
 ,
Simon P. Godehard,
Gottfried J. Palm,
Leona Berndt,
Christoffel P. S. Badenhorst,
Ann‐Kristin Becker,
Michael Lammers,
Uwe T. Bornscheuer
,
Simon P. Godehard,
Gottfried J. Palm,
Leona Berndt,
Christoffel P. S. Badenhorst,
Ann‐Kristin Becker,
Michael Lammers,
Uwe T. Bornscheuer
Promiscuous acyltransferase activity is the ability of certain hydrolases to preferentially catalyze acyl transfer over hydrolysis, even in bulk water. However, poor enantioselectivity, low transfer efficiency, significant product hydrolysis, and limited substrate scope represent considerable drawbacks for their application. By activity‐based screening of several hydrolases, we identified the family VIII carboxylesterase, EstCE1, as an unprecedentedly efficient acyltransferase. EstCE1 catalyzes the irreversible amidation and carbamoylation of amines in water, which enabled the synthesis of the drug moclobemide from methyl 4‐chlorobenzoate and 4‐(2‐aminoethyl)morpholine (ca. 20 % conversion). We solved the crystal structure of EstCE1 and detailed structure–function analysis revealed a three‐amino acid motif important for promiscuous acyltransferase activity. Introducing this motif into an esterase without acetyltransferase activity transformed a “hydrolase” into an “acyltransferase”.
Promiscuous acyltransferase activity is widespread in family VIII carboxylesterases. A detailed structure–function analysis improved understanding of this remarkable phenomenon and enabled us to rationally transform a hydrolase into an acyltransferase by introducing a single mutation.
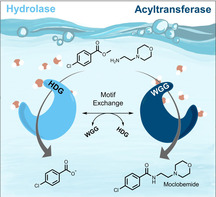
Enzymes are generally considered to be highly specific. However, exposure to non‐natural substrates can sometimes result in a reaction as well. “Substrate promiscuity” occurs if the chemical transformation complies with the enzyme's typical catalytic function. More interestingly, enzymes may also catalyze distinctly different chemical transformations. [1] This phenomenon is referred to as “catalytic promiscuity” and, along with gene duplication and divergence, marks the starting point for the evolution of new enzymes. [2] Combined with protein engineering, promiscuous enzymes represent attractive alternatives to conventional chemical catalysts. [3] Promiscuous acyltransferases for catalyzing bond‐forming reactions in aqueous solutions are of increasing interest as they represent an attractive alternative to conventional biocatalytic or chemical routes by making the use of expensive and less sustainable organic solvents dispensable. Moreover, they can be used in cascade reactions with other enzymes that are not active or stable in the presence of organic solvents. [4] Since being reported in 2007, the promiscuous acyltransferase MsAcT from Mycobacterium smegmatis had been unrivalled in terms of transfer efficiency.[ 5 , 6 , 7 ] However, we recently demonstrated that many esterases from the bacterial hormone‐sensitive lipase (bHSL) family have promiscuous acyltransferase activity, some comparable to that of MsAcT, suggesting that this phenomenon might be more widespread than previously thought. [8] However, enzymatic product hydrolysis and low transfer efficiency has always been a major drawback for application on industrial scale. In this study, we report the discovery that exceptional promiscuous acyltransferase activity is prominent in family VIII carboxylesterases.
We previously showed that the acyltransferase‐catalyzed formation of oligocarbonates from dimethyl carbonate and 1,6‐hexanediol opacifies an initially transparent, aqueous solution. [9] However, not all promiscuous acyltransferases are expected to produce oligocarbonates from these substrates. In this study, we used a general emulsion‐based assay, employing 2‐phenylethanol and vinyl acetate as substrates, to screen the hydrolases available in our laboratory. Acyltransferase activity is indicated by the occurrence of turbidity, which results from the low water solubility of the reaction product, 2‐phenylethyl acetate. [10] This approach led to the discovery that EstCE1, [11] a family VIII carboxylesterase, has promiscuous acyltransferase activity with a preference for aromatic acyl‐acceptor substrates (Figure S1).
We found that EstCE1 can catalyze not only the formation of esters, but also the formation of amides, carbonates, and carbamates in water (Figure 1). The ester formation from the reaction of benzyl alcohol with a fourfold excess of vinyl acetate is very fast, reaching >90 % conversion within seconds. However, the benzyl acetate formed is hydrolyzed again within two hours. Benzyl methyl carbonate formation is slower, even at higher excess of the acyl donor. However, the conversion is still at around 50 % after three hours. Furthermore, EstCE1 can catalyze the formation of N‐benzyl acetamide and methyl N‐benzylcarbamate. Almost full conversion is rapidly reached, and the reactions appear to be irreversible. Even after 24 hours, no product hydrolysis was detectable. Carbamates are essential intermediates in the synthesis of pharmaceuticals and are commonly used as protective groups. [12] The phosgene‐independent carbamoylation of amines presented here provides a much more environmentally friendly and more selective alternative to conventional routes. It is noteworthy that EstCE1′s synthetic potential goes beyond the structurally rather simple model compounds shown in Figure 1. As proof of concept, we used EstCE1 to synthesize the antidepressant moclobemide from methyl 4‐chlorobenzoate (200 mm) as acyl donor and 4‐(2‐aminoethyl)morpholine (50 mm) as acyl acceptor (Figure S4, ca. 20 % conversion).

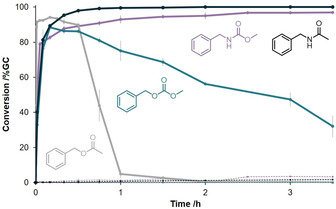
EstCE1‐catalyzed synthesis of benzyl acetate (gray), benzyl methyl carbonate (cyan blue), methyl N‐benzylcarbamate (violet), and N‐benzylacetamide (black) in aqueous buffer. The acceptor concentration was 50 mm for all reactions. For the ester formation, a fourfold excess of vinyl acetate was used. For amide synthesis, a tenfold excess of ethyl acetate was used. Carbonate and carbamate formation were performed using a tenfold excess of dimethyl carbonate. Dotted lines indicate the formation of the products in the absence of EstCE1.
Moreover, we used EstCE1 for the acetylation of several “difficult‐to‐resolve” secondary alcohols (Table S2). [13] EstCE1 exhibits enantioselectivity and, most strikingly, appears to be perfectly selective in the acetylation of (+)‐menthol. This finding is in good agreement with EstCE1′s high selectivity in the hydrolysis of (+)‐menthyl acetate. [11]
Interestingly, the hydrolysis of pNP‐esters by many family VIII carboxylesterases appears to be stimulated by methanol. [14] We suspect that these enzymes actually catalyze the acylation of methanol under the described conditions. The acyl‐enzyme intermediate of a promiscuous acyltransferase would be degraded more rapidly in the presence of an organic nucleophile like methanol, resulting in more rapid release of p‐nitrophenol and higher apparent hydrolysis activities. This is the principle behind our recently published pNP‐AcT assay. [8] For EstCE1, we could demonstrate an eightfold increase in apparent pNPA hydrolysis at only 6.3 % (v/v) methanol (Figure S1). To confirm that the accelerated release of pNP is a result of transesterification to methanol, we confirmed the EstCE1‐catalyzed formation of methyl butyrate from pNP‐butyrate and methanol by gas chromatography–mass spectrometry (GC–MS, Figure S5). This suggests that many of the methanol‐stimulated hydrolases reported in literature may actually be promiscuous acyltransferases, suggesting an interesting avenue for future research.
To investigate whether promiscuous acyltransferase activity is as common in family VIII carboxylesterases as we suspected, we expressed, purified, and assayed several EstCE1 homologs. This demonstrated that promiscuous acyltransferase activity is indeed widespread in this enzyme family (Figure 2). With two exceptions, the family VIII carboxylesterases showed significantly higher relative activities in the presence of benzyl alcohol than MsAcT and the previously characterized bHSLs.[ 6 , 8 ] EstCE1 catalyzes acetyl transfer to benzyl alcohol 66 times faster than hydrolysis of the acyl donor, thereby outperforming even one of the most efficient MsAcT variants (K97A; 50‐fold faster) recently reported by our group. [6] Strikingly, EstM2 [15] catalyzes the acetylation of benzyl alcohol 149 times faster than donor hydrolysis, making it roughly 20 times more efficient than wild‐type MsAcT. Only EstA (PDB 3ZYT) [16] did not show increased relative activity in the presence of benzyl alcohol, suggesting that it is not capable of catalyzing the acetylation of benzyl alcohol. Except for 3ZYT, none of the analyzed enzymes suffered from significant substrate inhibition or stability issues, even at 150 mm benzyl alcohol.
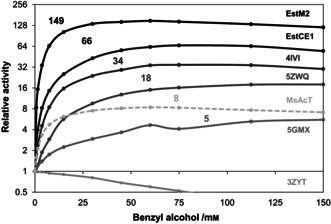
Relative acyltransferase activities of EstCE1 homologs, plotted on a logarithmic scale, as a function of benzyl alcohol concentration. Relative activities were determined in triplicates using the pNP‐AcT assay and the highest value for each enzyme is noted. In these reactions, pNPA is the acyl donor and benzyl alcohol is the acyl acceptor.
It is well known that the conformation of the acyl group in the acyl‐enzyme intermediate has a high impact on the efficiency of the deacylation step.[ 17 , 18 ] Therefore, the acyl donor scope of the family VIII carboxylesterases was investigated, using the pNP‐AcT assay and pNP‐esters of different chain lengths as acyl donors (Table 1 and Table S3). Almost all the enzymes examined turned out to be valuable for the acylation of benzyl alcohol with chain lengths ranging from C2 to C8. In general, the highest activities and AT/H ratios were measured for chain lengths from C2 to C6. EstCE1 and EstM2 have remarkably high acyltransferase activity in the kU/(mg enzyme) range (Table 1). EstSRT1 (PDB 5GMX) [19] and EstU1 (PDB 4IVI)[ 17 , 18 ] turned out to be superior in catalyzing transfer of a C8 chain, with AT/H ratios (4.6 and 5.6) in the range of the ratio of MsAcT for acetylation (Table S3). Extensive protein engineering was necessary for MsAcT to accept C8 chains, with even the best variant (F154A/I194V) having an AT/H ratio below 3. [6] While 3ZYT shows no transacetylase activity, moderate acyltransferase activity emerges in reactions with donors of longer chain lengths (Table S3). This may be explained by the hydrophobic aliphatic chains excluding water from the binding pocket, preventing hydrolysis and favoring binding of organic acceptors. [20]

|
Enzyme |
Donor |
ATmax [U mg−1] |
Hydrolysis [U mg−1] |
AT/H |
|---|---|---|---|---|
|
EstCE1 |
C2 C4 C6 C8 |
2605±86 1081±28 26.1±0.9 0.29±0.05 |
40.1±2.2 38.7±3.3 0.22±0.03 0.13±0.01 |
65 28 119 2.2 |
|
|
|
|
|
|
|
EstM2 |
C2 C4 C6 C8 |
5583±221 361±33 181.2±17.3 0.013±0.01 |
37.7±1.2 54.2±5.5 23.7±2.3 0.02±0.01 |
148 6.7 7.6 0.7 |
We solved the crystal structure of EstCE1 (PDB 7ATL), the first discovered and so far most versatile acyltransferase from this enzyme family, in order to gain a more in‐depth insight into structural features contributing to high acyltransferase activity. Typical for family VIII carboxylesterases, EstCE1 adopts a β‐lactamase fold (Figure 3 A) with the catalytic triad (S65, K68, and Y171) and the active site placed at the interface of an α/β‐subdomain and a structurally more flexible helical subdomain. As observed in several family VIII carboxylesterases, EstCE1′s active site pocket is covered by a so‐called Ω‐loop. The substrate‐binding cavity (Figure 3 A, gray transparent volume) can be divided into the R1 and R2 subsites. [18] In contrast to many structural homologs, the Ω‐loop in EstCE1 is significantly larger and therefore blocks the central part of the R1 subsite. Analysis of the ligand‐binding site revealed the presence of a hydrophobic cavity (Figure 3 B). Induced‐fit docking of benzyl alcohol into the modeled acetyl‐enzyme intermediate suggests a unique role of F243, which is part of the extended Ω‐loop in EstCE1 (Figure 3 B). Via π–π‐stacking, F243 helps to position the benzyl alcohol in a productive pose near the carbonyl of the acyl‐enzyme intermediate. Structural alignment revealed that there is no equivalent residue in Est‐Y29 (PDB 5ZWQ), [21] 4IVI, 5GMX, or 3ZYT. Homology modeling suggests that F244 of EstM2 corresponds to F243 of EstCE1 (Figure S9). This may explain why EstCE1 and EstM2 show significantly higher activity in the acetylation of benzyl alcohol compared to their homologs. Moreover, we used covalent docking to shed light on the donor chain length preference of EstCE1. In good agreement with the experimental data shown in Table 1, the modeling suggests that linear aliphatic chains with more than four carbons are likely to clash with the benzyl alcohol binding site (Figure 3 B).
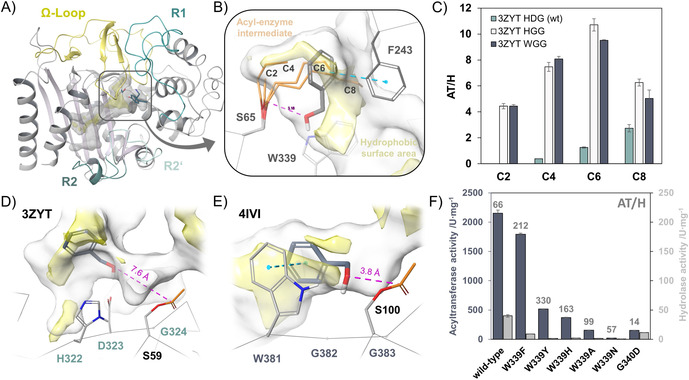
A) Ribbon diagram of EstCE1 with the Ω‐loop and the R1, R2, and R2′ segments highlighted. Hydrophobic volumes within the ligand‐binding site are shown in yellow with transparent surfaces. B) Active site of EstCE1. The acyl‐enzyme intermediates formed in the reactions summarized in Table 1 were modeled by covalent docking. The acyl acceptor, benzyl alcohol, was modeled into the structure using induced‐fit docking. Via π–π‐stacking with F243 in the Ω‐loop, benzyl alcohol is placed in a hydrophobic cavity near the nucleophilic center. The acyl‐enzyme intermediates of C6 and C8 are shown to clash with the putative benzyl alcohol binding site. C) Mutation of D323 to glycine heavily decreases the polarity of the substrate‐binding pocket (Figure S11) and introduces transacetylation activity into 3ZYT. D) and E) The acetyl‐enzyme intermediates of 3ZYT and 4IVI were modeled via covalent docking, and benzyl alcohol was placed in the acceptor‐binding site by rigid receptor docking. Hydrophobic regions are illustrated by yellow, transparent volumes. In the family VIII.1/2 esterase 4IVI, W381 of the WGG‐motif significantly contributes to the proper positioning of the acyl acceptor in a 3.8 Å distance to the acyl‐enzyme intermediate. In contrast, D323 in the HDG motif in 3ZYT leads to active repulsion of the acyl acceptor now placed more than 7 Å away from the acyl‐enzyme intermediate. F) Acyltransferase activity of several EstCE1 variants determined using the pNPA‐AcT assay, with benzyl alcohol as acyl acceptor. Specific acyltransferase activity refers to the maximum activity measured in a range of benzyl alcohol concentrations from 0 to 150 mm. The maximum AT/H ratios are shown for each variant.
Based on sequence alignments, the division of family VIII carboxylesterases into three subclasses was previously suggested. [15] EstCE1, EstM2, 5ZWQ, 5GMX, and 4IVI belong either to subclass VIII.1 or VIII.2, both sharing a conserved WGG motif (WSG for 5ZWQ) in the region where the so‐called KTG‐box exists in class C β‐lactamases (Figure S6). [22] Due to its proximity to the catalytic triad, this motif appears to largely influence the physical properties of the ligand‐binding site. Most interestingly, tryptophan in the first position (W339 in EstCE1) largely contributes to the formation of the hydrophobic cavity in family VIII.1 and VIII.2 carboxylesterases and thereby facilitates the binding and positioning of organic nucleophiles, as shown for EstCE1 and 4IVI (Figure 3 B,E). In 3ZYT, a member of the subclass VIII.3 (H‐x‐x motif), HDG is found in place of the WGG of EstCE1, causing a dramatic decrease in active site hydrophobicity (Figure 3 D). This may explain why 3ZYT is the only one of the investigated family VIII carboxylesterases that does not exhibit any transacetylase activity. Therefore, we had a closer look at the role of this motif in promiscuous acyltransferase activity. By mutation of the HDG in 3ZYT to HGG or WGG (Figure 3 C), we rationally transformed a “hydrolase” into an “acyltransferase” comparable to MsAcT, the current benchmark for promiscuous acyltransferase activity.[ 5 , 6 ]
To further improve our understanding of this motif on both hydrolase and acyltransferase activities, we constructed several EstCE1 variants which were investigated using the pNPA‐AcT assay (Figure 3 F). Strikingly, the W339F variant shows significantly reduced hydrolase activity while the acyltransferase activity remains comparable to that of wild‐type EstCE1. The AT/H ratio exceeds 200, clearly putting this variant above EstM2. However, the highest transfer efficiency was measured for the W339Y variant, reaching a remarkable AT/H ratio of 330. This variant is a roughly 50‐fold more efficient acyltransferase than MsAcT and acyl donor hydrolysis can almost be considered to be a negligible side reaction. Analogous to the HDG motif of 3ZYT, we introduced an aspartate residue into the second position of the motif in EstCE1 (giving WDG). As expected, this led to a drastically decreased AT/H ratio. These data underline the significance of this motif for promiscuous acyltransferase activity and acyl acceptor specificity in family VIII carboxylesterases.
The purpose of the current study was to identify novel biocatalysts for overcoming the synthetic limitations of available acyltransferases and to improve our understanding of promiscuous acyltransferase activity. We demonstrated that many family VIII carboxylesterases exceed the limits of what was previously thought possible by showing unprecedentedly high efficiency in the formation of esters, carbonates, carbamates, and amides like the antidepressant drug moclobemide. The identified three‐amino acid motif adjacent to the catalytic triad could be considered as an activity and specificity switch in this enzyme family. Most remarkably, it took only one rationally introduced mutation to transform 3ZYT, an esterase without promiscuous acetyltransferase activity, into an acetyltransferase. To the best of our knowledge, such a feat is unprecedented in the literature.
The authors declare no conflict of interest.
This work was funded by the Deutsche Forschungsgemeinschaft (German Research Foundation; 231396381/GRK1947) and the Bundesministerium für Bildung und Forschung (Grant No. 031B0354B). We thank the B.R.A.I.N. AG (Zwingenberg, Germany) for providing the expression plasmid for EstCE1. We also thank Prof. Wolfgang Streit (Univ. Hamburg) and Dr. Klaus Liebeton (B.R.A.I.N. AG) for valuable discussions. We thank Schrödinger Maestro for providing a trial license of their software. We acknowledge access to beamline BL14.2 of the BESSY II storage ring (Berlin, Germany) via the Joint Berlin MX‐Laboratory sponsored by the Helmholtz‐Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt‐Universität zu Berlin, the Max‐Delbrück Centrum, and the Leibniz‐Forschungsinstitut für Molekulare Pharmakologie. Open access funding enabled and organized by Projekt DEAL.
1
1b
1b
2
3
3a
3a
3b
4
5
6
7
8
8
9
10
11
12
13
14
14a
14c
14d
15
16
17
18
19
19b
20
20b
21
21a
 Discovery and Design of Family VIII Carboxylesterases as Highly Efficient Acyltransferases
Discovery and Design of Family VIII Carboxylesterases as Highly Efficient Acyltransferases
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
