 A convenient approach to synthesize substituted 5-Arylidene-3-m-tolyl thiazolidine-2, 4-diones by using morpholine as a catalyst and its theoretical study
A convenient approach to synthesize substituted 5-Arylidene-3-m-tolyl thiazolidine-2, 4-diones by using morpholine as a catalyst and its theoretical study
PLoS ONE


Competing Interests: The authors have declared that no competing interests exist.

- Altmetric
Thiazolidinediones are very important and used as a drug for the treatment of type 2 diabetes. Here, we report a convenient approach to synthesis 3-m-tolyl-5-arylidene-2,4-thiazolidinediones (TZDs) derivatives 7a-e in two steps with moderate to good yield using morpholine as a catalyst. All the structures were confirmed by their spectral IR, 1H NMR and 13C NMR data. The anti-diabatic activity of all synthesized molecules is evaluated by docking with peroxisome proliferator-activated receptor-γ (PPARγ). Preliminary flexible docking studies reveals that our compounds 7a, 7d and 7e showed better binding affinity with the protein and could be a potential candidate for the treatment of type 2 diabetes in near future.
Introduction
Heterocyclic compounds play an important role in medicinal chemistry. Specifically, nitrogen-containing heterocycles with a sulfur atom such as, thiazolidine-2,4-dione (TZDs, Fig 1). TZDs represent an important class of compounds showed a wide spectrum of biological and pharmacological activities [1,2]. In the past decades, TZDs have been the subject of extensive research because of their involvement in the regulation of different physiological processes which has been confirmed by numerous reviews [3,4]. A variety of compounds having TZDs core (Fig 1) were used for the treatment of type II diabetes and related diseases [5]. Thiazolidinediones showed antidiabetic activity by binding with gamma form of the peroxisome proliferator-activated receptor-γ (PPARγ). This stimulates peripheral adiposities to increase their uptake of free fatty acids, which leads to reduction in the fat stored in muscles, liver and visceral fat deposits. The TZDs also leads to an increase in the secretion of adiponectin and a decrease in the production of inflammatory mediators such as tumor necrosis factor-alpha (TNF-α), plasminogen activator inhibitor-1(PAI-1) and interleukin-6 (IL-6). This feature lead TZDs act as an aldose reductase inhibitor and tumor inhibitor [6,7]. Moreover, chemical modification on this heterocycle led to a class of compounds that possess several biological activities including, anti-inflammatory effects [8], antibacterial [9–12], antitubercular [13], and antifungal activity [9]. TZDs target vascular cells and monocytes/macrophages to inhibit the production of pro-inflammatory cytokines [14–17] as well as the development of oxidative stress [18] and cell adhesion molecules. They are also key intermediates for the synthesis of anti-HIV and anti-ischemic agents [19–21]. Therefore, simple structure and valuable pharmacological activity of these molecules gained special attention from synthetic chemists and pharmacologists [20].

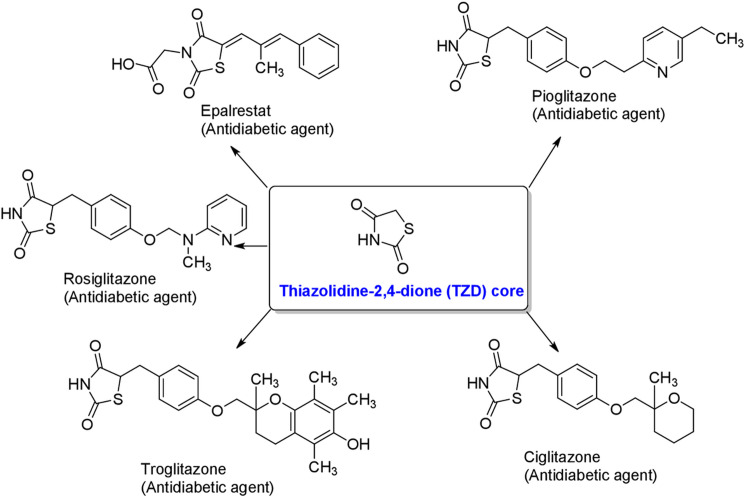
Thiazolidine-2,4-dione (TZD) and its derivatives.
Several methods have been reported up to date for the synthesis of thiazolidine-2,4-dione and its derivatives with an extensive range of catalysts including Alum [22], glycine, sodium carbonate, piperidinium acetate [23], piperidine [24], amines salts [25], baker’s yeast [26], ionic liquids [27–31] and nanoparticle-supported copper (II) catalysts [32]. However, several of these existing methods suffer from one or more drawbacks, such as low yields, long reaction times, environmentally unfavorable solvents, and a requirement for excess catalyst. Hence, a facile efficient process is still desirable. As a continuation our research goal to develop a convenient method to synthesize valuable synthon [33–36] from easily available starting materials, we developed a two-step method to synthesize thiazolidine-2,4-dione derivatives by using morpholine as a catalyst. We have performed Density functional theory (DFT) calculations in all our synthesized molecules to find out most active compound. To the best of our knowledge, the use of morpholine as a catalyst for the synthesis of 3-m-tolyl-5-arylidenthiazolidine- 2,4-diones as well as DFT calculations has not been previously reported.
Experimental
Chemistry
General
All products were characterized by IR, UV, 1H-NMR and 13C-NMR. Thin layer chromatography (TLC) was carried out on plates coated with silica gel (Merck, Silica Gel G) and spots were detected with iodine vapour. The UV spectra was recorded using SHIMADZU UV-160A spectrophotometer using chloroform as solvent. Infra-Red spectra was recorded using SHIMADZU IR-470A spectrophotometer by direct transmittance using KBr pellets. Multinuclear NMR (1H, 13C, DEPT-135) spectra were recorded on a BRUKER 400 MHz NMR spectrophotometer. Chemical shifts are reported in parts per million (ppm). Tetramethyl silane (TMS) served as an internal standard in 1H and 13C NMR (δ 0.00 ppm). All the chemicals used for the synthesis of target compounds have been purchased from Sigma Aldrich and were used as received. Scanned Spectra for all compounds are shown in S1–S29 Figs.
General procedure for the synthesis of 3-(m-tolyl)thiazolidine-2,4-dione (4)
A solution of m-tolylthiourea, (1, 5.0g, 30.0 mmol), 2-chloroacetic acid, (2, 2.8 g, 30.0 mmol) and 20mL hydrochloric acid 3 (30%, (ν/ν)) were stirred for half an hour and then refluxed for overnight at 120°C in a 250mL round-bottomed flask. After completion of the reaction, as monitored by TLC (2:1, Pet ether: Ethyl acetate), the reaction mixture was gradually cooled to room temperature. Then it was neutralized with dilute sodium hydroxide (0.1 M). Immediately a solid mass was formed which was filtered under reduce pressure, washed with cold water (50mL) and recrystallized from absolute alcohol to give a pure product 4 as a white crystal (2.49g, 40% yield).
General procedure for the synthesis of 5-arylidene-3-(m-tolyl)-thiazolidine-2,4-diones (7a-e)
For each experiment, 20mmol of compound 4 and 20 mmol of substituted benzaldehydes 5a-e was dissolved in absolute alcohol (10 mL) and refluxed with the Dean-Stark attachment for 2 hours by using morpholine 6 (10 mol%) as a catalyst. After completion of the reaction, as judged by TLC, the reaction was quenched with crushed ice. The crude product was filtered under reduced pressure, washed with cold water and purified by recrystallization using absolute alcohol. The yield of the products 7a-e ranged from 55–90%.
Spectral data
3-(m-tolyl) thiazolidine-2, 4- dione, Compound (4). Yield: 40%; Rf: 0.81; white crystal; UV (λmax nm): 274.50; IR (KBr) (νmax cm-1): 1760, 1688 (C = O, TZD ring), 1521, 1198 (-CH2 stretch), 761; 1H NMR (CDCl3, 400MHz, δ ppm): 7.43–7.32 (m, 3H), 7.13 (d, J = 7.6 Hz, 1H), 4.17 (d, J = 1.6 Hz, 2H), 2.203, (s, 3H); 13C NMR (CDCl3, 100MHz, δ ppm): 170.56, 170.43, 136.00, 131.97, 131.36, 130.09, 128.28, 127.25, 33.99, 17.52.
5-(2-Methoxybenzylidene)-3-m-tolyl thiazolidine-2, 4- dione (7a). Yield: 84% (morpholine 11%); Rf: 0.34; yellow crystals; UV (λmax nm): 358.50; IR (KBr) (νmax cm-1): 1742, 1690 (C = O, TZD ring); 1H NMR (CDCl3, 400 MHz, δ ppm): 8.37 (s, 1H), 7.54–6.99 (m, 7H), 3.94 (s, 3H), 2.24 (s, 3H); 13C NMR (CDCl3, 100 MHz, δ ppm): 167.43, 165.61, 158.56, 136.26, 132.39, 132.13, 131.29, 130.47, 130.33, 129.94, 129.53, 128.43, 127.14, 121.31, 120.94, 111.23, 55.54, 17.65.
5-(2-Chlorobenzylidene)-3-m-tolyl thiazolidine-2, 4- dione (7b). Yield: 88%; Rf: 0.50; green crystals; UV (λmax nm): 358.50; IR (KBr) (νmax cm-1): 1734, 1675 (C = O, TZD ring); 1H NMR (CDCl3, 400 MHz, δ ppm): 8.35 (s, 1H), 7.65–7.21 (m, 7H), 2.26 (s, 3H); 13C NMR (CDCl3, 100 MHz, δ ppm): 166.59, 164.92, 136.20, 136.06, 131.85, 131.80, 131.47, 131.37, 130.69, 130.54, 130.11, 128.35, 127.34, 127.22, 124.42, 17.66.
5-(2-Nitrobenzylidene)-3-m-tolyl thiazolidine-2, 4- dione (7c). Yield: 67%; Rf: 0.22; light brown crystals; UV (λmax nm): 332.00 and 243.50; IR (KBr) (νmax cm-1): 1690, 1607 (C = O TZD ring), 1352(-NO2); 1H NMR (CDCl3, 400 MHz, δ ppm): 7.99 (s, 1H), 7.50–7.20 (m, 7H), 2.45 (s, 3H); 13C NMR (CDCl3, 100 MHz, δ ppm): 166.97, 165.63, 141.47, 136.23, 134.60, 132.03, 131.33, 130.53, 130.39, 130.06, 130.01, 127.19, 120.07, 17.64.
5-(3-Hydroxybenzylidene)-3-m-tolyl thiazolidine-2, 4- dione, Compound (7d). Yield: 54%; Rf: 0.45; dark-brown powder; UV (λmax nm): 328.00;; 1H NMR (CDCl3, 400 MHz, δ ppm): 7.96 (s, 1H), 7.53 (s, 4H), 7.51–7.35 (m, 3H), 7.22–7.19 (d, J = 7.6 Hz, 1H), 2.24 (s, 3H); 13C NMR (CDCl3, 100 MHz, δ ppm): 166.36, 165.32, 136.83, 136.18, 132.95, 131.87, 131.73, 131.39, 130.13, 129.64, 128.36, 127.25, 121.97, 17.63.
5-(4-Chlorobenzylidene)-3-m-tolyl thiazolidine-2, 4- dione, Compound (7e). Yield: 74%; Rf: 0.12; off-white crystals; UV (λmax nm): 334.50; IR (KBr) (νmax cm-1): 1857 and 1783 (C = O TZD ring); 1H NMR (CDCl3, 400 MHz, δ ppm): 7.96 (s, 1H), 7.52 (s, 4H), 7.43–7.37 (m, 3H), 7.21–7.19 (d, J = 7.6 Hz, 1H), 2.24 (s, 3H); 13C NMR (CDCl3, 100 MHz, δ ppm): 166.37, 165.32, 136.85, 136.17, 132.96, 131.84, 131.72, 131.38,131.36, 130.12, 129.64, 128.34, 127.24, 121.96, 17.61.
Computational methods
DFT studies
All theoretical calculations were performed by using density functional theory (DFT), B3LYP (6-31G, d) basis set in Gaussian 09 Program suite. A full geometry optimization was performed for all structures, using this function and all geometries were visualized using Avogadro 1.2 software package.
Preparation of the protein
Crystal structure of human peroxisome proliferator-activated receptor gamma (PPARγ) [37] (PDB ID: 3GBK) was downloaded from RCSBPDB webpage (www.rcsb.org) and opened on PyMol (version 1.3) and waters and the agonist were removed from the crystal structure. The protein structure was then subjected to geometry and energy minimization in Swiss-PDBViewer (version 4.1.0) using GROMOS96 force field. The crystal structure of the protein after energy minimization was shown in Fig 2. The crystal structure was then saved as a.pdb file and used for molecular docking against the optimized structures of 4 and 7a-e.

The X-ray crystal structure of PPARγ (PDB ID: 3GBK).
Molecular docking
Docking of the optimized structures were performed against 3GBK using AutoDock Vina. As a control, the commercially available type 2 anti-diabatic drug, Epalrestat was used. Only flexible docking was performed where the ligands were flexible, but the protein was held rigid. The dimension of the grid-box for the protein was set to 73.5450, 60.6299, 79.5208 Å as the X-, Y- and Z-coordinates respectively. After docking, all generated files were collected and their non-covalent interactions with the energy minimized protein was evaluated in Accelerys Discovery Studio 4.
Results and discussion
Chemistry
The synthesis of the new TZD derivatives had two steps. At first phenyl thiourea 1 reacts with chloroacetic acid 2 in the presence of hydrochloric acid 3 (30 mol%, (ν/ν)) as a catalyst to produce the desired 3-(m-tolyl) thiazolidine-2,4-dione 4. Then the active methylene in position 5 of compound 4 undergo Knoevenagel condensations with various aromatic aldehydes 5a-e in the presence of morpholine 6 to form compounds 7a–e (S33 Fig). The yield in the first step is low because of the presence of unreacted starting material and formation of potential by product 3-toluidine (mechanism presented in S30 Fig). In second step, it should be noted that both electron-withdrawing and electron-donating groups in aromatic aldehydes are quite suitable and give the desired product good to moderate yield, except compound 7d, where the presence of hydroxy group, the reaction become sluggish and it is difficult to separate the product.
After completing the reaction, all the products are recrystallized by using suitable solvent and checked by TLC. All the structures were confirmed by their spectral data (IR, UV, 1H NMR, and 13C NMR). In compound 4, the IR spectra exhibited characteristic absorption bands at 1680–1690 cm-1 and 1755–1765 cm-1 due to the two C = O from the TZD heterocycle. The 1H NMR spectra showed characteristic doublets due to presence of C (5)-H in compound 4 at 4.169 and 4.173 ppm respectively which is further confirmed by DEPT spectra shown is S5 Fig. The condensation reaction between 4 and 5 formed compound 7, where The C (5)–H in thiazole disappeared and a characteristic peak appeared around 8.0 ppm indicate the conversion of the methylene group to C = C double bond. Both 13C NMR and DEPT-135 data supported the conversion of the methylene group to C = C double bond.
DFT studies: Thermodynamic results
Compounds 4 and 7a-e were optimized to obtain their most thermodynamically favorable configuration where Epalrestat was used as a control compound. The thermodynamic data of all synthesized compounds have negative electronic energy, enthalpy and Gibbs-free energy suggesting that all the synthesized molecules are thermodynamically stable (Table 1). Compound 4 is thermodynamically less stable than compared to the other compounds 7a-e including Epalrestat. In compounds 7a-e, all compounds have similar energy except 7b and 7e which have lower energy because of the presence of Cl atom, hence, more stable. It should be noted that compounds 7a-e after Knoevenagel condensation develop an alkene bond which stabilized the molecule by delocalized pi-orbitals hence favor lower energy. In order to find out the binding energy and hydrogen bonding capability, the dipole moment of all synthesized compounds was determined (S31 Fig and Table 1). The range of dipole moment for the synthesized molecules is in between 0.5–5.5 Debye, where 7e has the lowest dipole moment because of the presence chlorine atom at para-position and most for Epalrestat because it contains polar groups such as a carboxyl.

| Compound | Internal Energy, E | Enthalpy, ΔH | Free energy, ΔG | Dipole Moment, μ (D) |
|---|---|---|---|---|
| Epalrestat | -1333.010 | -1333.009 | -1333.080 | 5.5006 |
| 4 | -989.721 | -989.720 | -989.774 | 1.0328 |
| 7a | -1373.259 | -1373.258 | -1373.332 | 4.1495 |
| 7b | -1718.372 | -1718.371 | -1718.443 | 3.0389 |
| 7c | -1463.259 | -1463.258 | -1463.333 | 5.1613 |
| 7d | -1333.984 | -1333.983 | -1334.053 | 1.3643 |
| 7e | -1718.376 | -1718.375 | -1718.446 | 0.4355 |
Electrostatic results
To find out the reactivity of the molecule towards its receptor, HOMO-LUMO and hardness and softness of all synthesized molecules were calculated (S32 Fig and Table 2). In the Frontier molecular orbital, large energy gap between HOMO-LUMO indicates that the molecule is more stable and less reactive, where low energy gap suggests easier electronic transition and favor quicker reaction. All the synthesized compounds 7a-e except compound 4 have similar orbital energy gap suggesting similar reactivity towards its receptor.
| Compounds | HOMO (a.u) | LUMO (a.u) | Orbital energy gap (a.u) | Softness | Hardness |
|---|---|---|---|---|---|
| Epalrestat | -0.21876 | -0.08698 | 0.13178 | 15.177 | 0.06589 |
| 4 | -0.24558 | -0.03353 | 0.21205 | 9.4312 | 0.10603 |
| 7a | -0.21447 | -0.07913 | 0.13534 | 14.778 | 0.06767 |
| 7b | -0.23280 | -0.08768 | 0.14512 | 13.782 | 0.07256 |
| 7c | -0.24179 | -0.10205 | 0.13929 | 14.358 | 0.06965 |
| 7d | -0.22289 | -0.08649 | 0.13640 | 14.663 | 0.06820 |
| 7e | -0.22956 | -0.09098 | 0.13858 | 14.432 | 0.06929 |
Docking studies
Binding affinity is a measure of how strongly a molecule can fit into a receptor and interact with it through non-covalent bonding. To find out the binding affinity as well as protein interaction, we performed docking of all our synthesized compounds 4 and 7a-e with Peroxisome proliferator-activated receptor gamma (PPAR-γ or PPARG) by considering Epalrestat as a reference (Figs 3–9 and Table 3). In result, three compounds 7a, 7d and 7e showed better binding affinity with the protein having energy -8.2, -8.5 and -8.9 kcal/mol respectively than compared to Epalrestat which has a binding affinity of -7.9 kcal/mol and hence showed better non-covalent interactions. Compound 4 has -6.9 kcal/mol of energy when bound to the receptor. This result indicates that the binding pocket of the protein is quite large so the structure bigger than 4 would have more interactions when bound to the protein. This was true for all our compounds 7a-e which had higher binding affinity than 4. However, Compound 7b and 7c have lower binding affinity than Epalrestat which indicate that the ortho position may not be a good site for better binding affinity. Only exception was observed with 7a, where the methoxy group at ortho-position had no interaction with the protein. This result suggests that a good choice in a substituent may induce better binding. Compound 7e has better binding where p-Cl atom interacts with the protein. This finding suggests that adding larger groups in the para position may lead to higher binding affinity.
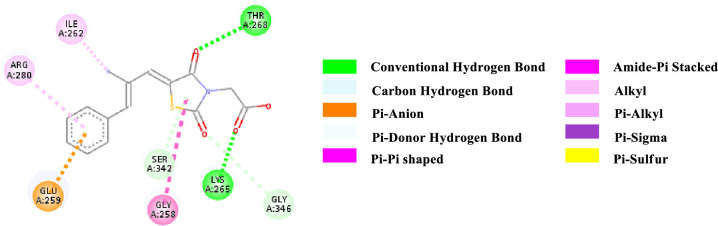
2D-representation of the non-covalent interactions of Epalrestat with receptor (3gbk) after flexible docking.
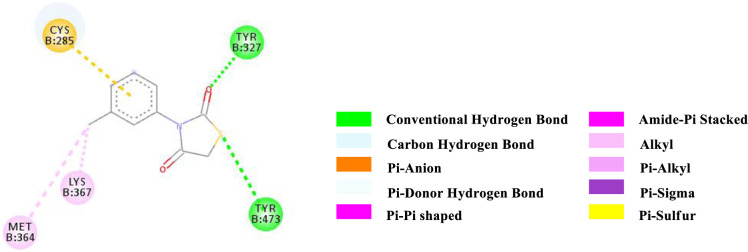
2D-representation of the non-covalent interactions of 4 and receptor (3gbk) after flexible docking.
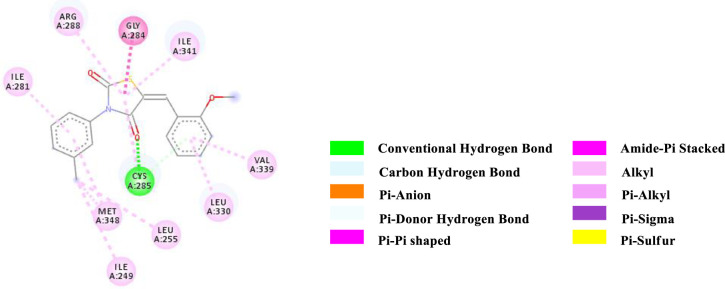
2D-representation of the non-covalent interactions of 7a with receptor (3gbk) after flexible docking.
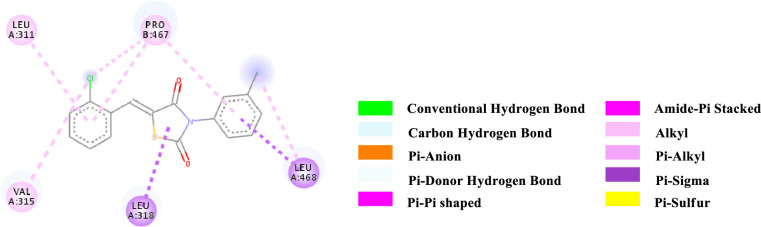
2D-representation of the non-covalent interactions of 7b with receptor (3gbk) after flexible docking.

2D-representation of the non-covalent interactions of 7c with receptor (3gbk) after flexible docking.
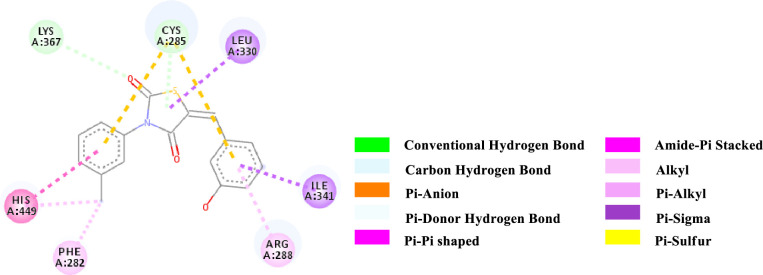
2D-representation of the non-covalent interactions of 7d with receptor (3gbk) after flexible docking.
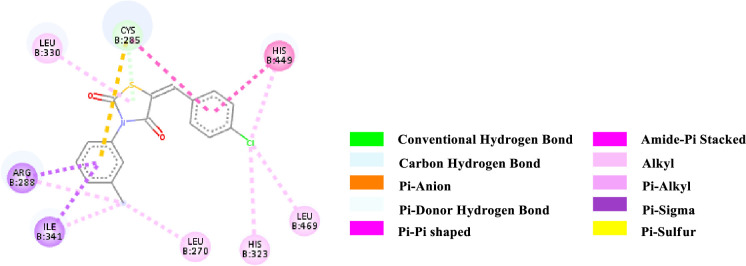
2D-representation of the non-covalent interactions of 7e with receptor (3gbk) after flexible docking.
| Compounds | Binding Affinity, kcal/mol | Type of Non-bonding Interaction | From | To | Bond Distance, Å |
|---|---|---|---|---|---|
| Epatrestat | -7.9 | HB | Lys 265 (NH) | C = O (ligand) | 3.06067 |
| HB | Thr 268 (OH) | C = O (4th position of TZD) | 3.21220 | ||
| CHB | Gly 346 (αCH) | C = O (2nd position of TZD) | 3.64643 | ||
| PA | Glu 259 (COO-) | Benzene (ligand) | 3.64293 | ||
| PDHB | Ser 342 (OH) | TZD core | 3.72507 | ||
| APS | Gly 258—Glu 259 (CONH) | TZD core | 4.20658 | ||
| Alkyl | Methyl (ligand) | Ile 262 (side chain) | 5.38888 | ||
| Pi-Alkyl | Benzene (ligand) | Arg 280 (side chain) | 4.88631 | ||
| 4 | -6.1 | HB | Tyr 327 (OH) | C = O (2nd position of TZD) | 3.12856 |
| HB | Tyr 473 (OH) | TZD (Sulfur) | 3.31594 | ||
| PS | Cys 285 (SH) | N-Aryl (ligand) | 4.25927 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Met 364 (side chain) | 5.29768 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Lys 367 (side chain) | 4.27732 | ||
| 7a | -8.2 | HB | Cys 285 (SH) | C = O (4th position of TZD) | 3.71078 |
| PDHB; PS | Cys 285 (SH) | 2-OMe (Aryl); TZD core (ligand) | 4.11671 | ||
| APS | Gly 284; Cys 285 (CONH) | TZD core (ligand) | 3.90759 | ||
| Alkyl | Methyl (Aryl; ligand) | Ile 249 (side chain) | 4.68460 | ||
| Alkyl | Methyl (Aryl; ligand) | Leu 255 (side chain) | 5.06828 | ||
| Alkyl | Methyl (Aryl; ligand) | Met 348 (side chain) | 5.05667 | ||
| Pi-Alkyl | 2-OMe (Aryl; ligand) | Cys 285 (side chain) | 5.40245 | ||
| Pi-Alkyl | TZD core (ligand) | Arg 288 (side chain) | 4.94846 | ||
| Pi-Alkyl | TZD core (ligand) | Ile 341 (side chain) | 4.27639 | ||
| Pi-Alkyl | 2-OMe (Aryl; ligand) | Leu 330 (side chain) | 4.87080 | ||
| Pi-Alkyl | 2-OMe (Aryl; ligand) | Val 339 (side chain) | 5.01580 | ||
| Pi-Alkyl | N-Aryl (ligand) | Ile 281 (side chain) | 4.99136 | ||
| Pi-Alkyl | N-Aryl (ligand) | Met 348 (side chain) | 5.24193 | ||
| 7b | -7.3 | PS | Leu 318 (Methyl, side chain) | TZD core (ligand) | 3.63093 |
| PS | Leu 468 (Methyl, side chain) | N-Aryl (ligand) | 3.63461 | ||
| PS | Leu 468 (Methyl, side chain) | N-Aryl (ligand) | 3.96613 | ||
| Alkyl | 2-Cl-Aryl (ligand) | Val 315 (side chain) | 5.14187 | ||
| Alkyl | 2-Cl-Aryl (ligand) | Pro 467 (side chain) | 4.68961 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Leu 468 (side chain) | 4.69788 | ||
| Pi-Alkyl | 2-Cl-Aryl (ligand) | Leu 311 (side chain) | 5.49394 | ||
| Pi-Alkyl | 2-Cl-Aryl (ligand) | Pro 467 (side chain) | 5.44119 | ||
| Pi-Alkyl | N-Aryl (ligand) | Pro 467 (side chain) | 5.10571 | ||
| 7c | -7.1 | HB | Asn 308 (NH2) | 2-NO2-Aryl (ligand) | 3.25728 |
| PS | Pro 304 (αCH) | 2-NO2-Aryl (ligand) | 3.77999 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Ala 213 (side chain) | 4.46163 | ||
| 7d | -8.5 | CHB | Lys 367 (side chain CH2NH2) | C = O (2nd position of TZD; ligand) | 3.70277 |
| PDHB; PS | Cys 285 (SH) | 3-OH-Aryl; TZD core (ligand) | 3.99461 | ||
| Pi-Sigma | Leu 330 (side chain) | TZD core (ligand) | 3.84019 | ||
| Pi-Sigma | Ile 341 (side chain) | 3-OH-Aryl (ligand) | 3.50391 | ||
| PS | Cys 285 (SH) | N-Aryl (ligand) | 5.46855 | ||
| PS | Cys 285 (SH) | 3-OH-Aryl (ligand) | 5.82541 | ||
| PPT | His 449 (Imidazole ring) | N-Aryl (ligand) | 4.96478 | ||
| APS | Cys 285; Gln 286 (CONH) | N-Aryl (ligand) | 5.20028 | ||
| Pi-Alkyl | 3-OH-Aryl (ligand) | Arg 288 (side chain) | 3.94502 | ||
| Pi-Alkyl | Phe 282 (Aryl) | Methyl (N-Aryl, ligand) | 4.90419 | ||
| Pi-Alkyl | His 449 (Imidazole ring) | Methyl (N-Aryl, ligand) | 4.80268 | ||
| 7e | -8.9 | PDHB; PS | Cys 285 (SH) | N-Aryl; TZD core (ligand) | 3.68179 |
| Pi-Sigma | Arg 288 (side chain) | N-Aryl (ligand) | 3.55725 | ||
| Pi-Sigma | Ile 341 (side chain) | Methyl (N-Aryl, ligand) | 3.77542 | ||
| PS | Cys 285 (SH) | N-Aryl (ligand) | 5.46193 | ||
| PPT | His 449 (Imidazole ring) | 4-Cl-Aryl (ligand) | 4.67448 | ||
| APT | Cys 285; Gln 286 (CONH) | 4-Cl-Aryl (ligand) | 4.81318 | ||
| Alkyl | 4-Cl-Aryl (ligand) | Leu 469 (side chain) | 4.17882 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Leu 270 (side chain) | 4.88063 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Arg 288 (side chain) | 4.06932 | ||
| Alkyl | Methyl (N-Aryl, ligand) | Ile 341 (side chain) | 4.55730 | ||
| Pi-Alkyl | TZD core (ligand) | Leu 330 (side chain) | 4.65177 | ||
| Pi-Alkyl | 4-Cl-Aryl (ligand) | Cys 285 (side chain) | 5.15119 | ||
| Pi-Alkyl | His 323 (Imidazole ring) | 4-Cl-Aryl (ligand) | 4.39467 | ||
| Pi-Alkyl | His 449 (Imidazole ring) | 4-Cl-Aryl (ligand) | 5.03461 |
The various types of non-covalent interactions of each molecule with the receptor was shown in Figs 3–9. Most of the amino acids from the protein that have interacted with the synthesized compounds are either hydrophobic or basic. Hydrophobic interactions usually occurred from alkyl/pi-alkyl to amino acids such as Leu, His, Arg etc. There are a few polar interactions (hydrogen bond, carbon hydrogen bond and Pi-Donor hydrogen bond) as well as other interactions also occurred dependent on the electronic environment (amide pi-stacked, pi-pi t stacked, pi-sigma, etc). It should be noted that binding site for protein is not similar for all synthesized molecules. All these data indicate that the core structure of our synthesized compounds is very cooperative with the protein.
Conclusion
In conclusion, we developed a convenient method to synthesize 3-m-tolyl-5-arylidenthiazolidine- 2,4-dione derivatives by using morpholine as a catalyst. Molecular flexible docking studies have shown that our synthesized compounds are very active and some of them shown better binding affinity with the protein than the commercially available drugs, Epalrestat. These results inspire us to study the moiety even further and test these molecules for their biological activity. To find out more potent compound we plan to do pharmacokinetics study in near future.
Acknowledgements
We are thankful to Wazed Miah Science Research Center (WMSRC), Jahangirnagar University for spectral data and Dr. Md. A. Halim, The Red-Green Research Centre for providing the computational studies.
References
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37