Aizaki, & Lai, 2005). It was shown that cholesterol reduction by methyl-β-cyclodextrin or mevastatin (H. Guo et al., 2017) disrupts lipid rafts that enable the binding of the virus to the host cell, thereby preventing its infection (Jeon & Lee, 2018; Y. Lu et al., 2008, Wang et al., 2020). Conversely, loading of cells with cholesterol (Hao Wang et al., 2020) or increasing the cholesterol concentration in extracellular solutions (C. Wei et al., 2020) increases viral entry. It appears that ACE2 and furin, a protease that cleaves the spike protein of SARS-CoV-2, are preferentially located in cholesterol-rich viral entry points that promote endocytic viral entry mechanisms and facilitate the efficient interaction of the spike protein with ACE2 (Glende et al., 2008; Hao Wang et al., 2020). Notably, a recent in vitro study investigating the SARS-CoV-2 protein-protein interactome identified the scavenger receptor BI (SR-BI), a cholesterol trafficking receptor, as a potential drug target (Gordon et al., 2020), and antagonists of SR-BI inhibited SARS-CoV-2 infectivity in Huh7 cell lines (C. Wei et al., 2020).
25-hydroxycholesterol (25HC) is the product of cholesterol oxidation by the enzyme cholesterol-25-hydroxylase (CH25H). Infection with SARS-CoV-2 has been shown to increase serum 25HC levels in mice and to induce the activity of CH25H in Caco-2 cells (Zu et al., 2020). Notably, 25HC significantly inhibited SARS-CoV-2 replication with an EC50 of 3.7 μM and reduced viral protein production in SARS-CoV-2-infected Vero cells. It also decreased the viral RNA load in both lung and trachea of infected mice (Zu et al., 2020). Another study also reported that the interferon-stimulated gene of CH25H is induced by SARS-CoV-2 infection in vitro and in COVID-19 patients (S. Wang et al., 2020). Furthermore, 25HC inhibited SARS-CoV-2 infection in lung epithelial cells and reduced viral entry in human lung organoids, presumably by preventing viral membrane fusion through activation of the ER-localized acyl-CoA:cholesterol acyltransferase, which leads to the depletion of cholesterol from the plasma membrane (S. Wang et al., 2020). Similar to 25HC, another cholesterol oxidation metabolite, 27-hydroxycholesterol, was shown to inhibit SARS-CoV-2 infection in Vero-E6 cells with an EC50 of 1.4 μM (Marcello et al., 2020). Interestingly, serum levels of 27-hydroxycholesterol were significantly decreased (50%) in SARS-CoV-2 infected patients, compared to the control group. In this context, high-density and low-density lipoprotein cholesterol and total cholesterol levels were reported to be significantly decreased in COVID-19 patients (Ressaire, Dudoignon, Moreno, Coutrot, & Dépret, 2020; G. Wang et al., 2020). Low cholesterol levels were correlated with a higher risk of developing severe events or longer recovery times in some studies (X. Ding et al., 2020; Ressaire, Dudoignon, Moreno, Coutrot, & Dépret, 2020; G. Wang et al., 2020), but not in another one (Tanaka et al., 2020).
It is well established that tissue cholesterol increases with age, and this accumulation is directly linked to disease pathologies, including atherosclerosis and inflammation. Remarkably, these diseases are highly comorbid with COVID-19 (Hao Wang et al., 2020). All these data support the potential use of statins to prevent or reverse host cell lipid raft alterations induced by COVID-19 infection, which could reduce both cell infection and viral replication. Thus, the pharmacological sequestration of cellular or viral cholesterol with statins has potential antiviral effects for preventing both virus attachment and internalization. Furthermore, fluvastatin decreased intracellular reactive oxygen species (ROS) by activating peroxiredoxin 1, a ROS scavenger, reduced proinflammatory responses in cultured cells and inhibited SARS-CoV-2 infection and replication in Vero E6 cells (H. Zhang et al., 2020). Pre-infection treatment with pravastatin reduced SARS-CoV-2 infection in Vero E6 cells (Mok et al., 2020). Finally, pretreatment with atorvastatin, pravastatin or fluvastatin impaired CD147 translocation to the cell surface, altered CD147 expression, structure and function by inhibiting protein isoprenylation and N-glycosylation in cultured monocytes (Sasidhar, Chevooru, Eickelberg, Hartung, & Neuhaus, 2017) and atherosclerotic plaques (X. Liang et al., 2017). CD147, also known as basigin, EMMPRIN or leukocyte activation antigen M6, is a receptor for the S protein of SARS-CoV and SARS-CoV-2 (K. Wang et al., 2020).
Lipid-lowering effects and some pleiotropic actions of statins, such as the downregulation of CD147 expression and function, disruption of lipid rafts, activation of autophagy, and attenuation of both the inflammatory response and the coagulation activation by these drugs, have been recently reviewed in the context of COVID-19 (Bifulco & Gazzerro, 2020; Katsiki et al., 2020; K. C. H. Lee, Sewa, & Phua, 2020; Radenkovic, Chawla, Pirro, Sahebkar, & Banach, 2020; Rodrigues-Diez et al., 2020). Noteworthy, the analysis of a randomized control trial in patients with acute respiratory failure suggested that treatment with statins at discharge following an episode of acute respiratory failure was associated with a significant reduction in one-year mortality (Noveanu et al., 2010). A retrospective cohort study with hospitalized pneumonia patients reported that prior and inpatient use of statins was associated with decreased mortality rates (Mortensen et al., 2012). However, in clinical studies with large cohorts of patients, statins were found to be ineffective in patients with ARDS (McAuley et al., 2014), sepsis-associated ARDS (Truwit et al., 2014), or ventilator-associated pneumonia (Papazian et al., 2013).
In older adults, a significant association between statin intake and the absence of symptoms during COVID-19 has been reported (De Spiegeleer et al., 2020). Notably, there are recent reports that the in-hospital use of statins reduced the mortality risk in 1,219 COVID-19 patients (X. J. Zhang et al., 2020) and in a small cohort of COVID-19 patients admitted to intensive care (Rodriguez-Nava et al., 2020). Furthermore, in a study with 151 hyperlipidemic COVID-19 patients, treatment with statins was independently associated with lower intensive care admission (Tan, Young, Lye, Chew, & Dalan, 2020). Similarly, a retrospective cohort study of 249 patients hospitalized with COVID-19 reports a significantly decreased risk of invasive mechanical ventilation in patients treated with statins (S. L. Song et al., 2020). In addition, in 170 hospitalized COVID-19 patients, the use of statins prior to admission was associated with a lower risk of developing severe COVID-19 and a faster time to recovery among patients without severe disease (Daniels et al., 2020). Similarly, in 983 diabetic COVID-19 patients, statin use was associated with reduced in-hospital mortality (Saeed et al., 2020). In addition, a lower SARS-CoV-2 infection-related mortality was observed in 581 patients treated with statins prior to hospitalization (Masana et al., 2020). However, another recent study with 2449 hospitalized COVID-19 patients with type 2 diabetes concluded that routine statin treatment is significantly associated with increased mortality (Cariou et al., 2020). The effects of antidiabetic and lipid-lowering drugs on the activity and expression of ACE2 are listed in Table 5 .
| Pharmacological agent/class | Experimental model /Tissue/Subject | Effect | Reference |
|---|---|---|---|
| Rosiglitazone/ Antidiabetic drug | 3T3-L1 murine adipocytes | Increased ACE2 mRNA expression | Gupte et al. (2008) |
| Rosiglitazone/ Antidiabetic drug | Male diabetic mice | Decreased urinary ACE2 activity due to decreased renal ACE2 shedding | Chodavarapu et al. (2013) |
| Pioglitazone/ Antidiabetic drug | Male Sprague-Dawley rats/ High fat diet-induced steatohepatitis model | Increased serum ACE2 levels and hepatic ACE2 mRNA and protein expression in control and high fat diet group | W. Zhang, C. Li et al. (2013) |
| Pioglitazone/ Antidiabetic drug | Rats/ Streptozotocin-induced diabetes model | Increased cardiac ACE2 protein expression | Weili et al. (2014) |
| Pioglitazone/ Antidiabetic drug | Male Sprague-Dawley rats/ High fat diet-induced steatohepatitis model | Increased ACE2 protein expression in liver, adipose tissue, and skeletal muscle in high fat diet group | W. Zhang et al. (2014) |
| Pioglitazone/ Antidiabetic drug | Male Sprague-Dawley rats/ Streptozotocin-induced diabetes model | Increased cardiac ACE2 immunostaining and mRNA expression | Qiao et al. (2015) |
| Rosiglitazone/ Antidiabetic drug | Male Wistar rats/ Aortic coarctation-induced hypertension model | Increased ACE2 protein expression | M. S. Aguilar et al. (2018) |
| Pioglitazone/ Antidiabetic drug | Male Sprague-Dawley rats/ Ischemia-reperfusion injury model | Renal ACE2 mRNA and protein expression was increased by injury and downregulated by alfacalcidol | Ali, Al-Shorbagy, Helmy, & El-Abhar (2018) |
| Rosiglitazone/ Antidiabetic drug | Male Sprague-Dawley rats/ Congestive heart failure model | ACE2 gene expression was upregulated by rosiglitazone in rats with heart failure | Goltsman et al. (2019) |
| Rosiglitazone/ Antidiabetic drug | Male Wistar rats/ Aortic coarctation-induced hypertension model | Aortic ACE2 protein expression was upregulated in hypertensive rats. | Sanchez-Aguilar et al. (2019) |
| Liraglutide/ Antidiabetic drug | Male Sprague-Dawley rats/ Streptozotocin-induced diabetes model | Pulmonary ACE2 mRNA expression was increased in control and diabetic groups. | Romani-Perez et al. (2015) |
| Liraglutide, linagliptin / Antidiabetic drugs | Male Sprague–Dawley rats/ Angiotensin II infusion model | Cardiac ACE2 activity decreased by Angiotensin II was upregulated by these drugs. | L. H. Zhang et al. (2015) |
| Exendin-4/ Antidiabetic drug | BALB/c mice/ unilateral ureter obstruction model | Increased renal ACE2 mRNA and protein expression. No effect on unobstructed kidney. | Le et al. (2016) |
| Liraglutide/ Antidiabetic drug | Female Sprague-Dawley rats/ Maternal food restricted pups | Lung ACE2 mRNA expression was increased in food restricted pups, but not in control group | Fandino et al. (2018) |
| Liraglutide/ Antidiabetic drug | Male C57BL/6J mice and HepG2 cell line/ High-fat-induced liver disease model | Increased ACE2 mRNA and protein expression in liver and HepG2 cells. | M. Yang et al. (2020) |
| Atorvastatin | Holtzman rats/ Streptozotocin induced diabetes model | Increased cardiac ACE2 mRNA expression | C. Aguilar, Ventura, & Rodriguez-Delfin (2011) |
| Rosuvastatin | Male Wistar rats/ Vascular balloon injury model | Vascular ACE2 mRNA and protein expression was decreased by the injury and the effect was partially reversed by rosuvastatin | Y. H. Li et al. (2013) |
| Atorvastatin | Rat aortic vascular smooth muscle cells | Decrease of ACE2 mRNA expression by TNF-α was restored by atorvastatin | Suski et al. (2014) |
| Atorvastatin | New Zealand White Rabbits/ High cholesterol diet, atherosclerosis model | Increased cardiac and renal ACE2 protein expression and mRNA increased only in cardiac tissue | Tikoo et al. (2015) |
| Clofibrate | Male Wistar rats/ Aortic coarctation-induced hypertension model | Clofibrate upregulated cardiac ACE2 mRNA expression. | Ibarra-Lara et al. (2016) |
| Fluvastatin | Male Lewis rats/ Streptozotocin-induced diabetes model | Decreased cardiac ACE2 protein expression in diabetic group was upregulated by fluvastatin | Y. H. Shin et al. (2017) |
| Rosuvastatin | Rats/ Nitric oxide inhibition and salt induced hypertension model | Increased renal ACE2 levels | Onat and ŞAhna (2018) |
| Pravastatin | Male Lewis rats/ Streptozotocin induced diabetes model | No effect on cardiac ACE2 protein expression. Increase ACE2 expression in the presence of insulin | Min et al. (2018) |
Glucocorticoids are mainly produced in the zona fasciculata of the adrenal cortex. When applied therapeutically, they have potent anti-inflammatory and immunosuppressive actions with additional metabolic and cardiovascular side effects, such as hypertension, hyperglycemia, and osteoporosis. Animals treated prenatally with glucocorticoids develop hypertension with decreased plasma ACE2 activity and diminished ACE2 expression in renal (P. C. Lu et al., 2016; Shaltout, Figueroa, Rose, Diz, & Chappell, 2009), cardiac (E. Kim et al., 2015), and placental (Ghadhanfar et al., 2017), but not adipose tissue (Massmann, Zhang, Seong, Kim, & Figueroa, 2017; H. R. Yu et al., 2018). This is associated with reduced Ang-(1-7) in the cerebrospinal fluid (Marshall et al., 2013). Maternal corticosterone exposure was reported to decrease renal ACE2 expression in females but to increase it in males (Cuffe, Burgess, O'Sullivan, Singh, & Moritz, 2016). Importantly, glucocorticoids, such as dexamethasone, potentiate Ang-II responses by upregulating the expression of AT1 receptors in cardiac (Xue, Patterson, Xiao, & Zhang, 2014) and vascular structures (Ullian, Walsh, & Morinelli, 1996). Interestingly, activation of the neutral amino acid transporter SLC6A19 (B0AT1), an accessory protein for ACE2 in the intestines, is regulated by the “serum and glucocorticoid inducible kinase” (SGK) isoforms 1-3 (Bohmer et al., 2010). Of note, budesonide, a glucocorticoid, activates ADAM17 in bronchial epithelial cells (Zijlstra et al., 2014). However, dexamethasone inhibited ADAM17 activity without affecting its expression level in lipopolysaccharide-activated RAW cells (Chuang et al., 2017).
Corticosteroid medications are commonly used in the treatment of several inflammatory pathologies, including asthma, inflammatory bowel disease (IBD), interstitial lung disease, ARDS, and systemic vasoplegic shock. The results of clinical studies attempting to correlate disease severity with ACE2 expression levels has not been conclusive. In IBD patients not using steroids, no significant change in ACE2 and TMPRSS2 gene expression was found in biopsy samples (Monteleone, Franze, & Laudisi, 2020). In another study, reduced ACE2 expression in biopsy samples from patients with Crohn’s disease was associated with inflammation and worse outcomes (Potdar et al., 2020). In 138 treatment naïve IBD patients, while ACE2 gene expression was decreased in the ileum, it was increased in colon samples (Krzysztof et al., 2020). In addition, in control patients, ACE2 expression was 25 times higher in the terminal ilium than in the colon, suggesting anatomical differences in ACE2 expression. In intestinal biopsies of IBD patients, treatment with glucocorticoids was associated with decreased ACE2 expression (Burgueno et al., 2020). Similarly, the use of corticosteroids, thiopurines and 5-aminosalicylate attenuated ACE2 and TMPRSS2 expression in inflamed colon and rectum (Suárez-Fariñas et al., 2020).
Clinical studies do not identify asthma as a risk factor of severe COVID-19-related illnesses (Z. Wu & McGoogan, 2020). Animal models indicate that ACE2 and Ang-(1-7) are protective in asthma (El-Hashim et al., 2012). In recent studies, ACE2 expression was not altered (Breidenbach et al., 2020; G. Li et al., 2020; Peters et al., 2020; Radzikowska et al., 2020) or reduced (Jackson et al., 2020; Kimura et al., 2020) in asthmatic patients; but increased expression of TMPRSS2, the enzyme facilitating SARS-CoV-2 entry into host cells, has been reported (Kimura et al., 2020; Radzikowska et al., 2020). However, in a large cohort study, increased ACE2 gene expression was reported in a sub-group of type 2 asthmatic patients (Camiolo, Gauthier, Kaminski, Ray, & Wenzel, 2020). Similarly, in bronchial brushings, biopsies and sputum-derived cells of patients with severe asthma, the gene expression of ACE2, TMPRSS2, and furin was positively correlated with asthma severity and glucocorticoid use (Kermani et al., 2020). However, in a recent study with 268 asthmatic patients, the use of glucocorticoids did not influence the gene expressions of ACE2, TMPRSS2, and furin in bronchial brushes and biopsy samples; and disease severity was not related to changes in these parameters (Bradding et al., 2020). In another study, the use of inhaled corticosteroids, but not of the synthetic corticosteroid triamcinolone acetonide, was associated with a lower expression of ACE2 and TMPRSS2 in asthmatic patients (Jackson et al., 2020). In patients with COPD, the administration of inhaled corticosteroids reduced sputum expression of ACE2 compared to controls (Finney et al., 2020). In this study, it was also shown that inhaled corticosteroids reduced ACE2 expression in airway epithelial cell cultures and mouse models, and the effect was reversed by interferon-β administration. Glucocorticoids, including hydrocortisone, prednisolone, dexamethasone, and methylprednisolone, significantly increased ACE2 protein expression in epithelial cell lines and reduced cytokine interleukin-6 production in human macrophages (Xiang et al., 2020). In bronchial epithelial cells from specimens of COPD patients, treatment with inhaled corticosteroids significantly decreased the expression of ACE2 and ADAM-17, and it was associated with decreased interferon type-1 gene expression (Stephen Milne et al., 2020).
In human epithelial cell cultures, the corticosteroid budesonide inhibits the replication of HCoV-229E, which uses aminopeptidase N as entry receptor (Yamaya et al., 2020). Recently, the glucocorticoid methylprednisolone was reported to increase the survival in a small cohort of COVID-19 positive ARDS patients (C. Wu et al., 2020). In earlier studies, methylprednisolone improved gastrointestinal pathology and accelerated recovery from HCoV infection (Rhoads, Macleod, & Hamilton, 1988), but it was also reported to promote the replication of HCoV-MHV-3 and to increase the mortality in mice (Fingerote, Leibowitz, Rao, & Levy, 1995). At clinically relevant concentrations, cortisone increases the replication of infectious bronchitis HCoV in tracheal organ cultures, whereas reproductive hormones, such as progesterone, estrogen, and testosterone, do not have this effect (Ambali & Jones, 1990). In a recent in vitro study, steroids (glycyrrhetinic, oleanolic acid) and bile acid derivatives inhibited binding of the SARS-CoV-2 spike protein to ACE2 (Carino et al., 2020). It was also reported that Ciclesonide, an inhaled corticosteroid, suppresses the replication of SARS-CoV-2 with an EC90 of 0.55 μM in human bronchial epithelial cells (Matsuyama et al., 2020).
During the previous SARS outbreak, it was reported that high-dose methyl prednisolone had beneficial effects (V. C. Cheng, Tang, Wu, Chu, & Yuen, 2004; Sung et al., 2004; Tsui, Kwok, Yuen, & Lai, 2003; Z. Zhao et al., 2003). However, a systematic analysis of clinical studies concludes that the results of corticosteroid therapies in SARS-CoV infections are inconclusive and that their application is not recommended (Stockman, Bellamy, & Garner, 2006). Furthermore, corticosteroid therapies reportedly decrease dendritic and T cells in the circulation (Z. Zhang et al., 2005), reduce cytokine releasing cells in the spleen (X. Zhang et al., 2008) and suppress cellular immune responses in the lungs (K. Jung et al., 2007). The use of high steroid doses is also associated with long lasting lipid disturbances (Q. Wu et al., 2017) and an increased risk of avascular necrosis (Sing, Tan, Wong, Cheung, & Cheung, 2020) in recovered SARS-CoV patients. During the recent COVID-19 outbreak, methylprednisolone therapies reportedly improved the clinical prognosis (Y. Wang, W. Jiang et al., 2020; F. Ye et al., 2020) or decreased the mortality rate in COVID-19 patients (Salton et al., 2020). Additional recent studies report that early, but not late phase (Mongardon et al., 2020), low-dose corticosteroids decrease mortality and improve COVID-19 clinical outcomes (Ji et al., 2020). However, another recent study indicates that corticosteroid use is associated with increased mortality and delayed SARS–CoV-2 coronavirus RNA clearance in 409 COVID-19 patients (J. Liu et al., 2020). While some of the meta-analyses conclude that the use of corticosteroids is associated with a higher rate of ARDS in COVID-19 patients (Z. Yang et al., 2020; J. J. Y. Zhang, Lee, Ang, Leo, & Young, 2020), a recent randomized study of several thousands of hospitalized COVID-19 patients reports that dexamethasone reduces the mortality among those receiving invasive mechanical ventilation or oxygen (Horby et al., 2020). In another study of 396 COVID-19 patients, the use of steroids significantly decreased in-hospital mortality (Fernandez Cruz et al., 2020), an observation that has been corroborated by several additional recent publications (Bani-Sadr et al., 2020; Chopra et al., 2020; Majmundar et al., 2020). Corticosteroid activation of glucocorticoid receptors has been suggested to suppress interleukin-6 release and mitigate multi-organ inflammation in some COVID-19 patients (Awasthi et al., 2020).
Non-steroid anti-inflammatory drugs (NSAIDs) inhibit the cyclooxygenase (COX)-dependent metabolism of arachidonic acid to prostaglandins. Treatments (50 μM, 48 hours) of arachidonic acid, octadecadienoic acid, and docosahexaenoic acid, but not eicosapentaenoic acid and stearic acid, significantly decreased ACE2 mRNA expression in porcine adipocytes (Tseng et al., 2010). Prostaglandins prevent hypertension and upregulate renal ACE2 protein expression in adult male rats exposed either prenatally to dexamethasone plus postnatally to high fat diet (P. C. Lu et al., 2016) or prenatally to high fructose intake (Tain, Lee, Wu, Leu, & Chan, 2016). Lipoxin A4, another product of arachidonic acid metabolism, attenuates lung injury and increases lung ACE2 levels and protein expression in a lipopolysaccharide-induced lung injury model (Q. F. Chen et al., 2018). Similarly, the lipoxin receptor agonist BML-111 also decreases lung and liver injuries, which are associated with increased ACE2 levels and protein expression in these tissues (Q. F. Chen et al., 2019; Hu et al., 2017). However, although pharmacological inhibition of soluble epoxide hydrolase, which metabolizes epoxyeicosatrienoic acids, improved disease pathology and increased epoxyeicosatrienoic acid levels, it did not reverse downregulation of cardiac ACE2 expression induced by high-fructose diet intake (Froogh et al., 2020). 20-Hydroxy-eicosatetraenoic acid (20-HETE), a cytochrome P450-derived arachidonic acid metabolite, increased blood pressure and vascular Ang-II expression. It also upregulated vascular ACE transcription without altering ACE2 expression (K. Sodhi et al., 2010). In addition, COX-2 and prostaglandin E2 (PGE2) activate ADAM17 (Al-Salihi et al., 2007). NSAIDs, through COX-independent mechanisms, promote shedding of L-selectin, a pro-inflammatory cell adhesion molecule, by activating ADAM17 and generating superoxide anions at the plasma membrane through NADPH-oxidase activation. They thereby interfere with neutrophil–endothelial cell adhesion (Dominguez-Luis et al., 2013; Gomez-Gaviro et al., 2002) and potentially decrease cell surface activity of ACE2.
Activation of both COX-1 and COX-2 mediates some of the Ang-II responses, such as hypertension, oxidative stress, and inflammation (Sriramula, Xia, Xu, & Lazartigues, 2015; R. Wu, Laplante, & de Champlain, 2005). Pharmacological inhibition or genetic deletion of COX-1 reduces the acute pressor effects of Ang-II in murine disease models (X. Cao et al., 2012; Z. Qi et al., 2002; Sriramula, Xia, Xu, & Lazartigues, 2015). COX-2 inhibition by rofecoxib and nimesulide attenuates Ang-II–induced oxidative stress, hypertension, and cardiac hypertrophy in rats (R. Wu, Laplante, & de Champlain, 2005). Thus, over-expression of ACE2 in the brain decreases blood pressure, oxidative stress and inflammation; it also down regulates COX expression in murine hypertension models (Sriramula et al., 2015). However, vasodilatatory and cardioprotective effects of Ang-(1-7) are also mediated by activation of COX, and these effects are inhibited by indomethacin, a non-selective COX inhibitor (X. Liao et al., 2011). Therefore, while some of the effects of NSAIDs potentiate Ang-II actions, others counteract Ang-II. Furthermore, all NSAIDs except ketoprofen, through COX-independent mechanisms, inhibit SIRT1 deacetylase (Dell'Omo et al., 2019), which counteracts upregulation of ACE2 by SIRT1 (Clarke et al., 2014). Importantly, SIRT1 expression was reported to be upregulated in the lungs of COVID-19 patients with comorbidities (Pinto et al., 2020).
Ibuprofen, a commonly used NSAID and non-selective COX inhibitor, attenuates cardiac fibrosis and upregulates cardiac ACE2 expression in streptozotocin-induced diabetic rats (Qiao et al., 2015). Other NSAIDs, including rofecoxib, meloxicam, celecoxib and flurbiprofen, at clinically relevant doses, were shown to induce modest increases in renal and cardiac ACE2 protein expression in adjuvant-induced arthritic rats (Asghar, Aghazadeh-Habashi, & Jamali, 2017).
The SARS-CoV has been shown to directly bind to the COX-2 promotor and to increase its expression (Yan et al., 2006). COX-2–dependent PGE2 was reported to attenuate the chronic antiviral lymphocyte response of unresolved viral infections (Schaeuble et al., 2019), suggesting that NSAIDs may have beneficial effects in the treatment of SARS-CoV-2 infection. However, two previous meta-analyses have shown that the use of NSAIDs, including ibuprofen, is associated with increased venous thromboembolism and increased risk of vascular events (T. Lee et al., 2016; Ungprasert, Srivali, Wijarnpreecha, Charoenpong, & Knight, 2015). NSAIDs increase the risk of thromboembolism (Schmidt et al., 2011), stimulate salt intake and enhance renal and pulmonary vasoconstriction (Cumhur Cure, Kucuk, & Cure, 2020; Harrington et al., 2008; Varga, Sabzwari, & Vargova, 2017), which are undesirable pharmacological actions in the treatment of COVID-19. In addition, observational studies suggest an association between pre-hospital NSAID exposure and a protracted and complicated course of pneumonia (Voiriot et al., 2019). Therefore, it has been recommended to use NSAIDs at the lowest effective dose for the shortest possible period, and, instead, in most cases, to use paracetamol (acetaminophen) as the first treatment option for fever or pain associated with infections (Zolk et al., 2020). Interestingly, indomethacin, a non-selective COX inhibitor, has antiviral effects with an EC50 of 5 μM for SARS-CoV, as demonstrated in human cell lines and by in vivo experiments (Amici et al., 2006). However, the effect of indomethacin is independent of COX inhibition, since high concentrations of aspirin do not have an antiviral activity. In clinical studies of small cohorts of COVID-19 patients, treatment with NSAIDs was associated with either adverse clinical outcomes (Jeong et al., 2020) or no effect on mortality rate (Abu Esba et al., 2020; Chandan et al., 2020; Lund et al., 2020) or a modest beneficial effect on survival rates (Bruce et al., 2020). The mechanisms of NSAID actions and their potential use in COVID-19 patients have recently been discussed (Cabbab & Manalo, 2020; Micallef, Soeiro, & Jonville-Béra, 2020). The effects of steroids, NSAIDs, and pharmacologically related compounds on ACE2 activity and expression are summarized in Table 6 .
| Pharmacological agent/class | Experimental model /Tissue/Subject | Effect | Reference |
|---|---|---|---|
| Betamethasone/ Glucocorticoid steroid | Male sheep/ Kidney and blood | Reduction in serum ACE2 activity. In isolated proximal tubules, ACE2 activity and expression were 50% lower in the treated sheep | Shaltout, Figueroa, Rose, Diz, and Chappell (2009) |
| Betamethasone/ Glucocorticoid steroid | Sheep/ Prenatal betamethasone exposed offspring’s | No change in choroid plexus ACE2 activity | Marshall et al. (2013) |
| Betamethasone/Glucocorticoid steroid | Female sheep/ Adipose tissue | No change in ACE2 mRNA expression | Massmann, Zhang, Seong, Kim, and Figueroa (2017) |
| Betamethasone/ Glucocorticoid steroid | Preterm, and term piglets/ Heart and kidney | Cardiac ACE2 expression was decreased in preterm piglets. Renal ACE2 expression was unaffected | E. Kim et al. (2015) |
| Dexamethasone/Glucocorticoid steroid | Sprague-Dawley rats/ Prenatal dexamethasone exposure | No change in renal ACE2 protein expression | P. C. Lu et al. (2016) |
| Dexamethasone/Glucocorticoid steroid | Sprague Dawley rats/ fetal tissue | Suppression of ACE2 mRNA and protein expression in placental tissue | Ghadhanfar et al. (2017) |
| Dexamethasone/Glucocorticoid steroid | Sprague-Dawley female rats/ Adipose tissue | No change in ACE2 gene expression | H. R. Yu et al. (2018) |
| Ibuprofen/ NSAID | Rats/ Streptozotocin-induced diabetes model | Increased cardiac ACE2 protein expression | Weili et al. (2014) |
| Ibuprofen/ NSAID | Male Sprague-Dawley rats/ Streptozotocin-induced diabetes model | Increased cardiac ACE2 immunostaining and mRNA expression | Qiao et al. (2015) |
| Rofecoxib, meloxicam, celecoxib and flurbiprofen/ NSAID | Male Sprague-Dawley rats/ Adjuvant induced arthritis model | All drugs increased cardiac and renal ACE2 protein expression arthritic rats with reduced ACE2 expression. | Asghar, Aghazadeh-Habashi, and Jamali (2017) |
Activation of vitamin D receptors (VDR) by 1,25-dihydroxyvitamin D (calcitriol) or pharmacologic VDR agonists is important for the control of phosphate and calcium homeostasis and bone remodeling but could also have beneficial effects by reducing the risk of cardiovascular morbidity and mortality, diabetes, autoimmune diseases, and cancer. Vitamin D3 supplementation was shown to upregulate cardiac ACE2 gene expression in normotensive rats; whereas vitamin D3 deficiency had no effect on ACE2 expression (Machado, Ferro Aissa, Ribeiro, & Antunes, 2019). Similarly, vitamin D deficiency did not affect serum ACE2 levels in a transgenic hypertension model (Andersen et al., 2015). In spontaneously hypertensive rats with an overactive RAS, calcitriol decreased oxidative stress, markedly reduced Ang-II formation, and upregulated brain ACE2 expression (C. Cui et al., 2019). Interestingly, in these experiments, calcitriol also upregulated ACE2 expression in the brains of normotensive rats, as well as in cultured BV2 retroviral immortalized microglial cells. Calcitriol, an active metabolite of vitamin D3, inhibited Ang-II and renin expression, decreased vascular permeability and cell death, and reversed lipopolysaccharide-induced downregulation of ACE2 in pulmonary tissue and vascular endothelial cells (J. Xu et al., 2017). Similarly, calcitriol increased renal ACE2 expression in diabetic rats with compromised ACE2 activity and counteracted glucose-induced downregulation of ACE2 by inhibiting p38 MAPK and ERK phosphorylation in NRK-52E cells (M. Lin et al., 2016). In type I non-obese diabetic rats, paricalcitol, a synthetic vitamin D analog, decreased serum ACE2 activity, renal oxidative stress, and circulating H2O2 levels. Although renal ACE2 activity was not altered, renal ADAM17 was reduced by paricalcitol (Riera et al., 2016). Importantly, in this study, paricalcitol upregulated ACE2 mRNA expression in epithelial cell lines in a dose-dependent manner. In line with these findings, paricalcitol inhibits aldosterone-induced upregulation of the ADAM17/TGF-α/EGF receptor pathway in cultured tubular epithelial cells (Morgado-Pascual et al., 2015). Vitamin D has been shown to suppress ADAM17 expression in A431 cell lines (Arcidiacono, Yang, Fernandez, & Dusso, 2015) and in parathyroid cells (Dusso, Arcidiacono, Yang, & Tokumoto, 2010).
A recent study reports that vitamin D attenuates lung injury by stimulating epithelial repair, reducing epithelial cell apoptosis, and decreasing TGF-β levels (Zheng et al., 2020). In animal experiments, it has been demonstrated that vitamin D reduces disease severity of coronaviruses (J. Yang et al., 2019) by regulating autophagy, enhancing cathelicidin production and inhibiting intestinal mucosa interleukin (IL)-6 and IL-8 mRNA expression, thereby lessening the severity of damage (Yuk et al., 2009). Vitamin D reduces the susceptibility to acute lung injury by inhibiting renin and consequently Ang-II biosynthesis (Zittermann et al., 2018). In addition, vitamin D reportedly reduces disease severity and decreases the risk of respiratory tract infections in a large cohort of adults (Zittermann, Pilz, Hoffmann, & Marz, 2016). In line with these findings, a recent meta-analysis study indicates that the use of vitamin D is associated with a reduced risk of acute respiratory infections, and administration of daily doses of 400-1000 IU vitamin D for up to 12 months was found to have a protective effect (Jolliffe et al., 2020). In a randomized controlled trial of school children, daily vitamin D intake resulted in a 58% reduction of the relative risk of influenza A, compared to the placebo group (Urashima et al., 2010). Importantly, post-infection treatment with 10 μM calcitriol significantly reduced SARS-CoV-2 infection in Vero E6 and human epithelial cells (Mok et al., 2020). In several recent studies on small cohorts of COVID-19 patients, vitamin D deficiency has been identified as an independent risk factor for increased mortality or a higher rate of intensive care admission and disease severity (Abrishami et al., 2020; Arvinte, Singh, & Marik, 2020; Baktash et al., 2020, Brenner et al., 2020; Carpagnano et al., 2020; Hernández et al., 2020; Macaya et al., 2020; Munshi et al., 2020; Panagiotou et al., 2020; K. Ye et al., 2020). Vitamin D deficiency was also associated with increased SARS-CoV-2 positivity rates and infection (Kaufman, Niles, Kroll, Bi, & Holick, 2020; Merzon et al., 2020). Conversely, vitamin D supplements were reportedly associated with a less severe disease progress and faster recovery rates in COVID-19 patients (C. Annweiler et al., 2020; G. Annweiler et al., 2020; Rastogi et al., 2020; Tan et al., 2020, Tan et al., 2020). Vitamin D supplementation has recently been reviewed (Grant et al., 2020; Malek Mahdavi, 2020; Tay, Mahajan, & Thornton, 2020) with the conclusion that it is effective in boosting the immune system, strengthening the lung epithelial barrier, and preventing an excessive inflammatory response and viral infections.
All-trans retinoic acid (atRA), a biologically active metabolite of vitamin A, modulates gene transcription and exerts its other effects by binding to the retinoic acid receptor, and interfering with transcription factors. atRA reduced the blood pressure, attenuated myocardial damage, and significantly upregulated cardiac and renal ACE2 expression in spontaneously hypertensive rats (Zhong et al., 2004). However, chronic atRA treatment did not have an effect on the expression of ACE2 in non-hypertensive rats (Zhong et al., 2004), suggesting that atRA can potentially be used in the treatment of hypertension. atRA decreased oxidative stress and Ang-II production; it also upregulated mitogen-activated protein kinase phosphatase (MKP)-1, MKP-2 and cardiac ACE2 expression in rats with pressure overload-induced cardiac remodeling (Choudhary et al., 2008). In rats with glomerulosclerotic lesions, atRA reduced glomerular lesions and Ang-II expression; it also markedly upregulated renal ACE2 mRNA and protein expression (T. B. Zhou, Drummen, Jiang, Long, & Qin, 2013). Treatment with atRA decreased the formation of reactive oxygen species, Ang-II expression and reversed the downregulation of ACE2 expression due to hypoxia-induced injury in renal tubular epithelial cells (T. B. Zhou, Ou, Rong, & Drummen, 2014). Importantly, vitamins D (Strycharz et al., 2018), C (Aşcı et al., 2016; M.-Z. Qi et al., 2018), A (A. N. Shin et al., 2018), and B3 (Hong et al., 2018) have been shown to activate SIRT1, suggesting that they can upregulate ACE2 expression (Clarke et al., 2014). In addition, atRA upregulates mRNA expression (Flannery, Little, Caterson, & Hughes, 1999) and promotes activation as well as translocation of ADAM17 to the cytoplasm (Koryakina, Aeberhard, Kiefer, Hamburger, & Kuenzi, 2009), suggesting that atRA can potentially modulate ACE2 shedding through ADAM17 as well. Effects of vitamins on the activity and expression of ACE2 are presented in Table 7 .
| Pharmacological agent/class | Experimental model /Tissue/ Subject | Effect | Reference |
|---|---|---|---|
| Calcitriol/1,25-dihydroxyvitamin D | Male Wistar rats and NRK-52E cells / Streptozotocin induced diabetes model | Increased ACE2 immunostaining and protein expression in kidney and NRK-52E cells | M. Lin et al. (2016) |
| Calcitriol/1,25-dihydroxyvitamin D | Male Wistar rats and pulmonary microvascular endothelial cells/ Lipopolysaccharide -induced lung injury model | Increased pulmonary ACE2 mRNA, immunostaining, and protein expression | J. Xu, Yang, et al. (2017) |
| Alfacalcidol//active metabolite of Vitamin D | Sprague-Dawley rats/ Ischemia-reperfusion injury model | Renal ACE2 mRNA and protein expression was increased by injury and downregulated by alfacalcidol | Ali, Al-Shorbagy, Helmy, and El-Abhar (2018) |
| Paricalcitol/ Synthetic vitamin D analog | Female NOD/ ShiLtJ and NOR/LtJ mice and MTC cells | Increased ACE2 immunostaining, mRNA and protein expression in kidney and MTC cells. | Riera et al. (2016) |
| Calcitriol/1,25-dihydroxyvitamin D | Male Spontaneously hypertensive rats and BV2 microglial cells | Increased ACE2 mRNA and protein expression in brain and BV2 microglial cells. | C. Cui et al. (2019) |
| All-trans retinoic acid/active metabolite of vitamin A | Spontaneously hypertensive and Wistar-Kyoto rats/cardiac tissue | Increased ACE2 mRNA and protein expression in heart and kidney | Zhong et al. (2004) |
| All-trans retinoic acid/active metabolite of vitamin A | Male Wistar rats/ Uninephrectomy and adriamycin induced glomerulosclerosis Model | Markedly increased renal ACE2 immunostaining, mRNA and protein expression | T. B. Zhou, Drummen, Jiang, Long, and Qin (2013) |
| All-trans retinoic acid/active metabolite of vitamin A | NRK-52E rat renal proximal tubular epithelial cell line/ Hypoxia-induced Injury model | Increased renal ACE2 mRNA and protein expression | T. B. Zhou, Ou, Rong, and Drummen (2014) |
| All-trans retinoic acid/active metabolite of vitamin A | Male / Aortic constriction induced pressure model | Increased cardiac ACE2 protein expression | Choudhary et al. (2008) |
Due to the pivotal role of ACE2 as entry receptor of SARS-CoV2, the prevention of SARS-CoV-2 spike protein-ACE2 interaction and subsequent viral infectivity is an important antiviral treatment strategy. In earlier studies, soluble ACE2 was able to block the replication of SARS-CoV in HEK-293T cells (Wenhui Li et al., 2003). Recombinant soluble human ACE2 fused to the Fc region of the human immunoglobulin IgG1 to increase short half-life of soluble ACE2 (Iwanaga et al., 2020; Lei et al., 2020) and human recombinant soluble ACE2 (Monteil et al., 2020) have been shown to inhibit SARS-CoV-2 infection in cell lines, engineered human blood vessels and kidney organoids. Recently, peptides mimicking the N-terminal helix of the human ACE2 protein, which contains most of the contacting residues for the S protein-binding site, were shown to block infection of human pulmonary cells with SARS-CoV-2, with IC50 values in the range of 60-800 nM (Karoyan et al., 2020). Similarly, ACE2 peptides optimized to SARS-CoV-2 spike protein binding regions using protein-engineering methods potently bound to the spike protein with a 170-fold higher affinity than wild-type ACE2 and inhibited SARS-CoV-2 infection (IC50 of 28 ng/ml) in cell lines (Glasgow et al., 2020). In another set of experiments, a fusion protein consisting of ACE2 and an immunoglobulin Fc protein effectively blocked SARS-CoV-2 infection in HEK-293T cells with an IC50 of 4 μg/mL (Y. Li et al., 2020).
In an earlier study, a compound coined SSAA09E2 has been shown to block the binding of the SARS-CoV spike protein to ACE2 and to inhibit SARS-CoV infection in ACE2 expressing HEK-293T cells with an EC50 of 3.1 μM (Adedeji et al., 2013). Chloroquine and hydroxychloroquine, in addition to their pH elevating effects in endosomes, bind to ACE2 (N. Wang et al., 2020), impair the terminal glycosylation of ACE2 (Vincent et al., 2005) and inhibit SARS-CoV replication (Al-Bari, 2017; Keyaerts, Vijgen, Maes, Neyts, & Van Ranst, 2004). They also prevent the entrance of SARS CoV-2 spike protein into ACE2 expressing cell lines (N. Wang et al., 2020). Results from recent studies reveal that chloroquine and, more effectively, hydroxychloroquine also inhibit the replication of SARS-CoV-2 in simian Vero cells (X. Yao et al., 2020). TAPI-2, an inhibitor of TNF-α converting enzyme (ADAM17), blocks SARS-CoV S protein-induced shedding of ACE2 and inhibits SARS-CoV cell entry (Haga et al., 2010). Recently, ceftazidime, an antibiotic, was shown to inhibit SARS-CoV spike protein-ACE2 interaction and to prevent SARS-CoV-2 pseudovirus infection of ACE2-expressing HEK-293T cells (C. Lin et al., 2020). Finally, various antiviral compounds, such as emodin, baicalin and green tea extracts found in Chinese herbs, are reviewed in other chapters of this review.
Drugs used for cardiovascular diseases and diabetes, as described in earlier sections, significantly interact with ACE2, due to important roles of the ACE2/Ang-(1-7)/Mas receptor axis in the pathogenesis of these diseases. However, some anticancer agents, antibiotics, and other drugs also modulate the activity and expression of ACE2. For example, propofol, an intravenous anesthetic, activates the phosphatidyl-inositol 3-kinase (PI3K)/Akt signaling pathway and upregulates ACE2 expression in human pulmonary endothelial cells (L. Cao, Xu, Huang, & Wu, 2012). Similarly, propofol prevents Ang-II-induced apoptosis and oxidative stress, increases NOS phosphorylation, and upregulates ACE2 protein expression in human umbilical vein endothelial cells (L. Zhang et al., 2018). Some other drugs, such as certain anticancer agents and antibiotics, also reportedly affect ACE2 activity, and a list of these drugs and pharmacological agents is provided in Table 8 .
| Pharmacological agent/class | Experimental model /Tissue/ Subject | Effect | Reference |
|---|---|---|---|
| Propofol/ Intravenous anesthetic | Human pulmonary artery endothelial cells | Increased ACE2 activity and mRNA expression | L. Cao et al. (2012) |
| Propofol/ Intravenous anesthetic | Human umbilical vein endothelial cell | Increased ACE2 protein expression | L. Zhang et al. (2018) |
| Pregabalin/ Neuronal calcium channel inhibitor. Treatment of epilepsy, neuropathic pain, fibromyalgia | Male Sprague-Dawley rats/ Pregabalin-induced cardiotoxicity | Decreased cardiac ACE2 protein expression | Awwad et al. (2020) |
| hValproic acid/ Sodium channel blocker, Treatment of epilepsy and bipolar disorder | Vascular endothelial cell cultures | Decreased ACE2 mRNA expression | Singh and Singh (2020) |
| Bleomycin/ Anticancer drug | C57BL/6J mice and male Wistar rats/ Bleomycin-induced lung fibrosis model | Decreased pulmonary ACE2 activity and protein expression. | X. Li et al. (2008) |
| Bleomycin/ Anticancer drug | Male Wistar rats/Bleomycin-induced lung fibrosis model | Increased pulmonary ACE2 immunostaining, protein and mRNA expression | Meng et al. (2014) |
| Bleomycin/ Anticancer drug | Male Sprague-Dawley rats/ Bleomycin-induced lung fibrosis model | Decreased pulmonary ACE2 immunostaining and mRNA expression | H. Wu et al. (2014) |
| Bleomycin/ Anticancer drug | Male C57BL/6 mice/ Bleomycin-induced lung fibrosis model | Decreased pulmonary ACE2 immunostaining and mRNA expression | L. Wang, Wang, Yang, Guo, & Sun et al. (2015) |
| Doxorubicin/ Anticancer drug | Male Wistar rats/ Doxorubicin-induced cardiomyopathy model | No change in cardiac ACE2 protein expression | H. Ma et al. (2017) |
| Etanercept/ Immunosuppressant | Male Sprague-Dawley rats/ Ang-II induced hypertension model | Increased brain ACE2 mRNA expression. No change in control rats. | Sriramula, Cardinale, & Francis (2013) |
| Cyclosporine/ Immunosuppressant | Human hepatoma HepG2 cell line | Decreased ACE2 expression through hepatic nuclear factor 4α | Niehof and Borlak (2011) |
| Cisplatin, gemcitabine/ Anticancer drugs | A549 lung cancer cell line | Both drugs increased ACE2 protein expression | Teng et al. (2018) |
| Ceftriaxone/ Antibiotic | Male Wistar and OXYS rats/ | Increased ACE2 in hypothalamus | Tikhonova et al. (2018) |
| Pirfenidone/ Antifibrotic drug | Male Sprague-Dawley rats/ Coronary artery ligation-induced myocardial infarction model | Increased cardiac ACE2 protein expression | C. Li et al. (2017) |
| Ulinastatin/ Treatment of pancreatitis | Male C57BL/6 mice/ Cerulean and lipopolysaccharide induced pancreatitis model | Increased pancreatic ACE2 immunostaining, mRNA and protein expression | R. Liu et al. (2014b) |
| Hydroxyurea/ Sickle cell anemia drug | C57BL/6 mice/ Sickle cell mice model | Reversal of decreased ACE2 mRNA expression in the kidneys of the sickle cell mice | dos Santos et al. (2014) |
| Fasudil/ Rho-kinase inhibitor and used for the treatment of cerebral vasospasm. | Male Sprague-Dawley rats/ Deoxycorticosterone-induced hypertension model | Increased vascular ACE2 immunostaining and mRNA expression | Ocaranza et al. (2011) |
| Fasudil/ Rho-kinase inhibitor and used for the treatment of cerebral vasospasm. | Male Sprague-Dawley rats/ Hypoxia exposure of pulmonary artery smooth muscle cells | Increased ACE2 protein expression | Y. X. Wang, Liu, Zhang, Fu, & Li et al. (2016) |
| Fasudil (HA-1077) / Rho-kinase inhibitor and used for the treatment of cerebral vasospasm. | Male Sprague-Dawley rats/ Pulmonary artery emboli model, Pulmonary artery endothelial cells | Marked increase in ACE2 mRNA and protein expression in cultured cells and pulmonary tissue from embolic rats | X. Xu et al. (2019) |
| Granulocyte colony stimulating factor/ Treatment of chemotherapy induced neutropenia | Male C57BL/6 mice/ Ang-II induced cardiac hypertrophy model | Increased cardiac ACE2 protein expression in Ang-II treated group. But, no effect in control group. | N. Jia et al. (2009) |
| Activated protein C/A serine protease used for treatment of severe sepsis | Male Sprague-Dawley rats/ Lipopolysaccharide induced kidney injury model | Increased renal ACE2 mRNA expression in injured kidneys | A. Gupta et al. (2007) |
| Activated protein C/A serine protease used for treatment of severe sepsis | Sprague-Dawley rats/ Cecal ligation and puncture induced polymicrobial sepsis model | Increased pulmonary ACE2 protein expression | Richardson et al. (2008) |
| Cinacalcet/1,25-dihydroxyvitamin D used for treatment of hyperparathyroidism | Male Wistar rats/ Adenine diet induced kidney disease model | No effect on renal ACE2 mRNA expression | Tormanen et al. (2017) |
| TRV027/ Biased agonist of AT1 receptor | Male spontaneously hypertensive and Wistar Kyoto rats | No effect on ACE2 activity and protein expression in HEK-293T cells | Carvalho-Galvão et al. (2018) |
| Adenine/ purine base | Male Wistar rats/ Adenine diet induced kidney disease model | Marked decrease in renal ACE2 mRNA expression | Tormanen et al. (2017) |
| Nanoparticles/ drug delivery vehicle | C57BL/6 J mice/ Effects of different size nanoparticles on lung injury model | Nanoparticle G5 decreased pulmonary ACE2 mRNA and protein expression. | Sun et al. (2015) |
In recent years, potential health and therapeutic benefits, nutritional values, and biological activities of phytochemicals, natural products and their bioactive compounds have been intensively studied. Among the vast number of these compounds, some phytochemicals can affect the activity and expression of ACE2.
Curcumin, a pigment extracted from the rhizomes of the turmeric plant Curcuma longa, exhibits diverse pharmacologic characteristics, such as anti-oxidant, anti-inflammatory, and anti-fibrotic properties. In rats subjected to Ang-II infusion, curcumin significantly decreased the arterial blood pressure, reduced AT1 receptor expression and upregulated the AT2 receptor. Along with these modulations, curcumin decreased the number of macrophages and myofibroblasts; it also inhibited collagen synthesis and tissue fibrosis, which were accompanied by reduced expression of TGF-β1 and phosphorylated-Smad2/3 (Pang et al., 2015). Importantly, curcumin upregulated ACE2 protein expression in cardiac tissue, suggesting beneficial effects of curcumin in cardiac fibrosis. In another study, treatment with a curcumin analog reduced serum creatinine, urea nitrogen and urine albumin; it decreased Ang-II, improved renal pathology and upregulated renal ACE2 protein and mRNA expression in diabetic rats (X. Xu, Cai, & Yu, 2018). In addition, curcumin and its amino acid conjugates upregulate ADAM17 expression in HEK-293 cells (Narasingappa et al., 2012), suggesting that curcumin can modulate ACE2 shedding. Curcumin, at a concentration of 20 μM, was found to inhibit SARS-CoV-induced cytopathogenic effects in Vero-E6 cells (Wen et al., 2007). Beneficial effects of curcumin in the context of COVID-19 have been reviewed recently (Zahedipour et al., 2020).
Embelin, a naturally occurring para-benzoquinone isolated from dried berries of false black pepper (Embelia ribes) plants with antioxidant, anti-inflammatory, antidiabetic, and analgesic effects, has been shown to inhibit ADAM17 expression and activity in cancer cell lines (Dhanjal et al., 2014), suggesting that embelin potentially upregulates ACE2 activity by inhibiting ADAM17 mediated shedding. Similarly, 4-Hydroxyisoleucine, a plant-derived antidiabetic compound extracted from the seeds of fenugreek (Trigonella foenum-graecum), has been shown to downregulate ADAM17 expression in 3T3-L1 adipocytes (F. Gao et al., 2015) and HepG2 cells (F. Gao et al., 2015).
Resveratrol, a stilbenoid and natural polyphenol that is found in high concentrations in the skins of red wine grapes (Vitis vinifera), in red wine and in sprouted peanuts (Arachis hypogaea), reportedly has beneficial cardiovascular and metabolic actions. Resveratrol decreased adipose tissue mass, improved insulin-sensitivity and glucose tolerance, lowered plasma levels of glucose and lipids and upregulated ACE2 mRNA expression through activation of SIRT1 in adipocyte cell cultures and adipose tissue from FVB/N mice fed on a high fat diet (Oliveira Andrade et al., 2014). The improved metabolic profile induced by resveratrol was associated with marked up-regulation of glucose transporter type 4 (GLUT4) in adipose tissue. GLUT4, a key protein in glucose metabolism, exerts its influence by stimulating protein AMP-activated protein kinase (AMPK) and phosphorylating forkhead/wingedhelix O (FoxO)1. Administration of resveratrol prevented the development of liver pathology in rats fed maternally and postnatally on a high fat diet. Antioxidant, anti-apoptotic, and lipid metabolism regulating actions of resveratrol are associated with upregulation of SIRT1, leptin and ACE2 mRNA and protein expression in the liver (Tiao et al., 2018). In thoracic aortas of aging rats, resveratrol reduced serum Ang-II, increased Ang-(1-7) levels, and upregulated protein expression of ACE2, along with expression of AT2 and Mas receptors (E. N. Kim et al., 2018). In apolipoprotein E-deficient mice fed on a high fat diet, resveratrol reduced the development of aortic aneurysms, elevated serum ACE2 levels and upregulated aortic tissue levels of ACE2 and SIRT1 activity, but decreased the phosphorylation of Akt and ERK1/2 (Moran et al., 2017). Since activation or increased expression of SIRT1 is associated with the induction of ACE2 expression (Clarke et al., 2014), activation of SIRT1 by phytochemicals, such as resveratrol (Borra, Smith, & Denu, 2005; E. N. Kim et al., 2018; Moran et al., 2017) and curcumin (Zendedel, Butler, Atkin, & Sahebkar, 2018), can potentially mediate ACE2 upregulation by these compounds. In addition, resveratrol has been shown to decrease inflammation and increase ADAM17 expression through SIRT1 activation in a colonic inflammation model (Sharma et al., 2014). It has also been demonstrated that resveratrol inhibits MERS-CoV infections (S. C. Lin et al., 2017), and some of its substituted derivatives possess antiviral activity against SARS-CoV (Y. Q. Li et al., 2006). Thus, resveratrol and its analogs may be effective against SARS-CoV-2 infection, too, as it was found to form highly stable bounds with the viral protein-ACE2 receptor complex in silico (Wahedi, Ahmad, & Abbasi, 2020). In a recent study, resveratrol showed an antiviral effect (IC50 = 66 μM), inhibiting SARS-CoV-2 replication and infection in Vero-E6 and human bronchial epithelial cells (Ellen ter et al., 2020). Another polyphenol. quercetin (IC50 = 4.5 μM) and its metabolites have been shown to inhibit the enzymatic activity of human ACE2 in a concentration-, time- and temperature-dependent manner (X. Liu, Raghuvanshi, Ceylan, & Bolling, 2020). Among other plant-based compounds, nicotianamine, isolated from soybean, was reported to be a potent inhibitor of human ACE2 activity with an IC50 of 84 nM (Takahashi, Yoshiya, Yoshizawa-Kumagaye, & Sugiyama, 2015). Recently, it was reported that organosulfur compounds, such as allyl disulfide and allyl trisulfide found in garlic, interact strongly with human ACE2 and the main protease PDB6LU7 of SARS-CoV (Thuy et al., 2020). GB-2, the formula from the Holy Heavenly Mother Peitian Temple in Puzi, Chiayi County, was widely used for the prophylaxis of SARS-CoV-2 infection in Taiwan. It was shown that GB-2 significantly decreased ACE2 protein and mRNA expression in HepG2 and HEK-293T cells in a concentration-dependent (10-250 μg/ml) manner (C. Y. Wu et al., 2020).
Biological activities of organic compounds used in Traditional Chinese medicine (TCM) have been the subject of considerable investigations, especially in the fields of cardiovascular and cancer research. Several commonly used TCM compounds have been reported to influence the activity and expression of ACE2. A detailed list of phytochemicals and naturally occurring substances is provided in Table 9 . The antiviral actions of these compounds on coronaviruses in general have been reviewed in detail recently (Islam et al., 2020; Mani et al., 2020). Astragaloside III, a triterpenoid saponin isolated from Astragali Radix, a widely used herb in TCM, has potent anti-inflammatory and anti-atherosclerotic effects. In endothelial cells, astragaloside III activates growth factor signaling through the p38 signaling pathway and upregulates ADAM17 (H. Wang et al., 2020), suggesting that cell surface ACE2 shedding can be modulated by astragaloside III. Similarly, paeoniflorin, another traditional Chinese medicine compound extracted from Paeoniae Radix, induces Src kinase dependent activation of ADAM17 in vascular endothelial cells (H. Wang et al., 2018). Tanshinones, a class of abietane diterpene phytochemicals isolated from Salvia miltiorrhiza, a well-known herb used in TCM, are known to have anti-inflammatory and anti-oxidant effects; they are used for the treatment of cardio- and cerebrovascular diseases (Z. Jiang, Gao, & Huang, 2019). Tanshinone IIA attenuates pulmonary fibrosis and lung injury; it also upregulates pulmonary ACE2 mRNA and protein expression (Y. Wang et al., 2018; H. Wu et al., 2014). Moreover, tanshinones inhibit SARS-CoV cysteine proteases, suggesting antiviral effects (J. Y. Park et al., 2012). In addition, abietane diterpenoids with a chemical structures similar to tanshinones, and labdane type diterpenoids potently inhibit SARS-CoV replication and virus-induced cytopathogenic effects in Vero-E6 cells with IC50 values ranging from 1.4 μM to 7.5 μM (Wen et al., 2007). Chemical components of the Lianhuaqingwen capsule, a commonly used antiviral TCM containing neochlorogenic acid, amygdalin, prunasin, forsythoside I, rutin, forsythoside A, and rhein, exhibited binding affinities to ACE2 with KD values ranging from 0.2 to 82.4 μmol/L, interrupted SARS-CoV-2 spike protein binding to ACE2 with different efficacies (X. Chen et al., 2020), inhibited SARS-CoV-2 replication in Vero-E6 cells and reduced pro-inflammatory cytokine expression in Huh-7 cells (Runfeng et al., 2020). Glycyrrhizin and its derivatives, active components of liquorice roots used in TCM, bind to ACE2 (KD of 4.4.μmol/L) and inhibit the interaction of SARS-CoV-2 spike protein with ACE2 (S. Yu et al., 2020). They also decrease the replication of SARS-CoV (Hoever et al., 2005) and SARS-CoV-2 in cell lines (X. Chen et al., 2020; S. Yu et al., 2020). Baicalin, a flavonoid isolated from the roots of Scutellaria baicalensis Georgi (Huang Qin) used in TCM, has anti-oxidative, anti-viral, anti-inflammatory, anti-HIV and anti-proliferative activities. Baicalin increased ACE2 mRNA and protein expression in human umbilical vein endothelial cells treated with Ang-II (X. Wei et al., 2015). Baicalin also showed an antiviral activity against SARS-CoV in fRhK4 and Vero-E6 cells (F. Chen et al., 2004). In addition, baicalin inhibited the replication of the porcine reproductive and respiratory syndrome virus, remotely related to SARS-CoV (Karuppannan, Wu, Qiang, Chu, & Kwang, 2012). It has been reported that green tea extracts, in a concentration range of 0.1to 0/8 mg/ml, are potent inhibitors of pseudotyped SARS-CoV and SARS CoV-2 infection by disrupting the binding of the spike protein to ACE2 (Joseph, T, Ajay, Das, & Raj, 2020). Emodin, another compound used in TCM, which can be isolated from rhubarb and buckthorn, reportedly inhibits the binding of SARS-CoV spike protein to ACE2 (Ho, Wu, Chen, Li, & Hsiang, 2007). Naringenin, a citrus fruit flavonoid, has been shown to inhibit SARS-CoV-2 infection in Vero-6 cells (Clementi et al., 2020), decrease the expression of the proinflammatory cytokines in Raw macrophage cells, and potentially bind to ACE2 (L. Cheng et al., 2020). In summary, several compounds used in TCM not only affect ACE2 expression, but also possess antiviral activities, which have been reviewed recently (Huang et al., 2020, Li et al., 2020). Elucidation of the full range of their pharmacological activities will not only benefit cardiovascular, diabetic, and cancer research, but may also aid in the development of new antiviral drugs.
| Pharmacological agent/class | Experimental model /Tissue/ Subject | Effect | Reference |
|---|---|---|---|
| Curcumin | Male Sprague Dawley rats/ Angiotensin II infusion | Increased myocardial ACE2 mRNA and protein expression | Pang et al. (2015) |
| Curcumin analog | Wistar rats/ High-fat-high-sugar- streptozotocin induced diabetes model | Increased renal ACE2 immunostaining and mRNA in diabetic mice | X. Xu, Cai, and Yu (2018) |
| Resveratrol | Male FVB/N mice/ High Fat induced obesity model | Increased adipose tissue ACE2 mRNA expression. | Oliveira Andrade et al. (2014) |
| Resveratrol | apolipoprotein E-deficient C57BL/6 mice and human aortic smooth muscle cells | ACE2 mRNA and protein expression was upregulated by resveratrol in all mice and cell models. | Moran et al. (2017) |
| Resveratrol | Male C57BL/6 mice/ | Increased aortic ACE2, immunostaining, protein and mRNA expression. | E. N. Kim et al. (2018) |
| Resveratrol | Sprague–Dawley rats/ High fat induced liver disease model | Increased liver ACE2 mRNA and protein expression | Tiao et al. (2018) |
| Quercetin | Recombinant Human ACE2 activity assay | Inhibition of ACE2 activity | X. Liu et al. (2020) |
| β-casomorphin-7/ Opioid-like peptide | Male Sprague-Dawley rats/ Streptozotocin induced diabetes model | Increased renal ACE2 mRNA expression in ACE2 downregulated diabetic rats, | W. Zhang, Miao, Wang, and Zhang (2013) |
| Geranium essential oil | HT-29 cells | Inhibition of ACE2 activity and protein and mRNA expression | Senthil Kumar et al. (2020) |
| Lemon essential oil | HT-29 cells | Inhibition of ACE2 activity and protein and mRNA expression | Senthil Kumar et al. (2020) |
| Caerulein/ Cholecystokinin analog | Male C57BL/6 mice/ Caerulein and lipopolysaccharide induced pancreatitis model | Marked increase in pancreatic ACE2 mRNA, protein expression and immunostaining | Y. Wang, J. Wang, et al. (2012) |
| Cerulein/ Cholecystokinin analog | Male C57BL/6, ACE2 knockout and ACE2 transgenic mice/ Caerulein and lipopolysaccharide induced pancreatitis model | Marked increase in pancreatic ACE2 mRNA and protein expression and immunostaining | R. Liu et al. (2014a) |
| Cerulein/ Cholecystokinin analog | Rat pancreatic acinar AR42J cells | Increase in ACE2 protein expression up to 6 hours, Decrease ACE2 at later time points | J. Wang et al. (2015) |
| Caerulein/ Cholecystokinin analog | Male C57BL/6 mice/ Caerulein induced pancreatitis model | No change in pancreatic and pulmonary ACE2 protein level but increased ACE2 activity in pancreatic tissue | Gaddam, Ang, Badiei, Chambers, & Bhatia (2015) |
| Taurine/ Naturally occurring organic compound and food additive | Male Wistar rats/ Stress induced hypertension model | Increased adrenal gland ACE2 mRNA and protein expression. No effect on hypothalamus and pituitary | Lv et al. (2015) |
| Esculetin/ Naturally occurring coumarin derivative | Male Wistar rats/ High fat and streptozotocin induced diabetes model | Increased vascular ACE2 immunostaining | Kadakol et al. (2015) |
| Osthole (natural coumarine derivative) | Male BALB/c mice/ lipopolysaccharide-induced acute lung injury | Increased pulmonary ACE2 immunostaining, mRNA and protein expression in lung injury group. | Shi et al. (2013) |
| Osthole (natural coumarine derivative) | Male Sprague-Dawley rats/ Bleomycin induced pulmonary fibrosis model | Increased pulmonary ACE2 immunostaining, mRNA and protein expression in fibrotic lung group | Y. Hao & Liu et al. (2016) |
| Sini decoction/ Traditional Chinese medicine | Male ICR mice / E. coli induced lung injury model | Increased lung ACE2 protein expression | J. Liu et al. (2018) |
| Sini decoction/ Traditional Chinese medicine | Male ICR mice / Lipopolysaccharide induced lung injury | Increased lung ACE2 immunostaining and protein expression in injured mice | Q. Chen et al. (2019b) |
| Eucommia ulmoides Oliv/ Traditional Chinese medicine | Male spontaneously hypertensive and Sprague Dawley rats | Increased renal ACE2 mRNA and protein expression in hypertensive rats | Z. J. Ding et al. (2020) |
| Qishenyiqi / Traditional Chinese medicine | Male Sprague-Dawley rats/ Coronary artery ligation induced disease model | Increased cardiac ACE2 mRNA expression | Y. Wang, C. Li, et al. (2012) |
| Puerarin (a natural hypertensive compound) | Male Sprague Dawley/ Goldblatt hypertensive rat model | No change in renal ACE2 mRNA expression in non-ischemic kidneys | S. Bai et al. (2013) |
| Baicalin (flavonoid) | Human umbilical vein endothelial cells/ Ang II treated cells | Increased ACE2 mRNA and protein expression. | X. Wei et al. (2015) |
| LRW (Pea derived natural peptide) | A7r5 cell line | Upregulated ACE2 protein expression | X. Wang et al. (2020) |
| IRW (Egg-white derived natural peptide) | Spontaneously hypertensive rats | Upregulation of mesenteric ACE2 gene expression | Majumder et al. (2015) |
| IRW (Egg-white derived natural peptide) | Spontaneously hypertensive rats and A7r5 cell line | Activates human ACE2 in vitro. Increases ACE2 activity, mRNA expression in A7r5 cells, and in kidney and aorta of hypertensive rats. | W. Liao, Bhullar, Chakrabarti, Davidge, & Wu (2018) |
| IRW (Egg-white derived natural peptide) | Male spontaneously hypertensive rats | Increased plasma ACE2 concentration and aortic ACE2 protein expression | W. Liao, Fan, Davidge, & Wu (2019) |
| Tanshinone IIA (Chinese herbal medicine) | Male Sprague-Dawley rats/ Bleomycin induced pulmonary fibrosis model | Increased pulmonary ACE2 immunostaining, mRNA and protein expression in rats with fibrotic lungs | H. Wu et al. (2014) |
| Tanshinone IIA (Chinese herbal medicine) | Male Sprague-Dawley rats/ Paraquat -induced lung injury model | Increased pulmonary ACE2 immunostaining, mRNA and protein expression in rats with lung injury | Y. Wang et al. (2018) |
| Naringenin (flavonoid) | Male Sprague Dawley rats/ 2-kidney, 1-clip hypertension model | Increased renal ACE2 immunostaining and mRNA expression | Z. Wang et al. (2019) |
| Ulmus wallichiana (multiple flavonoids) | Male Wistar rats/ Isoprenaline induced cardiac hypertrophy model | Increased cardiac ACE2 mRNA expression | Syed et al. (2016) |
| Rosmarinic acid (active ingredient of rosemary) | Male Sprague Dawley rats / Coronary artery ligation induced myocardial injury model | Increased cardiac ACE2 protein expression | Q. Liu et al. (2016) |
| Tsantan Sumtang (Tibetan medicine) | Male Sprague Dawley rats / Hypoxia-induced pulmonary hypertension model | Increased cardiac ACE2 immunostaining, mRNA and protein expression | Dang et al. (2020) |
| Ficus deltoidei (Herbal medicine) | Male spontaneously hypertensive rats | Threefold increase in serum ACE2 concentration | Azis et al. (2019) |
| Ginsenoside Rg3 | Male spontaneously hypertensive rats and C57BL/6 mice | Increased renal ACE2 immunostaining and mRNA expression in hypertensive groups | H. Liu et al. (2019) |
| Fugan Wan/ (Chinese herbal compound) | Waster rats/ Dimethyl nitrosamine induced hepatic fibrosis model | Increased liver ACE2 mRNA expression | S. Li, Zhao, Tao, and Liu (2020) |
| Red Liriope platyphylla extracts/ (Chinese herbal compound) | Spontaneously hypertensive and Wistar Kyoto rats/ Hypertension model | Increase in aortic ACE2 protein expression | Y. J. Lee et al. (2015) |
| Tempol/ Superoxide dismutase mimetic, antioxidant dietary supplement | Male Zucker rats/ Comparison of lean and obese rats | Increased renal ACE2 mRNA and protein expression in obese rats. No effect in lean rats | Luo et al. (2015) |
| Tempol/ Superoxide dismutase mimetic, antioxidant dietary supplement | Sprague-Dawley rats/ High salt induced hypertension model | Marked increase of renal ACE2 immunostaining and | G. Cao et al. (2017) |
| Ethanol | Wistar rats/ Maternal ethanol induced kidney injury and primary metanephric mesenchyme cells | Decreased renal ACE2 mRNA expression in offspring and in cell cultures | Zhu et al. (2018) |
| Perchlorate/ Environmental contaminant | Human chroriocarcinomic trophoblastic cell line BeWo | Increased ACE mRNA and protein expression | la Pena et al. (2018) |
Finally, the relationship between oxidative stress and ACE2 activity needs to be adressed briefly, since several naturally occurring compounds are known to have antioxidant properties, and ACE2 activity alleviates oxidative stress. Ang-II, as a known promoter of oxidative stress, increases the production of reactive oxygen species and superoxide levels; it also upregulates the expression and activity of enzymes involved in oxidative stress. ACE2, on the other hand, decreases oxidative stress by degrading Ang-II, and producing Ang-(1-7), which promotes antioxidant effects. Overexpression of catalase, a key antioxidant enzyme, decreases oxidative stress, hypertension, and renal fibrosis, normalizes renal functions, and upregulates renal ACE2 protein expression in diabetic Akita mice (Shi et al., 2013). Overexpression or pharmacological activation of ACE2 reduces oxidative stress, improves endothelial and vascular functions, increases Ang-(1-7) production, and promotes NOS phosphorylation (Y. Zhang et al., 2015). In another study, Ang II-induced hypertension, renal oxidative stress, and tubule-interstitial fibrosis were significantly reduced by treatment with recombinant human ACE2 (rhACE2) and, conversely, exacerbated in ACE2 knock out mice (J. Zhong et al., 2011). Similarly, in human smooth muscle cells, Ang-II induced superoxide generation and pro-inflammatory cytokine production. Activation of the JAK2–STAT3 and ERK1/2 signaling pathway was markedly reduced by rhACE2; conversely, they were aggravated by the ACE2 inhibitor DX600 (B. Song et al., 2013). Tempol, a superoxide dismutase mimetic, antioxidant and nutritional supplement, decreases high blood pressure in obese Zucker rats. In this animal model, it also reduces renal oxidative stress, improves renal functions and markedly upregulates renal ACE2 mRNA and protein expression (Luo et al., 2015). Tempol and another antioxidant, α-lipoic acid, reverse Ang-II- or deoxycorticosterone-induced down-regulation of ACE2 expression and upregulation of ADAM17 (de Queiroz, Xia, Filipeanu, Braga, & Lazartigues, 2015). Interestingly, α-lipoic acid treatment markedly downregulated the overexpression and the activity of ADAM17 in Neuro2A cells (de Queiroz, Xia, Filipeanu, Braga, & Lazartigues, 2015). Tempol upregulates renal ACE2 protein expression in both high salt intake and control rats (G. Cao et al., 2017). In cerebral arteries, tempol reduces vascular dysfunctions linked to increased oxidative stress promoted by genetic deficiency of ACE2 and aging (Peña Silva et al., 2012). Of note, tempol also decreases viral load, ameliorates encephalomyelitis and lessens the inflammation induced by CoV- MHV-59A (Tsuhako et al., 2010). Potential action mechanisms and the role of antioxidants in SARS-CoV-2 infection have been reviewed recently (Suhail et al., 2020).
In summary, ACE2 plays an important regulatory role in counteracting the deleterious effects of the Ang-II/ACE/AT1 receptor axis on cardiovascular and metabolic events. Thus, not surprisingly, a vast number of drugs, including some vasodilators, diuretics, steroids, NSAIDs, and some of the antidiabetic and cholesterol-lowering drugs, significantly affect the activity and expression of ACE2. In addition, some vitamins, phytochemicals, and various naturally occurring organic compounds employ the ACE2/Ang-(1-7)/Mas receptor axis to exert their beneficial effects. Considering the crucial role of ACE2 in coronavirus infections, potentially deleterious consequences of ACE2 upregulation by RAS inhibitors have been the subject of much recent debate, and, so far, evidence from clinical and epidemiological studies indicates that these drugs do not negatively affect the susceptibility and prognosis of COVID-19. It remains to be seen whether other drugs and pharmacologically active substances affect coronavirus susceptibility or disease prognosis.
Apparently, while some of the in vitro and in vivo experiments indicate no alterations, the majority of preclinical studies report a significant upregulation of ACE2 by RAS inhibitors in various tissues and organ preparations. Thus, there seem to be significant discrepancies between the results of these preclinical experiments and clinical studies. The following points can be considered to clarify some of these discrepancies. Peak plasma concentrations of ARBs at the daily adult therapeutic dose range are approximately 0.6 μM for losartan and 4.6 μM for valsartan. These values for ACE-Inhs., such as captopril, ramipiril, and lisinopril, are 3.4 μM, 0.1 μM, and 0.1 μM, respectively (Sriram & Insel, 2020), with considerably lower plasma concentrations attained during steady-state levels of maintenance therapies. In addition, typical treatment times for ACE-Inhs. and ARBs in animal studies are considerably shorter than clinical treatment durations for these drugs. Moreover, in the majority of these preclinical studies, concentrations were significantly higher than clinically advised. Thus, the data obtained from high-dose acute phase experiments may not be applicable to lower dose chronic phase experiments where receptor-dependent cellular adaptations can take place. Thus, experimental conditions in which mostly high drug concentrations are used for relatively short durations may not be comparable to the clinical use of these drugs, where lower maintenance concentrations are administered over several months or years. Numerous adaptation mechanisms, at the cellular, organ and organ-system levels, can develop, especially during long administration periods.
Importantly, in the majority of preclinical experiments, the regulatory actions of these drugs are described under disease situations. Thus, the reported effects on the expression or activity of ACE2 in animal studies could merely represent a normalization of ACE2 levels, rather than de novo expression of ACE2 in response to RAS inhibitors. Finally, often tissue-, species-, and gender–dependent effects of drugs can contribute to the observed pharmacological response. Therefore, vis-à-vis direct interpolation of preclinical data to clinical conditions may not be applicable for a given situation. For example, while rodents and humans share more than 80% sequence identity in their ACE2 protein, SARS-CoV cannot replicate through ACE2 in rodents (Gembardt et al., 2005; W. Li et al., 2004; McCray et al., 2007). Similarly, compounds efficiently activating ACE2 in humans can be ineffective in rodents or vice versa (Joshi, Balasubramanian, Vasam, & Jarajapu, 2016; Pedersen, Sriramula, Chhabra, Xia, & Lazartigues, 2011; Ye et al., 2012). Expression and activity of ACE2 shows significant variations among different cell types and organs. Expression of ACE2 in intestines and kidneys is more than two orders of magnitude higher than in lungs (Y. Chen, Guo, Pan, & Zhao, 2020; Hamming et al., 2004; Yiliang Wang et al., 2020). In fact, the activity of ACE2 in lungs is considerably low, and most of Ang-II conversion to Ang-(1-7) in pulmonary tissue is carried out through peptidases other than ACE2, whereas Ang-(1-7) formation in the kidney is mainly ACE2-dependent (Serfozo et al., 2020). It is unclear whether modest changes in ACE2 expression in lungs (< 2-fold, from most preclinical data) can impact the high infectivity of SARS-CoV-2 in host tissues (Sriram & Insel, 2020).
In addition, gender-dependent expression of the ACE2 gene, which is located on the X-chromosome, is suggested to impact the pathogenesis of diseases, such as hypertension (Ji et al., 2020) and COVID-19 (Klein et al., 2020). Moreover, the shedding process of ACE2 adds a further layer of complexity to the interpretation of ACE2 levels in biological fluids. Increased ACE2 levels in urine, cerebrospinal fluid, and plasma may in fact correspond to a decreased ACE2 activity at the tissue level (Anguiano, Riera, Pascual, & Soler, 2017; A. Gilbert et al., 2019; Palau, Pascual, Soler, & Riera, 2019; Úri et al., 2014). Nevertheless, both in vitro and in vivo experiments provide crucial information on the action mechanisms of drugs and guidance for further clinical studies. The knowledge of the pharmacological regulation of ACE2 by various drugs and compounds greatly helps to better understand how these molecules work at cellular and organ system levels.
As mentioned earlier, interruption of SARS-CoV-2 spike protein binding to ACE2 is a feasible treatment strategy against COVID-19. For this purpose, the application of soluble ACE2 fragments, ACE2 antibodies, recombinant ACE2, and Ang- (1-7) peptides have been proposed as alternative therapeutic approaches. Of note, pharmacological inhibition of ACE2 by MLN-4760 does not affect the interaction of SARS-CoV spike protein with ACE2 nor does it affect the SARS-CoV infection of HEK-293T cells expressing human ACE2 (W. Li et al., 2005). Furthermore, the SARS-CoV spike protein does not alter ACE2 activity (W. Li et al., 2005). Thus, not surprisingly, while some compounds that block spike protein binding to ACE2 do not inhibit the catalytic activity of ACE2 peptidase, some other compounds with strong inhibitory effects on the enzymatic activity of ACE2 have no antiviral actions (X. Chen et al., 2020). However, another ACE2 inhibitor, N-(2-aminoethyl)-1 aziridine-ethanamine, was found to be effective in blocking the SARS-CoV spike protein-mediated cell fusion (Huentelman et al., 2004), indicating an allosteric communication between the active site of ACE2 and its site of interaction with SARS-CoV spike protein. Furthermore, recently, binding of SARS-CoV-2 trimeric spike protein was shown to increase the proteolytic activity of ACE2 (J. Lu & Sun, 2020). Thus, it is conceivable that new drugs may be developed that bind to the active site of ACE2 and disrupt the interaction with the SARS-CoV-2 spike protein.
The authors declare that there are no conflicts of interest. I declare the above statement as corresponding author and on behalf of other authors.
 A comprehensive guide to the pharmacologic regulation of angiotensin converting enzyme 2 (ACE2), the SARS-CoV-2 entry receptor
A comprehensive guide to the pharmacologic regulation of angiotensin converting enzyme 2 (ACE2), the SARS-CoV-2 entry receptor
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
