 Responsive Emulsions for Sequential Multienzyme Cascades
Responsive Emulsions for Sequential Multienzyme Cascades
Angewandte Chemie (International Ed. in English)


These authors contributed equally to this work.

- Altmetric
Multienzyme cascade biocatalysis is an efficient synthetic process, avoiding the isolation/purification of intermediates and shifting the reaction equilibrium to the product side.. However, multienzyme systems are often limited by their incompatibility and cross‐reactivity. Herein, we report a multi‐responsive emulsion to proceed multienzyme reactions sequentially for high reactivity. The emulsion is achieved using a CO2, pH, and thermo‐responsive block copolymer as a stabilizer, allowing the on‐demand control of emulsion morphology and phase composition. Applying this system to a three‐step cascade reaction enables the individual optimal condition for each enzyme, and a high overall conversion (ca. 97 % of the calculated limit) is thereby obtained. Moreover, the multi‐responsiveness of the emulsion allows the facile and separate yielding/recycling of products, polymers and active enzymes. Besides, the system could be scaled up with a good yield.
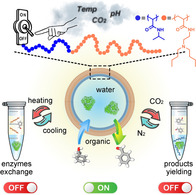
A CO2, pH, and thermo‐responsive emulsion was developed to perform multienzyme cascades without incompatibility and cross‐reactivity issues. This emulsion allows the on‐demand control of morphology and phase composition, and therefore exchanging enzymes and separate yielding/recycling of products and polymers. A three‐enzyme cascade was performed sequentially, each step was carried out under the optimal conditions of the corresponding enzyme.
Enzymes, the biologic catalysts from nature, have achieved enormous success in modern chemical and pharmaceutical industries. [1] In particular, they are increasingly explored for single‐step reactions including oxidation, reduction, and isomerization. [2] However, in nature, cells do not just employ single reactions alone but combine them for essential metabolic pathways, e.g., tricarboxylic acid cycle. [3] This natural design has initiated great efforts in bioinspired multienzyme reactions, e.g., one‐pot cascades, [4] to avoid the isolation of intermediates and overcome unfavorable reaction equilibrium, significantly saving time and production costs. [5]
However, operating multiple enzymes in abiological conditions often suffers from some intractable limitations. The first limitation, namely, incompatibility issue, stems from the fact that different enzymes often have different optimal reaction conditions, e.g., pH, temperature and co‐factors, and therefore cannot be simply combined in one system. [6] The second issue is the cross‐reactivity, which occurs when one enzyme accepts not only its substrates but also the intermediates/products in the whole cascade route, eventually leading to undesired side reactions. [7]
An elegant solution to the above issues is to compartmentalize multienzyme reactions in spatially different domains of one system. [8] Emulsions, the dispersion of one liquid phase within a second immiscible liquid, seem to be a good choice, [9] because they provide not only a large interfacial area for fast reactions but also heterogeneous phases to host different reactions. These benefits, especially their large interface, have promoted rapidly the field of “emulsion biocatalysis” to address a wide range of synthetic challenges to date. [10] For instance, microemulsions, [11] miniemulsions, [12] and Pickering emulsions [13] were employed to encapsulate enzymes for the single‐step enantioselective reduction, hydrolysis, and esterification. Recently, we further advanced the field by developing multi‐compartmentalized emulsion biocatalysis for benzoin condensation and carbonyl reduction. [14] In principle, polymeric emulsions are attractive because polymers are easily controlled for desirable application properties in addition to good recyclability. An example of such controllability is that polymeric emulsions can be made responsive to diverse external stimuli, e.g., pH, [15] temperature, [16] CO2, [17] magnetic field, [18] and light. [19] This stimuli responsiveness allowed the controllable emulsification and demulsification, which were successfully explored for oil recovery [20] and chemocatalysis. [21] However, until now, stimuli‐responsive emulsions for biocatalysis have been relatively underdeveloped,[ 13c , 22 ] and to the best of our knowledge, they have never been explored for multienzyme cascades despite their potential to improve reaction compatibility and reduce cross‐reactivity.
Herein, we present a scalable multi‐responsive emulsion as controllable liquid scaffolds to proceed efficient multienzyme cascades. A pH‐, CO2‐, and thermo‐responsive polymeric emulsion is obtained by utilizing a diblock copolymer, poly(N‐[2‐(dibutylamino)ethyl]acrylamide)‐b‐poly(N‐isopropylacrylamide) (PDBAEAM‐b‐PNIPAm), as the stabilizer. This multi‐responsiveness allows not only switching the biocatalysis on/off by on‐demand emulsification/demulsification, but also the control of phase composition in the demulsified status (Scheme 1). As a result, the exchange of enzymes and a facile yet high conversion of biocatalysis can be achieved by applying different stimuli. Furthermore, utilizing this system in a sequential approach, [23] each biocatalytic step undergoes under its optimal condition, and therefore, the performance of the multienzyme reactions can be improved without incompatibility and cross‐reactivity issues.

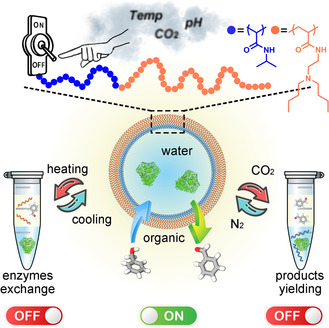
Multi‐responsive emulsion is constructed by a responsive polymer, PDBAEAM‐b‐PNIPAm, for controllable multienzyme cascades: Increasing temperature switches polymers from amphiphilic to hydrophobic, leading to the demulsification with polymers localized in the organic phase, which allows the exchange of enzyme solution for next biocatalysis. On the other hand, demulsification by lower pH or using CO2 results in a hydrophilic polymer and an organic phase without the polymer, which facilitates the product purification.
To design the multi‐responsive block copolymer, we opted to use PNIPAm and PDBAEAM as the temperature‐ and pH/CO2‐responsive segments because of their biocompatibility to enzymes [24] and orthogonal responsiveness in different stimuli state. [25] Moreover, due to their characteristic molecular structures, the hydrophilic PNIPAm and hydrophobic PDBAEAM can be respectively switched to the hydrophobic and hydrophilic segment in response to environmental changes, [26] thus allowing the precise control of the hydrophobicity of the block copolymer for multi‐responsive emulsions. The block copolymer was synthesized by reversible addition‐fragmentation chain‐transfer polymerization (see ESI), and the ratio of PDBAEAM/PNIPAm was controlled as 3:1 to stabilize a water‐in‐oil (W/O) emulsion. [27] As a result, PDBAEAM39‐b‐PNIPAm22 was synthesized.
The W/O emulsion, stabilized by the amphiphilic polymer, can be prepared by the facile mix of water and an organic solvent, such as toluene or dichloromethane (Figure S1). Typically, a 500 μL organic phase containing 40 mg mL−1 PDBAEAM39‐b‐PNIPAm22 was added to a 500 μL water phase, which is followed by handshaking to form a water/oil emulsion with good stability (Figures 1 a,b). The emulsification was tested under different pH conditions (Figure 1 c), and the results suggested that the emulsion could be successfully prepared when the pH value of the aqueous phase was higher than 6. In lower pH conditions, the whole diblock copolymer became hydrophilic and therefore cannot work as an emulsifier. The thermal stability of the emulsion was investigated at the temperature from 20 to 40 °C (Figure 1 d). When the temperature was lower than 25 °C, the emulsion was stable for more than 12 h (Figure S2). At 30 °C, slight phase separation was observed over 12 h. At 35 °C, the emulsion was stable for about 3 hours, while at 40 °C the emulsification was unsuccessful, which was due to the change of PNIPAm block from hydrophilic to hydrophobic over LCST.
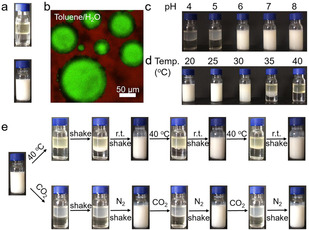
a) An organic phase containing PDBAEAM39‐b‐PNIPAm22 was added to an aqueous phase (upper), and the emulsion was obtained by handshaking (lower). b) Confocal laser scanning microscopy (CLSM) image of the emulsion, aqueous phase was in green and organic phase was in red. c, d) Emulsion's stability at different pH and temperatures. e) Responsiveness of emulsion to temperature change and CO2/N2.
To investigate the responsiveness of the emulsion, temperature change and CO2/N2 treatment were applied, respectively (Figure 1 e). Demulsification could be achieved by two methods via, for example, keeping the emulsion at 40 °C or bubbling CO2 to the emulsion for 10 minutes. Although in both cases demulsification was achieved, the location of the polymer was different, which was confirmed by the yellowish color of the toluene or water phase, respectively. After the demulsification, the system could not be emulsified anymore by shaking, thus suggesting the change of the block copolymer amphiphilicity. However, after cooling down the system to room temperature or bubbling N2, the emulsion could be obtained again by handshaking. The reversibility of emulsification/demulsification was demonstrated by repeating the heating/cooling and CO2/N2 cycles. Besides CO2 and temperature, the emulsion was also responsive to pH changing (Figure S3).
Three different enzymes, Candida antarctica Lipase B (CalB), thermophilic alcohol dehydrogenase (ADH‐ht), [28] and benzaldehyde lyase (BAL), [29] were obtained (see ESI) to study their catalytic performance in a 1 mL emulsion, where 2.5 mg mL−1 enzymes and 200 mM substrates resided in aqueous and organic phases, respectively. Three reactions, hydrolysis of acetic acid benzyl ester (AABE), oxidation of benzyl alcohol (BeOH), and benzoin condensation, were performed individually under their optimal pH conditions in the emulsion (Figures S5–7), using a biphasic system as a control. Product concentration was determined by GC measurements using an external standard method. [30] Figure 2 a showed that our emulsion system largely improved the catalytic efficiency of all reactions compared to the biphasic control due to its large interfacial area. Furthermore, the responsive emulsion contributed to facilitated purification. Most traditional emulsifiers end up in final product, bringing additional difficulty in product purification. [31] Even though some polymeric stabilizers can be precipitated from emulsions, [14a] it is usually at cost of using a large amount of antisolvents. Here, we showed the demulsification by CO2 treatment, the obtained colorless organic phase (Figure S4) contained no polymers and was sent directly for the follow‐up workup. Besides, the facile separation contributed to a higher conversion than the direct precipitation process (Figure 2 a) because precipitation could lead to a small amount of product embedded in the polymer sediments. The demulsification, however, offered clear phase separation and the entire product was in the organic phase and not wasted. Therefore, the responsive emulsion provided an easy, economical (less solvent needed) method with the high bioconversion. The final conversion of the biocatalytic reactions was about 95 % (hydrolysis by CalB), 80 % (oxidation by ADH‐ht), and 40 % (benzoin condensation by BAL), respectively.
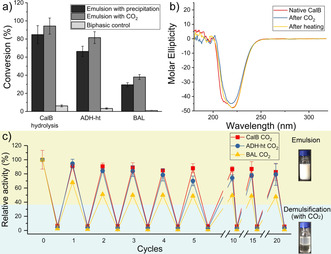
Single enzymatic reactions in the 1 mL multi‐responsive emulsion. a) Detected conversion of hydrolysis of acetic acid benzyl ester (AABE), oxidation of benzyl alcohol (BeOH) and benzoin condensation in the biphasic system, emulsion system and after demulsification by CO2. b) CD spectrum of native CalB, CalB recycled from the emulsion system by temperature and CO2. c) Relative activity of CalB, ADH‐ht, and BAL in emulsification/demulsification cycles by N2 and CO2. In each cycle, the catalytic performance was investigated in both emulsion and demulsified state.
Next, the influence of demulsification on enzymes and their reusability was investigated. Circular dichroism (CD) spectrum (Figure 2 b) showed negligible changes in enzyme secondary structure after demulsification by both CO2 treatment and heating. This positive result encouraged us to perform 20 cycles of emulsification/demulsification, in which the enzyme activity in emulsion and demulsified status was compared. In the case of demulsification by CO2 (Figure 2 c), the activity of both CalB and ADH‐ht had an only slight decrease, while BAL lost 30 % and 50 % activity after the first and second round and finally retained 45 % after 20 cycles. This was because the introduction of CO2 lowered the pH value of the system, which demoted BAL performance. On the other hand, when using temperature change to run the cycles (Figure S8), the activity of CalB and BAL decreased gradually after each circle, and finally reached 70 % and 60 % after 20 cycles, respectively. In comparison, as a thermophilic enzyme, ADH‐ht was almost not influenced in the first 15 cycles and eventually retained more than 80 % activity. These results suggested that the multi‐responsiveness of our emulsion not only allowed for a facile recycling of products but also provided more opportunities for enzyme reuse under their favourable conditions.
The accomplishment of single enzyme reactions encouraged us to further investigate their cascade reaction (Figure 3 a). To this end, initially, we were curious about whether our responsive emulsion would be useful for avoiding multienzyme cross‐reactivity. Interestingly, no cross‐reactivity could be observed in the one‐pot cascade reaction (Figure S15), which may be either due to the substrate specificity of enzymes[ 28a , 32 ] or because of side‐products below our detection limit. However, one big challenge of this cascade was that the optimal condition for every step differed largely from each other (Figures S16–18). For example, CalB and ADH‐ht preferred higher temperatures, while BAL was only stable at the temperature from 20 to 30 °C (Figure S18). Considering that the polymeric emulsion was not stable at high temperature, 30 °C was, therefore, set for the one‐pot cascade reaction in further studies. In addition to the temperature, pH also had a great influence on all enzymatic performance (Figure S19). The three‐step cascade reaction was carried out under both pH 6 and 8 for 24 hours. In the pH 6 experiment (Figure S20), the first step of hydrolysis by CalB was fast and almost fully completed. However, due to the low activity of both ADH‐ht and BAL under an acidic condition, the production of benzaldehyde (25 mM) and benzoin was largely limited (4.6 mM, overall conversion 9 %). In comparison, in the case of one‐pot cascade at pH 8 (Figure 3 b), although the first step was slower, the latter two steps were carried out under suitable pH and were able to shift the reaction equilibrium. Eventually, the overall conversion was higher (5.6 mM, overall conversion 11 %).
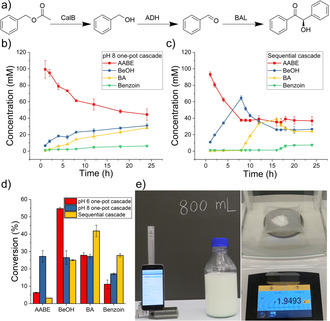
a) Reaction Scheme of the cascade reaction. b) Time course of the concentration of AABE, BeOH, benzaldehyde (BA), and benzoin in a one‐pot cascade at pH 8, 30 °C. c) Time course of the concentration of the four compounds in the sequential cascade. d) Overall conversion of compounds in long‐term pH 6 and pH 8 one‐pot cascade, and sequential cascade. e) 800 mL reaction experiment and yielded product.
To improve the overall catalytic efficiency, the above three‐step reaction was then performed in a sequential fashion and operated separately under the individual optimal pH and temperature conditions of the corresponding enzyme, which was achieved by the stepwise heating to allow on‐demand phase separation for changing enzyme solution from one to another (Scheme 1). Initially, every single biotransformation was carried out for 8 hours. Figure 3 c showed the concentration change of substrates, intermediates, and final products. AABE was hydrolyzed rapidly by CalB, and after 8 hours, about 65 % was converted to BeOH. CalB was then replaced by ADH‐ht for the following oxidation reaction. In the next 8 hours, more than half of BeOH was consumed to produce about 40 mM benzaldehyde. Demulsification and exchange of the enzyme were then performed again to introduce BAL for benzoin condensation. In the last 8 hours, 7.3 mM benzoin was obtained as the final product (single‐step conversion near 35 %). The final conversion in this sequential cascade approach was slightly higher than that of 24‐hour one‐pot cascade experiments, and it was 15 %.
In order to further improve the reaction conversion, we provided enough reaction time to each step of the cascade. Specifically, the first‐step reaction was conducted for 20 hours, while the second and third steps were operated for 24 hours. This prolonged reaction time eventually contributed to 29 % conversion (Figure 3 d), which is ca. 1.5‐fold greater than the best one‐pot cascade reaction (i.e., pH 8 one‐pot). The calculated maximum theoretic overall conversion of this three‐step cascade in a sequential way was 30 % (calculation see ESI), therefore, 29 % was already very close (ca. 97 %) to the limit. This improved biocatalysis was attributed to the controllable emulsions providing each reaction with optimal conditions and facile purification.
Furthermore, an 800 mL reaction experiment was carried out under the same condition as the small‐scale. The large‐scale emulsion remained the thermo‐ and CO2‐responsiveness, and 1.95‐gram pure final product was yielded (Figures 3 e, S24–26). Therefore, the increase of reaction volume from 1 to 800 mL did not change emulsion properties and bioconversion, thus indicating that our system is not only scalable but also independent of the reaction volume.
In summary, we have designed an amphiphilic block copolymer to stabilize a water‐in‐oil emulsion with pH‐, CO2‐, thermo‐responsiveness. The obtained multi‐responsive emulsion allows for not only the encapsulation of three active enzymes but also a facile product purification via stimuli‐responsive phase separation. This finding is significant because it provides a simple solution to the longstanding separation problem in the field of emulsion catalysis. Furthermore, the responsive emulsion allows us to proceed a three‐step enzymatic cascade with each step performed sequentially under an optimal condition, eventually leading to a 29 % final conversion. In addition, our emulsion can be scaled up 800 times for the gram‐scale production. This proof‐of‐principle example, on one hand, suggests the fascinating perspectives of responsive emulsions for cascade reactions by avoiding catalyst incompatibility. On the other hand, its exploration for different cascades, ranging from multienzyme to chemoenzymatic reactions, is envisioned.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We acknowledge financial support from the German Research Foundation and the Independent Research Fund Denmark. We acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. We thank Prof. Marion B. Ansorge‐Schumacher for valuable discussions and support. We thank Prof. Wolfgang Kroutil for providing the plasmid of ADH‐ht. Z.S. thanks the support from China Scholarship Council (CSC). Q.Z. thanks Dahlem Research School for financial support. Open access funding enabled and organized by Projekt DEAL.
References
1
1e
1e
2
3
3b
3d
4
4a
4c
4e
4e
4f
4g
5
6
6b
8
8
9
9a
9a
9b
10
10b
10b
11
12
13
13b
13b
13c
14
14b
15
15a
15b
16
16a
16a
16b
16c
17
17a
17a
17b
18
18a
18b
19
20
21
22
24
24a
24b
25
25b
26
27
28
28b
29
30
30a
30b
30b
30c
30d
30e