
 ,
Henning Klaasen,
Melanie C. Witteler,
Johannes Neugebauer,
Hong‐Ying Gao,
Armido Studer,
Harald Fuchs
,
Henning Klaasen,
Melanie C. Witteler,
Johannes Neugebauer,
Hong‐Ying Gao,
Armido Studer,
Harald Fuchs
Theses authors contributed equally to this work.
The formation of azo compounds via redox cross‐coupling of nitroarenes and arylamines, challenging in solution phase chemistry, is achieved by on‐surface chemistry. Reaction products are analyzed with a cryogenic scanning tunneling microscope (STM) and X‐ray photoelectron spectroscopy (XPS). By using well‐designed precursors containing both an amino and a nitro functionality, azo polymers are prepared on surface via highly efficient nitro‐amino cross‐coupling. Experiments conducted on other substrates and surface orientations reveal that the metal surface has a significant effect on the reaction efficiency. The reaction was further found to proceed from partially oxidized/reduced precursors in dimerization reactions, shedding light on the mechanism that was studied by DFT calculations.
Herein we disclose the on‐surface redox coupling of arylamines and nitroarenes on Ag(111). Starting from well‐designed monomeric precursors, the cross‐coupling yielding azo bridged polyphenylenes was investigated by STM and XPS. Further, first steps toward copolymerization were performed with two comonomers. Mechanistic studies and supportive DFT calculations are provided.
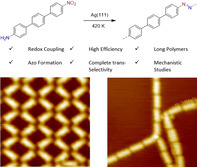
During the last decade, on‐surface chemistry has attracted extensive attention as a bottom‐up strategy for the synthesis of materials with interesting properties and functions. Various organic reactions have been successfully realized on metal surfaces[ 1 , 2 , 3 , 4 ] and most of them aim for carbon−carbon bond formation. Only a few carbon−nitrogen and nitrogen−nitrogen bond forming processes have been reported, such as imine,[ 5 , 6 , 7 ] imide, [8] azo group formation[ 9 , 10 , 11 ] and azide–alkyne cycloaddition.[ 12 , 13 ] Among these N‐based functional groups, the azo group shows valuable properties in photo switching.[ 14 , 15 , 16 ] In solution phase chemistry, azo compounds are obtained through an azo coupling using diazonium salts, but the cross‐coupling of nitro and amino compounds remains challenging. Herein, on‐surface azo formation by nitroarene–arylamine redox cross‐coupling (path 1 in Scheme 1 a) is successfully realized on the Ag(111) surface under ultrahigh vacuum (UHV) conditions. On‐surface azo formation via reductive nitroarene or oxidative arylamine homocoupling (paths 2 and 3 in Scheme 1 a) shows poor efficiency. Low‐temperature scanning tunneling microscopy (LT‐STM) and X‐ray photoelectron spectroscopy (XPS) were applied for product analysis. The chemical structures of organic precursors used are depicted in Scheme 1 b (for their synthesis, see the Supporting Information, SI). By using well‐designed precursors, azo polymers and alternating azo oligomers are accessible on‐surface. In addition, the effect of the metal surface on the azo formation is investigated.

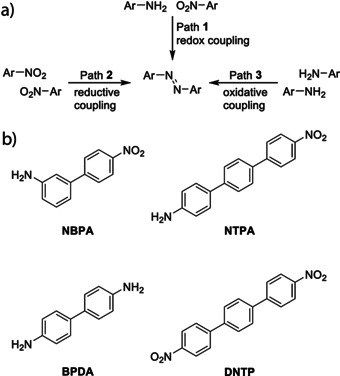
a) Investigated coupling pathways and b) bifunctional monomers towards on‐surface azo group formation.
To distinguish the three possible coupling reactions (paths 1–3 in Scheme 1 a), the unsymmetrical bifunctional 4′‐nitrobiphenyl‐3‐amine (NBPA) was studied on the Ag(111) surface (Figure 1). After adsorption on Ag(111) kept at room temperature (RT), NBPA assembled into a well‐ordered monolayer (Figure 1 a). The self‐assembly exhibits a hexagonal honeycomb motif composed of hexamer units, in which H‐bonding is assumed between the nitro and amino groups. [17] After annealing (393 K), azo groups were formed (as further supported by XPS analysis, see below) resulting in cyclic oligomers (Figure 1 c) and long chains (Figure 1 d). According to the STM images, most of the azo groups were formed via nitro–amino coupling (black arrows in Figure 1 c,d), as estimated by the angle at the connection points between the monomeric units. Few azo groups (ca. 15 %) were formed through nitro–nitro homocoupling (white arrows in Figure 1 b,d) and azo groups resulting from amino–amino coupling were rarely observed. For NBPA, the reaction efficiency in Scheme 1 a could be ranked as path 1≫ path 2> path 3. In line with this finding, reductive homocoupling of 4,4′′‐dinitro‐p‐terphenyl (DNTP), a monomeric precursor designed to follow reaction pathway 2, showed low efficiency on Ag(111) (Figure S1) and only short azo chains were obtained. To further validate the selectivity, monofunctional aromatic precursors were tested in cross‐dimerization experiments (see SI). In agreement with the reactivity trend observed for NBPA, nitro–amino and nitro–nitro couplings were noted, whereas products derived from arylamine homocouplings following path 3 were rarely identified (Figure S2–S5). In case of the attempted aniline homocouplings, we identified amides coordinating to surface adatoms. Importantly, it was found that electron‐withdrawing groups attached to the aniline reduce the amount of these adatoms. We assume that amine metalation suppresses oxidative homocoupling. On the other hand, the adatoms might lead to a more reactive substrate and be beneficial for side reactions such as reductive homocoupling of nitro groups or branching. Further, it was found that rigid compounds engage in these cross‐couplings more efficiently. Monomers with fixed conformation and planar structure showed a better cross‐coupling selectivity (Figure S3) and the statistical analysis revealed that NBPA, substituted with an electron withdrawing group and low degree of conformational flexibility, afforded the highest tendency toward the redox coupling product (Figure S6). Homo‐dimerization of anilines following path 3 was rarely observed without the presence of the nitro functionality in accordance with previously reported studies[ 18 , 19 ] (Figure S7).
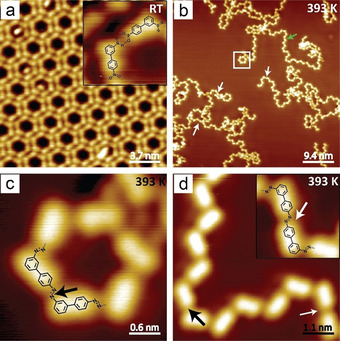
Azo formation on Ag(111) surface. a) STM image of NBPA self‐assembled on Ag(111) surface after deposition at RT. Inset: zoomed‐in STM image of (a). b–d) STM images after annealing the sample at 393 K for 30 min. Black arrow: azo groups formed by nitro–amino coupling. Green arrow: azo chain. Inset in (d): zoomed‐in image of azo group formed by nitro–nitro coupling (white arrows). Set point: a) and inset: U=−1.2 V, I=80 pA. b–d) U=−1.4 V, I=70 pA. Size of inset in (a): 2.0×2.0 nm2, (d): 2.3×2.3 nm2.
To achieve long azo polymer chains and guided by the identified reactivity trend, the linear 4′‐nitro‐p‐terphenyl‐4‐amine (NTPA) bearing an amino as well as a nitro functionality was prepared. The stability of NTPA during evaporation was confirmed with in situ mass spectrometry. Upon adsorption on the Ag(111) surface at RT, NTPA molecules assembled into regular sieve structures (Figure 2 a,b). H‐bonds are assumed to connect neighboring compounds. Resulting from the substrate symmetry, the neighboring NTPA molecules are not perfectly perpendicular to each other within the assembly. After annealing at 440 K for 30 min, terphenyl polymers linked by azo bonds were formed (Figure 2 c). The high‐resolution STM image showed that azo groups were formed with trans‐geometry (Figure 2 d). The distance between the centers of the nearest terphenyl groups is 1.51±0.03 nm, which agrees with a theoretical model (Figure S8) and an ex situ prepared model azo compound that further exhibits a similar STM topography (Figure S9). Tip manipulations confirmed the covalent bonding between the polymer units (Figure S10).

Synthesis of linear azo polymers on Ag(111). a) STM image of NTPA assembly. b) Zoomed‐in STM image of (a). c) STM image of polymers obtained after annealing the sample at 440 K for 30 min. d) Zoomed‐in image of the polymer. Set point: a) U=−2 V, I=50 pA; b) U=−1 V, I=50 pA; c,d) U=−1 V, I=70 pA.
Next, we tested the effect of the substrate on the reaction outcome and NTPA was investigated on various surfaces. For comparison, STM results recorded after annealing at 440 K are shown in Figure 3. Images showing the self‐assembled structure of NTPA after deposition and the reaction products at other temperatures are presented in the SI (Figure S11). On Ag(110), polymers with a similar contrast and length of the recurring motif as observed on Ag(111) were obtained after annealing at 440 K (Figure 3 a,b). They tend to align along the [1 0] direction of the surface, but multiple defects (green arrows in Figure 3 b) were noted at the azo linkers. On Au(111), NTPA self‐assembled in long 1D chains along the reconstructed herringbones after deposition (Figure S11 e). Coupling did not occur at 440 K and the self‐assembled structure remained (Figure 3 c,d). The distance between the recurring motif in the chain is 1.71±0.03 nm, clearly indicating linkage via H‐bonding. Au(110) was more reactive and deposition of NTPA caused the thermodynamically unstable (1×3) reconstruction of Au(110) (Figure S11 j). Annealing at 440 K led to a more pronounced formation of the (1×3) reconstruction as well as to polymerization of NTPA inside the (1×3) channels (Figure 3 e,f). Some depressions (green arrow in Figure 3 f) were observed on the polymers, which were ascribed to the missing atoms underneath. A similar phenomenon was also noted on Ag(110) (Figure S11 d). The distance between two recurring moieties indicates the same outcome as found on Ag(111). Overall, the (110) surfaces provide a distinct confinement for the products, which were observed with higher order than on the (111) surfaces. Finally, reactivity of NTPA was studied on Cu(111). Instead of the azo formation, activation of N−H, N−O, and C−H bonds occurred. After deposition and at different annealing temperatures, clusters and short chains connected via coordination bonds (Figure 3 g,h, Figure S11 m–p) were the main products, in agreement with previously described dehydrogenation of aniline derivatives on Cu(111).[ 18 , 19 ] Overall, our experiments indicate that the azo formation reaction is highly influenced by the metal type as well as the surface structure.

NTPA at different surfaces. a,b) STM images recorded after annealing a sub‐monolayer of NTPA on Ag(110) at 440 K. c,d) STM images recorded after annealing a sub‐monolayer of NTPA on Au(111) at 440 K. e,f) STM images recorded after annealing a sub‐monolayer of NTPA on Au(110)at 440 K. g,h) STM images recorded after annealing a sub‐monolayer of NTPA on Cu(111) at 440 K. Set points: a) U=−1.5 V, I=160 pA; b) U=−0.2 V, I=370 pA; c,d) U=−1.1 V, I=47 pA; e,f) U=−1.3 V, I=130 pA; g) U=−0.85 V, I=400 pA; h) U=−0.14 V, I=570 pA.
Attempting alternating copolymerization, DNTP and biphenyl‐4,4′‐diamine (BPDA) were co‐adsorbed on Ag(111) (Figure 4). After deposition, ordered and disordered phases were observed, denoted by black dashed rectangle and white dashed ellipse in Figure 4 a. Consistent with previous works,[ 18 , 19 ] the BPDAs were found connected by adatoms (Figure 4 b,c, also see Figure S7). In the disordered phases, azo‐connected oligomers were already formed at RT (white and blue arrow in Figure 4 c), as also found for other monomers, usually after long‐time annealing (Figure S12 and S13). Annealing (343 K) improved reaction efficiency (Figure 4 d) and alternating azo chains with up to five units were identified (Figure 4 e,f). A strictly alternating polymerization was difficult, as the nitro–nitro coupling of DNTP would introduce defects. Furthermore, BPDA could easily desorb from the surface at high temperature, perturbing the 1:1 stoichiometry of the two reaction partners. To achieve longer alternating polymers, strategies of surface confinement and specially designed precursors need to be considered.
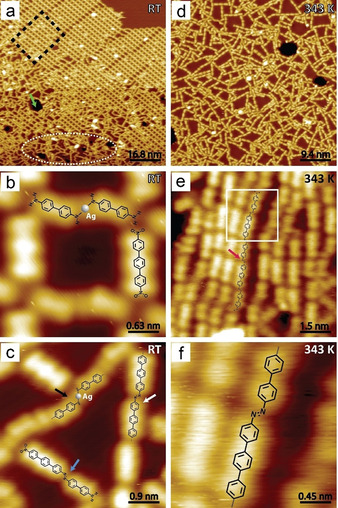
Towards alternating polymers on Ag(111) surface. a–c) Co‐adsorption of DNTP and BPDA. a) Overview image with ordered (black rectangle) and disordered (white ellipse) structures. b) Zoomed‐in ordered assembly. c) Zoomed‐in disordered phase. Blue arrows: azo bond formed by nitro and amino. White arrow: terphenyl chains. Black arrow: BPDA chains via N−Ag−N bonds. d–f) STM images recorded after annealing at 343 K for 30 min. d) Overview image with surface hollows. e) Zoomed‐in image of alternating chains. Red arrow: azo bonds. f) High resolution image of azo‐linkage. Set point: a,b) U=−1.7 V, I=50 pA; c) U=−1.7 V, I=60 pA; d) U=−1 V, I=50 pA; e,f) U=−1.3 V, I=45 pA.
To understand the chemical states, XPS experiments were conducted. The spectra recorded were analyzed using a commercial processing software (CasaXPS). Figure 5 shows the N 1s spectra recorded on NTPA samples prepared at different conditions. In the region of N 1s, the clean silver surface shows a strong background. The spectrum of the clean surface was fitted by two optimized components (not shown), whose peak positions and full width at half maxima were used to reckon the surface contribution in the analysis of the spectra on other samples. Since the coupling between nitro and amino groups can be performed at RT (Figure S12 and S13), a monolayer was deposited onto Ag(111) at 170 K to avoid a partial reaction during the XPS measurement. In the XP spectrum, a peak at a binding energy of 399.6 eV was observed and could be assigned to the amino groups.[ 20 , 21 , 22 ] Unfortunately, even under these conditions the nitro group could not be observed in the spectrum. After annealing at 390 K, the amino peak vanished and two new peaks were observed at 400.1 and 397.7 eV. In consistency with the STM results, the peak at 400.1 eV indicates the formation of azo groups.[ 23 , 24 , 25 ] The N 1s peaks observed on Ag(111) agree well with those of powders and reported data (Table S1). The peak at 397.7 eV is ascribed to deoxygenated nitro [26] and the dehydrogenated amino groups,[ 7 , 27 , 28 ] before azo group formation. On the multilayer sample, the N 1s peak arising from the nitro groups is observed at 406 eV.[ 21 , 29 , 30 ] The absence of the peak of nitro groups in the monolayer samples could result from the strong background as well as the easy reduction of nitro groups on the silver surface (O 1s spectra in Figure S14). The signals of intact nitro and amine groups in a monolayer of NTPA were only observed on the Au(111) surface (Figure S14), revealing the very low activity of these functional groups in line with their poor reaction efficiency found in STM experiments. The XP spectra concede a possible mechanistic model in which the nitro groups are deoxygenated and reduced stepwise, while the amino groups are partially oxidized and metalated. This causes the net comproportionation before azo formation. We therefore assumed that arylhydroxylamines, as a result of the weak N−O bond, should easily be metalated like amines and thus lead to reactive intermediates of the sequence. Indeed, by using N‐(naphthalene‐1‐yl)hydroxylamine as starting material, dimerization towards 1,1′‐azonaphthalene at 383 K was achieved at Ag(111) (Figure S15), in line with our mechanistic model as discussed below. Furthermore, nitrosoarenes could either be intermediates or act as precursors for intermediates of our reaction sequence. To validate our assumption of stepwise reduction of the nitro group we tested p‐decyloxynitrosobenzene on Ag(111) and found a clean dimerization at 420 K (Figure S16).
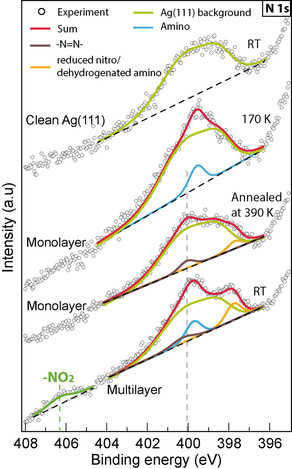
N 1s XP spectra recorded on a clean Ag(111) surface, NTPA monolayer adsorbed on cold Ag(111) (170 K), NTPA monolayer annealed at 390 K for 30 min, and multilayers deposited onto Ag(111) held at RT. Dashed line: azo peak.
With these mechanistic indications in hand, we wanted to gain further insights into the reaction mechanism. Coupling of aniline and nitrobenzene on Ag(111) to form trans‐azobenzene was studied as a model reaction using DFT calculations. A four‐layered slab model with a calculated lattice constant of 4.07 Å was used to sample the Ag(111) surface. The calculations were performed with the PBE functional,[ 31 , 32 ] the wave functions were expanded in a plane‐wave basis with an energy cut off of 500 eV and a 3×3×1 k‐point grid was used to sample the Brillouin Zone (see SI for further details). 18 structures possibly involved in this model reaction were calculated on the Ag(111) surface (also see Figure S17–S19 and following discussion). These structures range from the starting compounds (chosen as reference state with a relative energy of 0 kJ mol−1, in Figure 6 referred to as Ph‐NH2+Ph‐NO2) to trans‐azobenzene (co‐adsorbed with two oxygen atoms and one H2 molecule, Ph−N=N−Ph+2 O+H2). The resulting energy profile can be seen in Figure 6. Please note that no reaction barriers or transition states were calculated (also see the discussion following Figure S19 for further details). [33] Two pathways connecting the intermediates with the lowest energies are highlighted in green color. As first steps, an H atom and an O atom are removed from aniline and nitrobenzene at the cost of approximately 133 kJ mol−1 and 102 kJ mol−1, respectively. These two processes lead to the intermediate structure Ph−NH+Ph−NO+O+H, which is 235 kJ mol−1 higher in energy. Next, an N−N single bond can be formed, releasing approximately 34 kJ mol−1. The intermediate Ph−NH−NO−Ph can then lose the nitrogen‐bound H and O atoms in a slightly endothermic stepwise procedure to form the azo product. Since hydrogen atoms form H2 with a very low reaction barrier of <25 kJ mol−1 [34] on Ag(111), it can be expected that the two abstracted H atoms can recombine at any time during the reaction, which presumably suppresses the formation of other by‐products apart from H2 and adsorbed O atoms. This recombination is exothermic by approximately 25 kJ mol−1. Considering that the reactions are performed under UHV conditions and that the adsorption energy of H2 molecules on Ag(111) surfaces is very small (<10 kJ mol−1), it can be assumed that newly formed H2 molecules will be quickly and continuously removed from the equilibrium. Naturally, this enhances the formation of H2 and will contribute to the driving force of the overall endothermic on‐surface reaction. The gain in entropy resulting from the desorption of H2 has been identified as driving force in other on‐surface reactions,[ 35 , 36 ] further supporting the formation and desorption of H2. Contrary to hydrogen, the abstracted oxygen atoms are observed to remain—at least partially—adsorbed on the surface, even at elevated temperatures (Figure S14). This agrees well with literature, where the recombinative desorption of O2 on the Ag(111) surface is observed at temperatures higher than 597 K. [37]
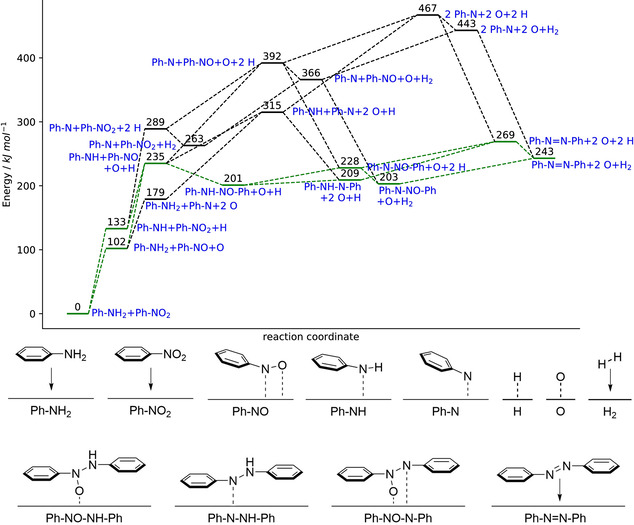
Calculated energy profile of the reaction of aniline and nitrobenzene on Ag(111) and corresponding structures. Two possible pathways via the most energetically favorable intermediate structures are highlighted in green color. All alternative routes are shown as dashed black lines.
In summary, on‐surface azo group formation was realized on silver surfaces. STM results reveal that the azo group formation by nitro–amino redox cross‐coupling (path 1) is the most efficient reaction on the silver surface in comparison with nitro–nitro (path 2) and amino–amino homocoupling (path 3). XPS measurements, as well as theoretically and experimentally analyzed model compounds further supported the correct structural assignment after reaction on silver surface. Using well‐designed molecular precursors, azo polymers were successfully prepared. Experiments at different surfaces revealed that the metal types and the surface structures have a significant influence on the reaction outcome. A mechanistic model was developed based on the XPS study and dimerization reactions of possible intermediates as model compounds. Based on a DFT study a stepwise splitting of N−H and N−O bonds from the starting material along N−N bond formation was suggested as the most likely reaction path. Our work provides a new bottom‐up strategy to synthesize functional materials through on‐surface reactions.
The authors declare no conflict of interest.
We thank the Deutsche Forschungsgemeinschaft (SFB 858, TRR 61, FU 299/19, GA2430/1‐1, MO 2345/4‐1, and AM 460/2‐1) for financial support. H.‐Y.G. acknowledges financial support by NSFC within Grant 21972104 and “1000‐Youth Talents Plan”. X.M. thanks the Alexander von Humboldt foundation for financial support. H.K. wants to thank Dr. Anup Bhunia and Niklas Radhoff for helpful discussions. Open access funding enabled and organized by Projekt DEAL.
1
3
5
5
6
6
7
9
9
10
11
11
12
13
14
15
15
16
16
17
18
19
20
21
24
25
26
27
28
29
30
32
33
34
35
37
38
 Azo bond formation on metal surfaces
Azo bond formation on metal surfaces
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
