 Gm14230 controls Tbc1d24 cytoophidia and neuronal cellular juvenescence
Gm14230 controls Tbc1d24 cytoophidia and neuronal cellular juvenescence
PLoS ONE


Competing Interests: The authors have declared that no competing interests exist.

- Altmetric
It is not fully understood how enzymes are regulated in the tiny reaction field of a cell. Several enzymatic proteins form cytoophidia, a cellular macrostructure to titrate enzymatic activities. Here, we show that the epileptic encephalopathy-associated protein Tbc1d24 forms cytoophidia in neuronal cells both in vitro and in vivo. The Tbc1d24 cytoophidia are distinct from previously reported cytoophidia consisting of inosine monophosphate dehydrogenase (Impdh) or cytidine-5’-triphosphate synthase (Ctps). Tbc1d24 cytoophidia is induced by loss of cellular juvenescence caused by depletion of Gm14230, a juvenility-associated lncRNA (JALNC) and zeocin treatment. Cytoophidia formation is associated with impaired enzymatic activity of Tbc1d24. Thus, our findings reveal the property of Tbc1d24 to form cytoophidia to maintain neuronal cellular juvenescence.
Introduction
We previously identified juvenility-associated genes (JAGs) that were selectively highly expressed in juvenile tissues [1]. By extending our analysis to the noncoding element of the transcriptome, we further identified juvenility-associated long noncoding RNAs (JALNCs) as lncRNAs that were predominantly expressed in juvenile [2]. Among the JALNCs, Gm14230 maintains cellular juvenescence that is characterized by cellular properties of growth, differentiation capacity and resistance to cellular senescence [2]. It is not completely understood how Gm14230 depletion leads to the loss of cellular juvenescence. Here, we show that Gm14230 depletion causes formation of a cellular macrostructure called cytoophidia consisting of Tbc1d24 protein.
Tbc1d24 is expressed abundantly in the cerebral cortex in juvenile mice and encodes a GTPase activating protein (GAP) that regulates cytoskeletal reorganization, vesicular transportation and cellular migration [3]. Tbc1d24 possesses functional domains such as a TBC (Tre2-Bub2-Cdc16) domain with Rab GAP activity [4, 5] and a TLDc (domain catalytic) domain that mediates resistance to oxidative stress [6, 7]. Tbc1d24 GAP regulates small GTPases Arf6 [3] and Rab35 [8]. Arf6 is implicated in membrane trafficking between plasma membrane and endocytic compartments through activation of lipid-modifying enzymes phospholipase D and phosphatydylinositol-4-phosphate 5 kinase (PIP5K) [9, 10]. Rab35 mediates membrane trafficking in synapses and between plasma membrane and early endosomes [11]. Rab35 and Arf6 antagonize each other in membrane trafficking [12]. Tbc1d24 interacts with EphrinB2 and plays a role in locomotion of neural crest cells [8]. However, it remains to be completely understood how the enzymatic function of Tbc1d24 is regulated in a cell.
Mutations in TBC1D24 cause the refractory seizure syndrome called epileptic encephalopathy (EE). TBC1D24 mutations have been reported in patients with DOORS (deafness onychodystrophy, osteodystrophy, mental retardation and seizures [13]) syndrome, progressive myoclonic epilepsy [14], early infantile EE [15] and malignant migrating partial seizures of infancy (MMPSI, [16]). A truncating mutation of TBC1D24 results in severe neurodegeneration [17]. Consistently, Tbc1d24 knockout mice displayed spontaneous seizures and abnormal behaviors with compromised axonal growth [18], vesicle trafficking [19, 20] and dendritic spine formation in cortical and hippocampal neurons [21].
The cytoophidium is a macromolecular ring- or rod-like structure found in the cytoplasm [22–24]. Cytoophidia have been reported to be formed by polymerization of the enzymatic proteins cytidine-5’-triphosphate synthase (Ctps, [25–29]), inosine monophosphate dehydrogenase (Impdh, [30–36]), asparagine synthetase [37] and phosphofructokinase-1 (Pfk1, [38]).
We here show that Tbc1d24 forms cytoophidia in neuronal cells both in vitro and in vivo. Tbc1d24 cytoophidia were induced by loss of cellular juvenescence caused by Gm14230 depletion and zeocin-induced genotoxicity. Tbc1d24 cytoophidia were distinct from the previously characterized cytoophidia formed by Impdh and Ctps. The GAP activity of Tbc1d24 is suppressed by forming cytoophidia. The loss of cellular juvenescence enhances formation of the cytoophidia that has a protective role against the cellular stress. Thus, Tbc1d24 cytoophidia underlie the physiological functions of juvenile neuronal cells.
Materials and methods
Mouse experiment
All animal experiments were approved by the institutional animal care and use committee at Shiga University of Medical Science. All experiments were performed in accordance with the relevant guidelines and regulations. The brains of C57BL/6N mice perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) on the postnatal day 11 (P11) were dissected and post-fixed with 4% PFA in PBS. The brains were cryoprotected in 15% sucrose in PBS for 3 days at 4°C. The brains were embedded in optimum cutting temperature (OCT) compound (Sakura Finetek, Tokyo, Japan), frozen and stored at -80°C until use. Brains were sectioned at 20 μm thickness using a cryostat (CM3050, Leica) and mounted onto MAS-coated slide glass (Matsunami glass, S9441, Osaka, Japan). The sections were washed 3 times in 0.1% Triton X-100 in PBS (PBS-T), and nonspecific staining was blocked by treatment with 3% bovine serum albumin (BSA) in PBS-T at room temperature (RT) for 1 hour (hr). The sections were incubated with primary antibodies overnight at 4°C, washed 3 times in PBS-T, and then incubated with secondary antibodies overnight at 4°C. The information for the antibodies were shown below. After washing 3 times in PBS-T, the slides were mounted with Prolong Gold Antifade mountant with DAPI (Invitrogen).
Cell culture
Neuro2a and SH-SY5Y cells were cultured in Eagle’s minimal essential medium supplemented with 10% fetal bovine serum (FBS) and Ham’s F12:EMEM (1:1) supplemented with 10% FBS, respectively. To model loss of cellular juvenescence, 200 μg/ml zeocin was added to the culture medium for 72 hrs or 96 hrs as indicated in Figure legend. For detection of dead cells, Sytox blue dye (Thermo Fischer, S11348, Massachusetts, US) was added to the culture medium at 5 μM. For evaluation of Impdh or Ctps cytoophidia, Acivicin or 6-diazo-5-oxo-L-norleucine (DON) was added to the culture medium at 2 mM for 24 hrs. For induction of Impdh cytoophidia, cells were treated with 2 μM mycophenolate (MPA) for 24 hrs. Cell images were obtained with EVOS FL cell imaging system (Thermo Fisher Scientific). The number of cells per field was counted with Fiji software (https://imagej.net/Fiji, [39]).
Plasmid construction
The Gm14230 sequence was amplified by PCR with the primers (Forward: 5’-GTTTCCGGAGCGCCTACT-3’, Reverse: 5’-CTGTTCTGAGTCGCTCTTGC-3’) and a cDNA library derived from mouse NIH3T3 cells. The PCR product was first cloned into the pMD20-T vector (Takara BIO, Shiga, Japan) for sequence confirmation. The cloned product was then subcloned into pc.DNA-3.1(-) using HindIII and BamHI sites.
Transfection in cultured cells
For knockdown experiments, Lipofectamine RNAiMAX (Invitrogen, California, US) was used to transfect siRNA duplexes at 20 nM: mouse Tbc1d24 siRNA1 (Sigma-Aldrich, Mission siRNA SASI_Mm01_00145836, Missouri, US), Tbc1d24 siRNA2 (Sigma-Aldrich, Mission siRNA SASI_Mm01_00145838), mouse Gm14230 siRNA1 (5’-GGUGCUAAAGGACCAGUUGdTdT-3’), Gm14230 siRNA2 (5’-GCUCAGUCGGCUCAGAGUAdTdT-3’) and siRNA negative control (Invitrogen, AM4611). The knockdown efficiency was assessed 48 hrs after transfection by qPCR or western blot analysis.
In overexpression experiments, the Gm14230 plasmid was transfected into Neuro2a cells with Lipofectamine 2000 (Invitrogen). The extent of forced expression was assessed 48 hrs after transfection by qPCR.
Real-time quantitative PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific). The extracted RNA was quantified using a NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific) and reverse transcribed using a high-capacity RNA to cDNA kit (Applied Biosystems, California, US) and PCR thermal cycler Dice (Takara BIO) according to the manufacturer’s instructions. qPCR was performed using a LightCycler 480 SYBR Green I Master Kit on a LightCycler 480 instrument (Roche, Basel, Switzerland) using reverse transcribed cDNA as a template. The specificity and quality of qPCR amplification was assessed by agarose gel electrophoresis and a melting curve analysis. The expression levels were normalized to mouse Tubb5 or Polr2a as indicated in the Figure legends. The primers used for qPCR are mouse Tbc1d24 (Forward: 5’-AGCACTGAGGCAGAAGGGTA-3’, Reverse: 5’-CATCTCCTTCACGCTGACAA-3’), Gm14230 (Forward: 5’-CTTACCACGTGTGCCAGTGT-3’, Reverse: 5’-CTGGGGTCACTGGTGGAAT-3’), Tubb5 (Forward: 5’-GATCGGTGCTAAGTTCTGGGA-3’, Reverse: 5’-AGGGACATACTTFCCACCTGT-3’) and Polr2a (Forward: 5’-GAGTCCAGAACGAGTGCATGA-3’, Reverse: 5’-ACAGGCAACACTGTGACAATC-3’).
Western blot analysis
Cell lysates were prepared using RIPA lysis buffer containing 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS. The lysates were centrifuged at 15,000 rpm for 10 min. The lysates were mixed with Laemmli sample buffer and boiled at 98°C for 2 min. The protein samples were run on SDS-PAGE along with protein markers for SDS-PAGE (Nacalai Tesque, 02525–35, Kyoto, Japan) and blotted onto a PVDF membrane (GE healthcare, Illinois, US). The following primary antibodies were used: Tbc1d24 (ProteinTech, 1:1000, Illinois, US; LSBio, 1:1000, Illinois, US), p21 Waf1 (Abcam, ab109199, 1:1000, Cambridge, UK), p27 Kip1 (Cell Signaling Technology, #3698, 1:1000, Massachusetts, US), Impdh (ProteinTech, 12948-1-AP, 1:100), Cpts1 (ProteinTech, 15914-1-AP, 1:100), Arf6 (Cell Biolabs, STA-407-6, 1:500, California, USA) and beta-actin (Cell Signaling Technology, #5174, 1:4000). HRP-conjugated anti-mouse IgG (Thermo Fisher Scientific, 32430) and anti-rabbit IgG (Thermo Fisher Scientific, 32460) were used as secondary antibodies at 1:1000 dilution. For detection of immunoreactive bands, Chemi-Lumi One L or Chemi-Lumi One Ultra (Nacalai Tesque) were used. Uncropped blots were shown in S1 Raw images. Densitometric analyses for blotting data were performed using Fiji [39].
Arf6 activity assay
The active form of Arf6 was detected using a GTP-bound Arf6-specific pulldown assay kit (Cell biolabs, STA-407-6). Cells were lysed in Arf6 lysis buffer containing 25 mM HEPES, pH 7.5, 150 mM NaCl, 1% NP-40, 10 mM MgCl2, 0.2 mM EDTA and 2% glycerol, followed by centrifugation at 15,000 rpm for 10 min at 4°C. The supernatant was incubated with Golgi-localized γ-ear containing Arf-binding protein 3 protein binding domain (GGA3 PBD) agarose beads for 1 hr at 4°C. Beads were washed 3 times in Arf lysis buffer. The pellets were added to 2x Laemmli sample buffer and boiled at 95°C for 5 min. The supernatants were analyzed with SDS-PAGE and western blot analysis with Arf6 antibody (Cell Biolabs, STA-407-6, 1:500). HRP-conjugated anti-mouse IgG (Thermo Fisher Scientific, 32430) was used as the secondary antibody at 1:1000 dilution.
Cycloheximide chase assay
To access whether cytoophidia formation affected the half-life of Tbc1d24 protein, Tbc1d24 protein expression levels were analyzed in Neuro2a cells treated with zeocin at 200 μg/ml or control distilled water for 72 hrs in the presence of cycloheximide (CHX) at 20 μg/ml. Cell lysates were prepared at 0, 4, 8, 12 hrs after the CHX treatment and analyzed for the expression of Tbc1d24 protein by western blots as described above.
Co-immunoprecipitation assay
To analyze potential interactions of Tbc1d24 with Ctps and Impdh, Neuro2a cells were transfected with FLAG tagged TBC1D24 plasmid (GenScript). Forty-eight hrs after transfection, cells were lysed with NETN buffer containing 20 mM Tris-HCl (pH 8.0), EDTA 1 mM, NaCl 100 mM and 0.5% NP-40, followed by centrifugation at 20,800x g for 15 min at 4°C to remove debris. Protein G Sepharose beads (Invitrogen) were incubated with 2 μg anti-FLAG antibody or control rabbit IgG (Cell Signaling Technology, #2729) for 1 hr at RT, and then incubated with the cell lysates overnight at 4°C. The beads were then washed with NETN200 wash buffer containing 20 mM Tris-HCl, 1 mM EDTA, 200 mM NaCl and 0.5% NP-40 four times, followed by elution with Laemmli sample buffer and denaturation at 95°C for 5 min. The samples were analyzed by western blots with Impdh, Ctps and FLAG antibodies.
Senescence-associated beta-galactosidase activity
For staining of senescence-associated beta-galactosidase (SA-β-gal) activity, Neuro2a cells transfected with Gm14230 siRNA were cultured for 7 days followed by fixation with 3% (v/v) formaldehyde. The cells were then washed with PBS twice and incubated with the staining solution containing 5 mM K3Fe(CN)6, 2 mM MgCl2, 150 mM NaCl, 30 mM citric acid/phosphate buffer, 5 mM K4Fe(CN)6, and 1 mg/ml X-Gal for 16 hrs at 37°C.
Immunocytochemistry
Cells were fixed with 4% PFA at RT for 10 min and permeabilized with 0.1% Triton X-100 in PBS for 2 min. For blocking, cells were incubated with 2% FBS in PBS for 1 hr at RT. Cells were then incubated at 4°C overnight with the following primary antibodies: Tbc1d24 (ProteinTech, 25254-1-AP, 1:200, LSBio, LS-C679739, 1:200, Washington, US), Vimentin (Santa Cruz, sc-6260, 1:100, Texas, US), Impdh (Santa Cruz, sc-36-5171, 1:100, ProteinTech, 12948-1-AP, 1:100), Cpts1 (ProteinTech, 15914-1-AP, 1:200 and LSBio, LS-C197001, 1:100). After washing with PBS, cells were incubated with anti-mouse IgG conjugated with Alexa Fluor 546 (Invitrogen, A11030, 1:1000) or anti-rabbit IgG conjugated with Alexa Fluor 488 (Invitrogen, A11008, 1:1000) for 1 hr at RT. Prolong Gold Antifade mountant with DAPI (Invitrogen) was used for mounting and nuclear staining. Cellular images were taken with a confocal microscope (Leica, TCP SP8, Wetzlar, Germany) or a super-resolution fluorescence microscope (GE healthcare, DeltaVision Elite). The length (μm) of cytoophidia was measured using Fiji [39].
Analyses of cytoophidia in various conditions
To assess the Acivicin-responsibility of Tbc1d24 cytoophidia, Neuro2a cells were treated with 2mM Acivicin or control distilled water for 24 hrs. Acivicin is a known inducer of Impdh and Ctps cytoophidia. Immunofluorescence analyses for Tbc1d24, Impdh and Ctps were conducted to assess the influences on cytoophidia formation.
To assess reversibility of Tbc1d24 cytoophidia formation, Neuro2a cells were treated with 2 μM MPA or dimethyl sulfoxide (DMSO) for 24 hrs before washing out MPA. The cells were then further cultured for 24 or 48 hrs and analyzed for Tbc1d24 cytoophidia by immunocytochemistry described as above.
For the assessment of a protective effect of Tbc1d24 in loss of cellular juvenescence, Neuro2a cells were transfected simultaneously with Gm14230 (or control) siRNA and pcDNA3.1-FLAG-TBC1D24 (or empty) plasmid. Cell growth were analyzed 48 hrs after the transfection.
To assess the effect of forced expression of Tbc1d24 in zeocin-induced cellular juvenescence loss, Neuro2a cells were transfected with pcDNA3.1-FLAG-TBC1D24 plasmid or control empty pcDNA3.1 plasmid. Twenty-four hrs later, the cells were treated with zeocin at 200 μg/ml or distilled water for 72 hrs for the assessment of cell growth.
To assess a role of Tbc1d24 in loss of cellular juvenescence induced by Gm14230 depletion, Neuro2a cells were transfected simultaneously with Tbc1d24 and Gm14230 siRNAs. The cell growth and viability were analyzed 48 hrs after the transfection. For the viability assessment, Sytox blue staining was performed described as above.
Gene expression analysis in mouse cerebral cortexes
Tbc1d24 expression in mouse cerebral cortexes at P1 and P56 was analyzed by the strand-specific RNA-seq dataset we previously deposited with the accession number DRA009510. The RNA-seq data were processed using BioLinux8 [40]. The expression was visualized using Integrative Genomic Viewer (http://software.broadinstitute.org/software/igv/). Gene expression levels show fragments per kilobase of transcript per million fragments sequenced (FPKM) at P1 and P56.
Statistical analysis
For all quantified data, the mean ± standard error of the mean (SEM) was presented. Statistical significance between two experimental groups was indicated by an asterisk, and comparisons were made using Student’s t-test. P-values less than 0.05 were considered significant.
Results
Tbc1d24 forms cytoophidia
We previously performed a comprehensive transcriptomic analysis to elucidate mechanisms for the juvenile properties of animals and identified JAGs and JALNCs. Among the JAGs, we focused on Tbc1d24, the gene predominantly highly expressed in juvenile compared to adult in the cerebral cortex (Fig 1A, S1 Table). We validated the juvenile-predominant expression of Tbc1d24 by real-time qPCR (Fig 1B). We performed immunostaining of Tbc1d24 in Neuro2a mouse neuroblastoma cells and found that Tbc1d24 took a ring-like macrostructure in the cytoplasm (Fig 1C). Similar macrostructures, called as cytoophidia, have been reported with proteins such as Impdh and Ctps. Super-resolution imaging further revealed Tbc1d24 took a string-like structure (Fig 1D). The Tbc1d24 cytoophidia was also detected with the other antibody developed against the distinct region of Tbc1d24 protein (Fig 1E). To examine whether Tbc1d24 cytoophidia was formed in the mouse cerebral cortex, we performed immunohistochemistry for Tbc1d24 and observed the similar string structure in the cerebral cortex of mice (Fig 1F). Tbc1d24 cytoophidia was observed also in SH-SY5Y, human neuroblastoma cells (Fig 1G), indicating Tbc1d24 cytoophidia was conserved across species.

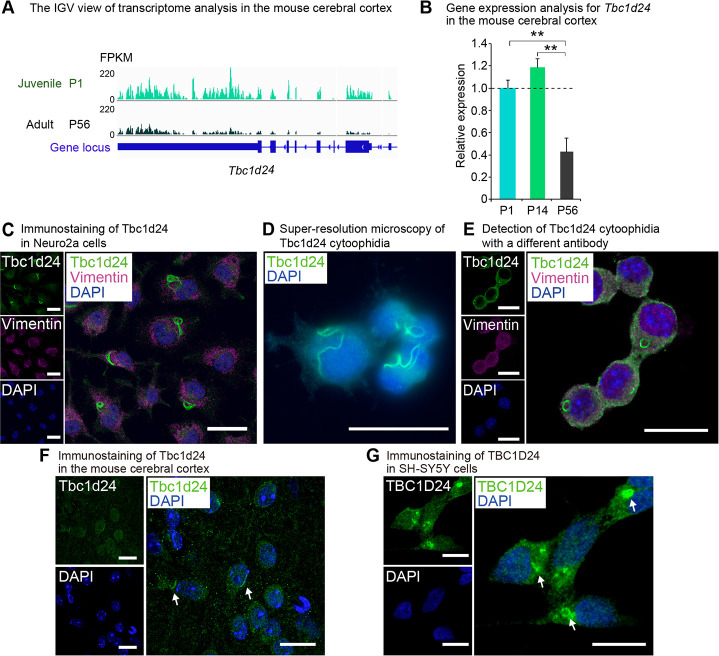
Tbc1d24 is expressed predominantly in juvenile in the cerebral cortex and forms cytoophidia.
(A) IGV view of the transcriptome analysis in the mouse cerebral cortex at postnatal day (P) 1 and P56. Numbers next to the read patterns (0 and 220) indicate fragments per kilobase of exon per million reads mapped (FPKM) values. (B) qPCR analysis of mouse Tbc1d24 in the postnatal mouse cerebral cortex at P1, P14 and P56. Data were normalized to Polr2a (n = 3). (C) Immunofluorescence analysis of Tbc1d24 in Neuro2a cells. Cells were also stained with the acetylated tubulin antibody and DAPI. Scale bar = 25 μm. (D) Super-resolution microscopy of Tbc1d24 cytoophidia in Neuro2a cells. Scale bar = 25 μm. (E) Immunofluorescence analysis of Tbc1d24 and Vimentin with a different antibody against Tbc1d24 (LSBio, LS-C679739) in Neuro2a cells. Scale bar = 25 μm. (F) Immunofluorescence analysis of Tbc1d24 in the mouse cortical tissue. The arrows indicate Tbc1d24 cytoophidia. Scale bar = 25 μm. (G) Immunofluorescence analysis of TBC1D24 in SH-SY5Y cells. The arrows indicate TBC1D24 cytoophidia. Scale bar = 10 μm. **p<0.01; Student’s t-test. The data were presented as the means ± standard error of the mean (SEM).
Tbc1d24 cytoophidia is distinct from Impdh and Ctps cytoophidia
To examine whether Tbc1d24 cytoophidia was distinct from previously known structures, we investigated a contribution of proteins that have been reported to form cytoophidia. The expression levels of the cytoophidia-related genes Impdh and Ctps were also higher in juvenile than in adult (S1 Table). Immunostaining with Impdh (Fig 2A) or Ctps (Fig 2B) did not show any colocalization to Tbc1d24 cytoophidia. Co-immunoprecipitation assay with Neuro2a cells expressing FLAG-Tbc1d24 did not show the appreciable interactions of Tbc1d24 protein with Impdh and Ctps protein (S1 Fig). The distinction of Tbc1d24 cytoophidia from Impdh and Ctps cytoophidia were further assessed as follows. Impdh cytoophidia is known to be induced by 6-diazo-5-oxo-L-norleucine (DON), Acivicin [41] and mycophenolate (MPA). DON and Acivicin are glutamine analogs and inhibit glutamine-dependent enzymes [42]. Both are strong inducer of Impdh and Ctps cytoophidia [22, 43]. MPA reversibly inhibits Impdh activity that converts IMP to guanosine monophosphate (GMP) [44]. MPA induces Impdh cytoophidia [43]. Tbc1d24 cytoophidia were decreased significantly by DON (Fig 2C and 2D) and MPA (Fig 2E and 2F), clarifying the distinct response of Tbc1d24 to DON and MPA compared to Impdh. Transient treatment with MPA showed that Tbc1d24 cytoophidia reformed 48 hrs after washing out MPA, revealing the reversible nature of Tbc1d24 cytoophidia formation (S2 Fig). Acivicin treatment also resulted in significant suppression of Tbc1d24 cytoophidia (Fig 2G and 2H), making a contrast to Impdh cytoophidia that were markedly induced (S3C and S3D Fig).
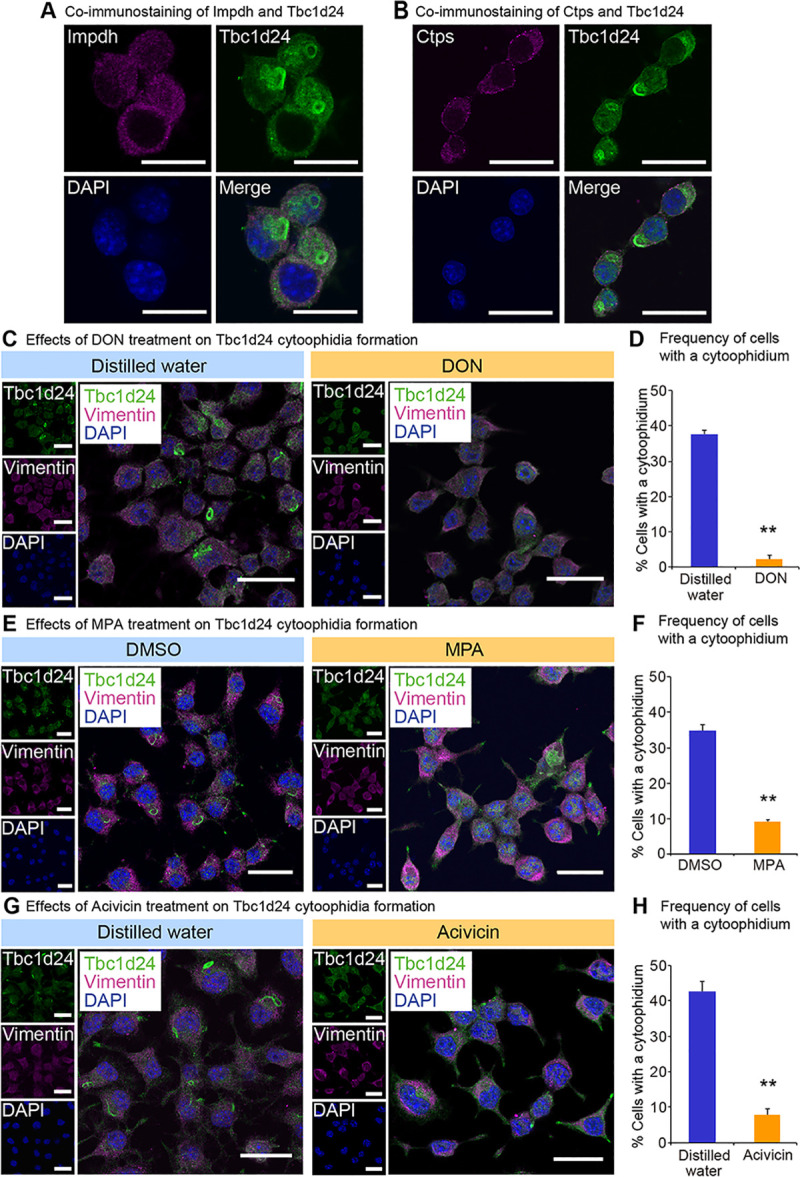
Tbc1d24 cytoophidia are distinct from Impdh and Ctps cytoophidia.
(A) Costaining of Impdh and Tbc1d24 in Neuro2a cells. Scale bar = 25 μm. (B) Costaining of Ctps and Tbc1d24 in Neuro2a cells. Scale bar = 25 μm. (C) Immunofluorescence analysis of Tbc1d24 in Neuro2a cells treated with control distilled water or 2 mM 6-diazo-5-oxo-L-norleucine (DON) for 24 hours (hrs). Scale bar = 25 μm. (D) Frequency of Tbc1d24 cytoophidium-positive Neuro2a cells treated with 2 mM DON for 24 hrs. (E) Immunofluorescence analysis of Tbc1d24 in Neuro2a cells treated with control dimethyl sulfoxide (DMSO) or 2 μM mycophenolate (MPA) for 24 hrs. Scale bar = 25 μm. (F) Frequency of Tbc1d24 cytoophidium-positive Neuro2a cells treated with 2 μM MPA for 24 hrs. (G) Immunofluorescence analysis of Tbc1d24 in Neuro2a cells treated with control distilled water or 2 mM Acivicin for 24 hrs. Scale bar = 25 μm. (H) Frequency of Tbc1d24 cytoophidium-positive Neuro2a cells treated with 2 mM Acivicin for 24 hrs. **p<0.01; Student’s t-test. The data were presented as the means ± SEM.
Ctps cytoophidia formation is known to be augmented by DON and Acivicin [22]. Tbc1d24 cytoophidia were decreased significantly by DON (Fig 2C and 2D) and Acivicin (Fig 2G and 2H), revealing the distinct responses of Tbc1d24 compared to Ctps. To examine whether Tbc1d24 protein expression was suppressed by these compounds, we performed western blot analysis that showed Tbc1d24 protein was not decreased significantly by these treatments, indicating that the decrease of cytoophidia was not due to suppression of Tbc1d24 protein (S4 Fig). These findings indicate Tbc1d24 cytoophidia are distinct from Impdh and Ctps cytoophidia.
Tbc1d24 cytoophidia is increased by loss of cellular juvenescence
We next investigated physiological relevance of Tbc1d24 cytoophidia. Because Tbc1d24 was expressed in a juvenile cell, we evaluated the role of the cytoophidia for cellular juvenescence. The loss of cellular juvenescence was induced by depletion of Gm14230. Gm14230 is a JALNC and is essential for cellular juvenescence [2]. The siRNA-mediated depletion of Gm14230 (Fig 3A) induced cytoplasmic extensions and impairment of cellular growth (Fig 3B and 3C), hallmarks of juvenescence loss. The Gm14230-depleted Neuro2a cells exhibited positivity for senescence-associated beta-galactosidase (SA-β-gal) activity (Fig 3D and 3E). The expression of senescence-associated proteins was enhanced by Gm14230 depletion, corroborating that Gm14230 depletion led to the loss of cellular juvenescence (Fig 3F and 3G). The loss of cellular juvenescence resulted in more frequent formation of cytoophidia, revealing that Tbc1d24 cytoophidia formation was inducibly formed by the loss of cellular juvenescence (Fig 3H and 3I). Based on the finding that Gm14230 depletion induces the cytoophidia, we next asked whether the forced expression of Gm14230 suppressed the cytoophidia formation. Transfection of Gm14230 (Fig 4A) enhanced the growth of Neuro2a cells significantly (Fig 4B and 4C). This enhancement of the cellular growth resulted in the significant suppression of the cytoophidia formation, suggesting the cytoophidia formation was impaired in the rigorously growing cells (Fig 4D and 4E). These findings indicate that Tbc1d24 cytoophidia formation is negatively regulated by Gm14230.
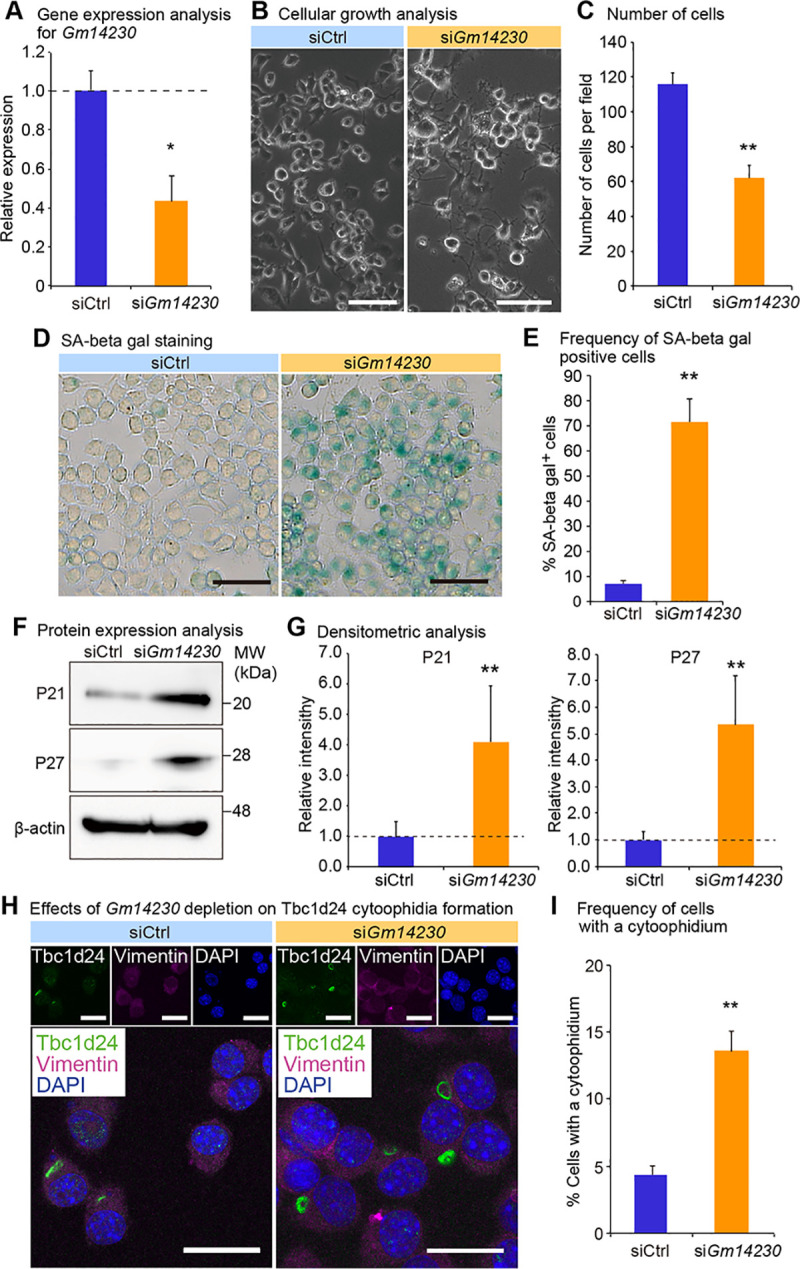
Gm14230 controls cellular juvenescence and Tbc1d24 cytoophidia.
(A) qPCR analysis of Gm14230 in Neuro2a cells transfected with control siRNA (siCtrl) or Gm14230 siRNA. Data were normalized to the expression of Tubb5 (n = 3). (B) The appearance of Neuro2a cells 72 hrs after transfection of control siRNA or Gm14230 siRNA. Scale bar = 100 μm. (C) Number of cells per field 72 hrs after transfection with control siRNA or Gm14230 siRNA. (D) Senescence-associated (SA) beta-galactosidase activity staining in Neuro2a cells 72 hrs after transfection with control siRNA or Gm14230 siRNA. Scale bar = 100 μm. (E) Number of SA beta-gal-positive cells per field 72 hrs after transfection with control siRNA or Gm14230 siRNA. (F) Western blot analysis of Neuro2a cells 72 hrs after transfection with control siRNA or Gm14230 siRNA. β-actin (Actb) was used as a loading control. MW, molecular weight. (G) The densitometric analysis for western blot analyses from which the representative images were shown as Fig 3F. The intensity of the bands was quantified and normalized to those of Actb. The ratios to Actb were further normalized to siCtrl. (H) Immunofluorescence analysis of Tbc1d24 and Vimentin in Neuro2a cells transfected with control siRNA or Gm14230 siRNA. Scale bar = 25 μm. (I) Frequency of Tbc1d24 cytoophidia in Neuro2a cells transfected with control siRNA or Gm14230 siRNA. *p<0.05 and **p<0.01; Student’s t-test. The data were presented as the means ± SEM.
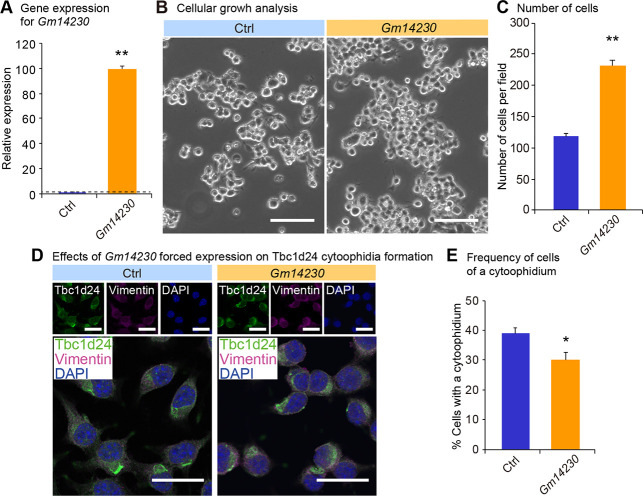
Influence of Gm14230 expression on Tbc1d24 cytoophidia formation. (A) qPCR analysis of Gm14230 in Neuro2a cells transfected with the empty control or the Gm14230 plasmid. Data were normalized to Tubb5 (n = 3). (B) The appearance of Neuro2a cells 72 hrs after transfection of the empty control or the Gm14230 plasmid. Scale bar = 100 μm. (C) Number of cells per field 72 hrs after transfection of the empty control or the Gm14230 plasmid. (D) Immunofluorescence analysis of Tbc1d24 and Vimentin in Neuro2a cells transfected with the empty control or the Gm14230 plasmid. Scale bar = 100 μm. (E) Frequency of Tbc1d24 cytoophidia in Neuro2a cells transfected with the empty control or the Gm14230 plasmid. *p < 0.05 and **p < 0.01; Student’s t-test. The data were presented as the means ± SEM.
Inspired by the finding that cytoophidia formation was induced by the loss of cellular juvenescence, we next asked the biological role of cytoophidia in the juvenescence-losing cells. Forced expression of Tbc1d24 ameliorated the growth impairment caused by Gm14230 depletion, implying the role of Tbc1d24 to resist the progression of the loss of cellular juvenescence (S5 Fig). To further assess the resistive role of Tbc1d24 in the juvenescence loss, we next asked the effect of Tbc1d24 suppression in the context of Gm14230 depletion. Simultaneous knockdown of Gm14230 and Tbc1d24 resulted in synergistic effects on the cell growth impairment (S6A and S6B Fig) and cell toxicity (S6C and S6D Fig), corroborating that Tbc1d24 constitutes a protective machinery against the juvenescence loss.
Zeocin-induced loss of cellular juvenescence increases Tbc1d24 cytoophidia
To further evaluate the role of cytoophidia in the cellular juvenescence, we utilized the assay system in which cells were treated with a genotoxic agent zeocin to induce the loss of cellular juvenescence [2, 45]. Zeocin treatment provoked cell growth impairment and cytoplasmic enlargement, suggesting that cells lost cellular juvenescence (Fig 5A and 5B). Tbc1d24 cytoophidia were more frequently observed in zeocin-treated cells compared to control cells (Fig 5C and 5D). Kinetics for zeocin treatment-induced cytoophidia were analyzed by the time course experiment as follows. Longer incubation with zeocin resulted in more obvious cellular shape changes that were accompanied by more frequent (S7A and S7B Fig) and longer (S7A and S7C Fig) cytoophidia formation, indicating the positive correlation of the juvenescence loss and cytoophidia formation. The finding of time-dependent increment of the cytoophidia led us to a question whether the Tbc1d24 protein half-life was affected by the cytoophidia formation. The cycloheximide (CHX) chase assay for Tbc1d24 protein showed elongated half-life of Tbc1d24 protein in zeocin-treated cells compared to control cells, suggesting that the enhanced stability of protein contributed to the larger cytoophidia (S8 Fig).
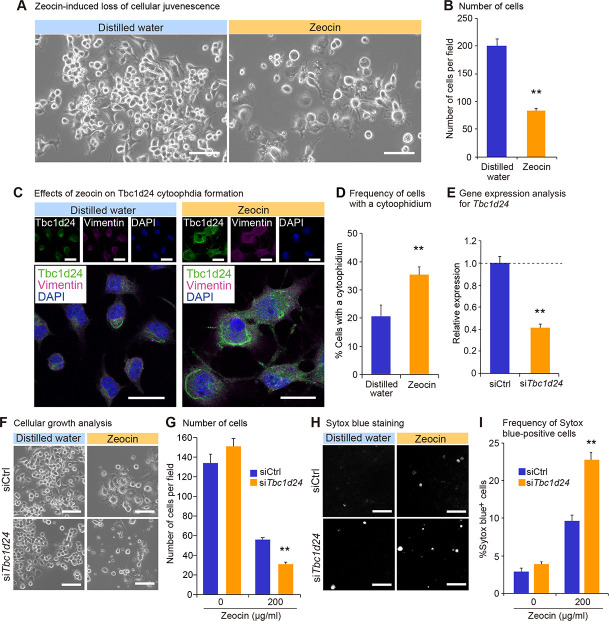
Tbc1d24 cytoophidia formation is enhanced in zeocin-induced loss of cellular juvenescence.
(A) The appearance of Neuro2a cells 72 hrs after treatment with zeocin. Scale bar = 100 μm. (B) Number of cells per field 72 hrs after treatment with or without zeocin. (C) Immunofluorescence analysis of Tbc1d24 and Vimentin treated with or without zeocin for 72 hrs. Scale bar = 25 μm. (D) Frequency of cytoophidia in Neuro2a cells with or without zeocin treatment for 72 hrs. (E) qPCR analysis of Tbc1d24 in Neuro2a cells transfected with control siRNA or Tbc1d24 siRNA. Data were normalized to Tubb5 (n = 3). (F) The appearance of Neuro2a cells treated with or without zeocin for 96 hrs and simultaneously transfected with control siRNA or Tbc1d24 siRNA. Scale bar = 100 μm. (G) Number of cells per field 96 hrs after treatment with or without zeocin. (H) The appearance of cells with Sytox blue staining in the same fields as in (F). The gray color dots indicate dead cells stained with Sytox blue. (I) Frequency of Sytox blue-positive cells. **p < 0.01; Student’s t-test. The data were presented as the means ± SEM.
To assess a resistive role of Tbc1d24 in zeocin-induced juvenescence loss, we first tested the effect of Tbc1d24 forced expression. The transfection of Tbc1d24 alleviated cellular growth impairment and morphological changes induced by zeocin treatment, indicating Tbc1d24 plays a protective role against the cytotoxicity provoked by zeocin-induced juvenescence loss (S9 Fig). Furthermore, we evaluated the effect of the depletion of Tbc1d24 on the zeocin-induced juvenescence loss (Fig 5E). The impairment of cellular growth induced by zeocin was exacerbated by Tbc1d24 depletion, corroborating the cytoprotective function of Tbc1d24 (Fig 5F and 5G). The detection of dead cells by Sytox blue staining revealed higher toxicity with Tbc1d24-depleted cells compared to control siRNA-transfected cells, suggesting Tbc1d24 protected cells from the cellular stress given by the zeocin-induced juvenescence loss (Fig 5H and 5I). These findings suggested the cytoprotective role of Tbc1d24 cytoophidia in the zeocin-induced loss of cellular juvenescence.
Tbc1d24 GAP enzymatic activity is impaired by cytoophidia formation
Lastly, we asked whether the cytoprotective role of Tbc1d24 was a consequence of alteration of GAP enzymatic activity. We interrogated the GAP activity of Tbc1d24 by examining GTP-bound status of Arf6, a substrate of Tbc1d24 [3]. The increased Tbc1d24 cytoophidia induced by Gm14230 depletion was correlated with impaired GAP activity of Tbc1d24, indicating that Tbc1d24 GAP activity was suppressed by cytoophidia formation (Fig 6A and 6B). We next evaluated the correlation of the magnitude of cytoophidia formation and the GAP activity. To assess the correlation between the cytoophidia size and the GAP activity, cells were analyzed for cytoophidia formation and GAP activity in a time course-dependent manner after Gm14230 depletion. This analysis revealed the time course-dependent impairment of Tbc1d24 GAP activity (S10A and S10B Fig) that was accompanied with cytoophidia growth in number (S10C and S10D Fig) and size (S10C and S10E Fig). These findings suggest that the cytoophidia formation was associated with impairment of Tbc1d24 activity.
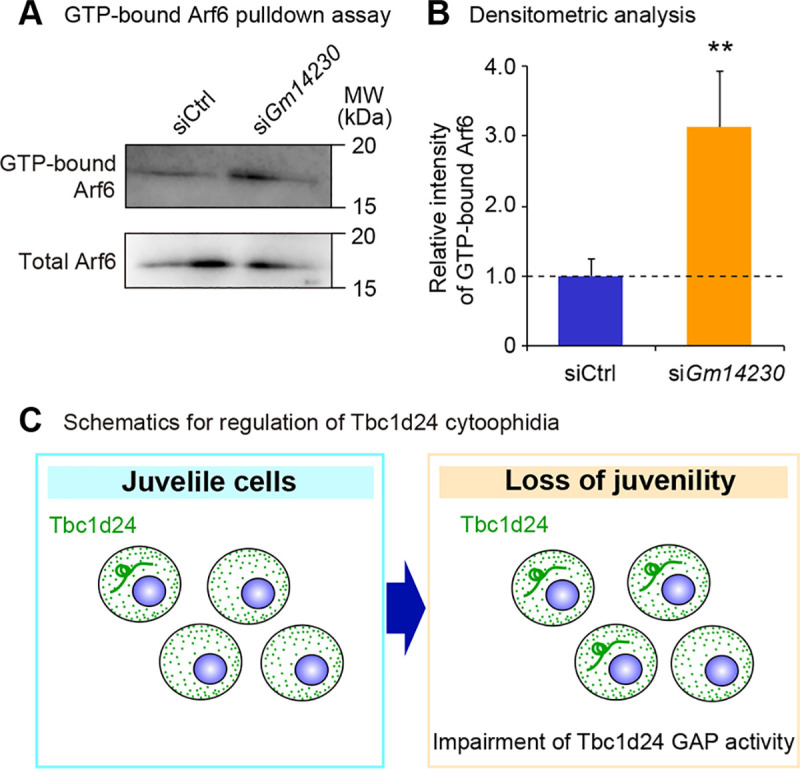
Lower GAP activity of Tbc1d24 is associated with cytoophidia formation.
(A) GTP-bound Arf6 pulldown assay in Neuro2a cells transfected with control siRNA or Gm14230 siRNA. Total Arf6 levels were examined to evaluate the equal expression of Arf6 protein after Gm14230-depletion. (B) The densitometric analysis for western blot analyses from GTP bound Arf6 pulldown assay. The intensity of the bands was quantified and normalized to those of total Arf6. The ratios to total Arf6 were further normalized to siCtrl. **p < 0.01; Student’s t-test. Data were represented as the means ± SEM. (C) Schematics for the role of Tbc1d24 cytoophidia to control GAP activity in a cellular juvenescence-dependent manner.
Together, our analyses reveal the Tbc1d24 protein forms cytoophidia in neuronal cells both in vitro and in vivo. The cytoophidia formation is controlled by JALNC Gm14230 and is induced upon the loss of cellular juvenescence caused by Gm14230 depletion and zeocin treatment. Depletion of the cytoophidia exacerbates the cell toxicity provoked by loss of the cellular juvenescence. The cytoophidia formation was associated with impaired enzymatic activity of Tbc1d24 (Fig 6C).
Discussion
We in this paper described that Tbc1d24 protein formed cytoophidia in neuronal cells and that Tbc1d24 cytoophidia possessed a protective role against the cell stress provoked by loss of cellular juvenescence. The formation of Tbc1d24 cytoophidia was augmented by the loss of cellular juvenescence induced by depletion of JALNC Gm14230 and treatment with zeocin, a genotoxic agent. The cytoprotective function of Tbc1d24 was revealed by synergistic effect of Tbc1d24 depletion on the cell toxicity caused by Gm14230 depletion and zeocin treatment. The GAP activity of Tbc1d24 was impaired by cytoophidia formation, indicating that cytoophidia formation was associated with suppressed Tbc1d24 activity. Thus, Tbc1d24 constitutes the cell stress-coping machinery under the control of Gm14230.
The cytoprotective property of Tbc1d24 cytoophidia was demonstrated by the experiment in which cells depleted of Tbc1d24 were more susceptible to zeocin toxicity. Loss of cellular juvenescence caused by Gm14230 depletion or zeocin treatment significantly increased cytoophidia formation, indicating cytoophidia formation was suppressed in juvenile cells. To assess the physiological function of Tbc1d24 cytoophidia, the cell viability was tested in Tbc1d24-depleted cells after zeocin treatment. More dead cells were found among Tbc1d24-depleted cells compared to control siRNA-transfected cells, suggesting a protective role of Tbc1d24 when cells were induced to lose cellular juvenescence. Gm14230 may function as a stress sensor that mediates the stress signals to the cytoophidia formation. As the protein that is highly expressed in juvenile cells, Tbc1d24 may function to maintain cellular juvenescence by forming the cytoophidia.
As above, Tbc1d24 cytoophidia may function as the stress-response element and protect cells from zeocin treatment- and Gm14230-depletion-induced loss of the cellular juvenescence. This idea was further supported by the findings that Tbc1d24-depletion increased cellular toxicity upon Gm14230 loss (S6 Fig) and that TBC1D24 forced expression rescued the cell toxicity of zeocin treatment (S9 Fig).
The filamentous macrostructure of Tbc1d24 cytoophidia were similar to that of Impdh and Ctps cytoophidia, but we demonstrated the Tbc1d24 cytoophidia were distinct from the Impdh and Ctps cytoophidia by a series of experiments. Treatment with DON, MPA and Acivicin suppressed Tbc1d24 cytoophidia formation without affecting Tbc1d24 protein levels. These compounds decrease intracellular GTP through Impdh inhibition. The lower GTP levels may affect cytoophidia formation through modulating the Tbc1d24 enzymatic activity. Further experiments are required to assess the association with intracellular GTP levels and Tbc1d24 molecular behaviors.
We consider that Tbc1d24 cytoophidia are distinct from mere protein aggregations and function as the reservoir of the enzyme to cope with cellular stresses. Our work suggests that Tbc1d24 cytoophidia function as a reservoir of Tbc1d24 protein to adjust GAP activity. Ctps cytoophidia were reported to correlate with both negative [27, 46] and positive [47] activities. Impdh cytoophidia were correlated with positive activity [41]. Our data show a negative correlation between Tbc1d24 cytoophidia formation and GAP activity. This finding suggests that Tbc1d24 cytoophidia functions as a Tbc1d24 reservoir to store Tbc1d24 in an inactive form in the context of the cellular stress response.
Mutations in TBC1D24 have been discovered in patients with EE, a severe brain syndrome that develops in juvenile in humans. Patients with EE exhibit cognitive, behavioral and neurological deficits. The exact genotype-phenotype correlation is under investigation [48]. Most of the affected individuals possess homozygous mutations in TBC domain or compound heterozygous mutations in TBC or TLDc domains. Our findings imply a therapeutic potential of targeting Tbc1d24 cytoophidia in neuronal cells for the treatment of EE. Mutations in TBC1D24 may affect cytoophidia formation, compromising the cellular stress response. It might be possible to induce TBC1D24 cytoophidia via a compound to cope better with the cellular stresses, which may lead to a new therapeutic approach for the detrimental neurological disorders.
We revealed the property of Tbc1d24 protein to form cytoophidia in response to loss of cellular juvenescence. Tbc1d24 cytoophidia formation was augmented by depletion of JALNC Gm14230 and the genotoxic agent zeocin. Depletion of Tbc1d24 cytoophidia enhanced cellular toxicity against zeocin treatment, indicating the cytoprotective role for Tbc1d24 cytoophidia. Cytoophidia formation correlated with impairment of GAP activity, thus implying that Tbc1d24 cytoophidia function as the repressive machinery for Tbc1d24 enzymes. Collectively, our findings illustrate the new role of Tbc1d24 for cytoophidia formation and stress response, illuminating a novel therapeutic approach for the neurological disorders.
Acknowledgements
We would like to thank all the researchers of National Cerebral and Cardiovascular Center Research Institute for their helpful discussions and sincere cooperation. This study was supported by the Central Research Laboratory at Shiga University of Medical Science.
References
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48