
Competing Interests: The authors have declared that no competing interests exist.
Dendritic cells are sentinels of the immune system and represent a key cell in the activation of the adaptive immune response. Hypoxia-inducible factor 1 alpha (HIF-1α)–a crucial oxygen sensor stabilized during hypoxic conditions–has been shown to have both activating and inhibitory effects in immune cells in a context- and cell-dependent manner. Previous studies have demonstrated that in some immune cell types, HIF-1α serves a pro-inflammatory role. Genetic deletion of HIF-1α in macrophages has been reported to reduce their pro-inflammatory function. In contrast, loss of HIF-1α enhanced the pro-inflammatory activity of dendritic cells in a bacterial infection model. In this study, we aimed to further clarify the effects of HIF-1α in dendritic cells. Constitutive expression of HIF-1α resulted in diminished immunostimulatory capacity of dendritic cells in vivo, while conditional deletion of HIF-1α in dendritic cells enhanced their ability to induce a cytotoxic T cell response. HIF-1α-expressing dendritic cells demonstrated increased production of inhibitory mediators including IL-10, iNOS and VEGF, which correlated with their reduced capacity to drive effector CD8+ T cell function. Altogether, these data reveal that HIF-1α can promote the anti-inflammatory functions of dendritic cells and provides insight into dysfunctional immune responses in the context of HIF-1α activation.
Hypoxia inducible factor 1 (HIF-1) is a master regulator of the cellular response to hypoxia, and regulates multiple processes including angiogenesis, protein synthesis and glycolytic and oxidative metabolism [1]. HIF-1 belongs to a family of proteins that includes HIF-1α, HIF-1β (also known as aryl nucleotide receptor; ARNT) and HIF-2α, and functions as a heterodimeric transcription factor of HIF-1α and HIF-1β [2]. HIF-1β is constitutively expressed, while the expression of HIF-1α (and also HIF-2α) are tightly regulated. The regulation of HIF-1α is multi-faceted, with the most well-established pathway centred on the constitutive hydroxylation of HIF-1α on conserved, prolyl residues during normoxic conditions. HIF-1α hydroxylation by the PHD family of enzymes [3–6] facilitates its recognition by pVHL [7], which targets HIF-1α for degradation. During hypoxia, HIF-1α is no longer hydroxylated, and is able to translocate to the nucleus to dimerize with HIF-1β and carry out transcription of target genes. The cellular functions of HIF-1 in response to hypoxia have been well studied, particularly in the context of the tumor microenvironment. However, the characterization of its role in the immune system has been limited.
Several studies have shown that HIF-1α has important, but at times, opposing, effects on immune cell function. The loss of HIF-1α in macrophages and neutrophils reduces the severity of cutaneous inflammation and passive arthritis models [8], and HIF-1α deficient DCs were shown to have decreased expression of co-stimulatory markers compared to wildtype DCs and a reduced capacity to induce T cell proliferation [9]. HIF-1α has also been shown to be required for myeloid cell motility and bactericidal activity [10, 11]. Conversely, conditional deletion of HIF-1α in a subsequent study, using the same model as Cramer et al., resulted in exacerbated airway allergy in an allergic asthma model [12]. These data were attributed to the role of HIF-1α in promoting anti-inflammatory functions of interstitial macrophages, a population that was also affected when using the LysM-Cre conditional knockout model. HIF-1α upregulation has also been observed in macrophages following exposure to heat-killed bacteria in normoxic conditions [11], and several studies have shown that HIF-1α is stabilized downstream of TLR or LPS stimulation [13–16].
In dendritic cells (DCs), opposing functions for HIF-1α have also been reported by various groups. One study showed that DCs exposed to hypoxic conditions, increased the production of pro-inflammatory cytokines [17], while another study showed that the loss of HIF-1α in DCs unexpectedly resulted in increased pathology in a DSS-colitis model [18]. Additional studies have suggested that HIF-1α inhibits pro-inflammatory DC function and downstream induction of CD8+ T cell responses in vitro [19], and dampens dendritic cell function in a Leishmania infection model [20, 21].
HIF-1α has been shown to be a crucial mediator of glycolytic re-programming in pro-inflammatory DCs [16]. Metabolic re-programming of DCs towards glycolysis is suggested to be necessary for CCR7-dependent migration of DCs to draining lymph node, and CCR7 stimulation has also been found to further drive DC metabolic reprogramming towards glycolysis in a HIF-1α-dependent manner [22].
In our studies, we examined the consequences of either constitutive HIF-1α expression or HIF-1α deletion on DC function. For constitutive HIF-1α expression, we used a model where a form of HIF-1α that cannot be hydroxylated was expressed in DCs. This form of HIF-1α escapes recognition by pVHL and cannot be degraded through this pathway. These DCs were then tested in vitro and in vivo to investigate the functional outcome of sustained HIF-1α expression in DCs on the immune response.
To study the consequences of HIF-1α expression in dendritic cell function, we obtained loxP-stop-loxP-Hif1a-dPA transgenic mice that possess a mutant form of HIF-1α with P402A and P564A substitutions, placed downstream of a floxed transcriptional stop cassette [23]. Mutation of these proline residues in HIF-1α abrogates their hydroxylation and subsequent recognition by pVHL, and therefore prevents the degradation of HIF-1α through this pathway. We crossed these loxP-stop-loxP Hif1a-dPA mice to Vav-Cre mice to generate loxP-stop-loxP-Hif1a-dPa Vav-Cre+ (herein referred to as HIF-1α-Tg) mice, enabling constitutive expression of HIF-1α in all hematopoietic cells (Fig 1A). We generated BMDC by culturing bone marrow from HIF-1α-Tg mice in the presence of GM-CSF and did not observe any differences between WT and HIF-1α-Tg in either CD11c+MHCII+ BMDC growth, frequency or viability (Fig 1B and 1C, S1A Fig). As expected, we confirmed increased expression of HIF-1α in HIF-1α-Tg BMDCs compared to WT (Fig 1D). Interestingly, levels of HIF-1α were further upregulated following CpG-stimulation of HIF-1α-Tg BMDCs. In Hif1afl/fl Cd11c-Cre-Gfp (HIF-1α KO) BMDCs, some HIF-1α signal was unexpectedly observed in HIF-1α KO BMDCs following 18h of CpG stimulation (Fig 1D). However, expression of HIF-1α target genes including Ldha and Slc2a1 were significantly reduced in HIF-1α KO BMDCs (S2 Fig), as expected. Consistent with this data, we observed increased expression of several HIF-1α target genes including Slc2a1 [24] and Nos2 [25] in HIF-1α-Tg compared to WT BMDCs following CpG stimulation (S2 Fig). Expression of other genes including Adora2b, Arg1 and Epo were also elevated, but not significantly increased in HIF-1α-Tg BMDCs following stimulation, while Arg2 was not differentially altered in HIF-1α-Tg BMDCs. We further confirmed increased protein levels of iNOS in HIF-1α-Tg DCs (Fig 1E). Next, the surface expression of conventional activation markers on HIF-1α-Tg BMDCs was examined to determine whether any changes could be detected compared to WT BMDCs. No major differences in the levels of CD80, CD86, CD40, MHC II or MHC I were found between WT and HIF-1α-Tg BMDCs following stimulation with CpG (Fig 1F). The expression levels of other markers such as CD103, OX40L, CCR7 or inhibitory molecules including PD-L1 (a predicted HIF-1α target [26]) and PD-L2, were also found to be similar between WT and HIF-1α-Tg BMDCs (Fig 1G). Additionally, no differences in antigen uptake or processing were detected (S1B–S1D Fig). These data indicated that constitutive and functional HIF-1α expression in BMDCs was induced, and that sustained expression of HIF-1α did not alter the number of BMDCs nor their surface levels of activation or inhibitory markers.

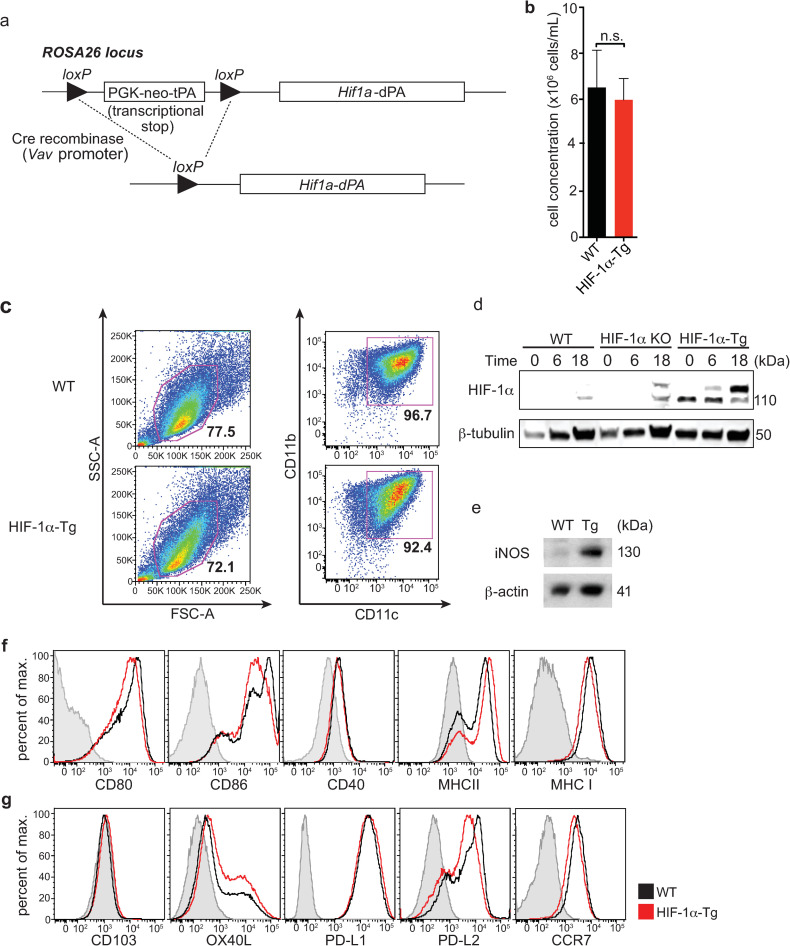
Constitutive expression of HIF-1α in BMDCs leads to increased iNOS expression without affecting surface levels of co-stimulatory or co-inhibitory molecules.
(a) Hif1a-dPA loxP-stop-loxP mice express a transgene containing a floxed transcriptional stop cassette upstream of a mutated HIF-1α with two proline to alanine substitutions (P402A and P564A; HIF-1α-dPA). (b) Average cell concentration following 10 days of BM culture in the presence of GM-CSF (data shown is representative of 4 independent experiments). (c) BMDC frequencies, based on FSC/SSC and CD11c+CD11b+ profiles, are similar between WT and HIF-1α-Tg cultures. (d) Constitutive HIF-1α expression was confirmed in HIF-1α-Tg BMDCs by Western blot before or after stimulation with CpG for 16-20h. (e) Expression of iNOS (a direct HIF target) by Western blot in CpG-stimulated, WT and HIF-1α-Tg BMDCs. (f) BMDCs were left unstimulated or stimulated with CpG, and stained for various DC activation markers and (g) other surface molecules including CD103, OX40L, PD-L1, PD-L2, and CCR7. For all plots, the solid black line indicates WT BMDCs and the solid red line indicates HIF-1α-Tg BMDCs. FMO controls are indicated by the shaded grey histogram. For experiments with CpG stimulation of BMDC, stimulation was performed by adding CpG to BMDCs at a concentration of 10μM and incubating for 16-20h. Western blot data are representative of two independent experiments, and flow cytometry data are representative of at least five independent experiments. For (b), n.s.: not significant using Student’s t-test (two-tailed).
A series of experiments were performed to evaluate the immunostimulatory capacity of HIF-1α-Tg BMDCs in vivo. We have developed an assay to evaluate DC function in vivo using the RIP-gp model. In this transgenic model, the lymphocytic choriomeningitis virus glycoprotein (LCMV-gp) is expressed by the β-islet cells of the pancreas using the rat insulin promoter (RIP). Studies have shown that the T cells specific for the LCMV-gp remain ignorant or naïve and can be activated by a variety of stimuli [27]. We have shown that DCs can be generated from the bone marrow, pulsed with gp peptides, and matured with various TLR stimuli to induce an LCMV gp-specific cytotoxic response that leads to the destruction of β-islet cells and diabetes [28, 29]. Therefore, we examined whether HIF-1α-Tg DCs could stimulate a functional CD8+ T cell response in the RIP-gp model (Fig 2A). As expected, CpG-stimulated, WT BMDCs pulsed with LCMV peptides were able to induce diabetes in RIP-gp mice after approximately 8–10 days. In contrast, RIP-gp mice receiving CpG-matured HIF-1α-Tg BMDCs showed reduced incidence of diabetes compared to mice immunized with wildtype BMDCs (Fig 2B and 2C), while HIF-1α KO CpG-stimulated BMDCs had similar diabetes induction compared to WT (S3 Fig). Reduced CD8+ infiltration was observed in the pancreas from RIP-gp mice that were given HIF-1α-Tg BMDCs; however, this difference was not statistically significant (Fig 2E). Therefore, HIF-1α-Tg DCs have a limited capacity to induce a gp-specific response in vivo compared to wildtype DCs.
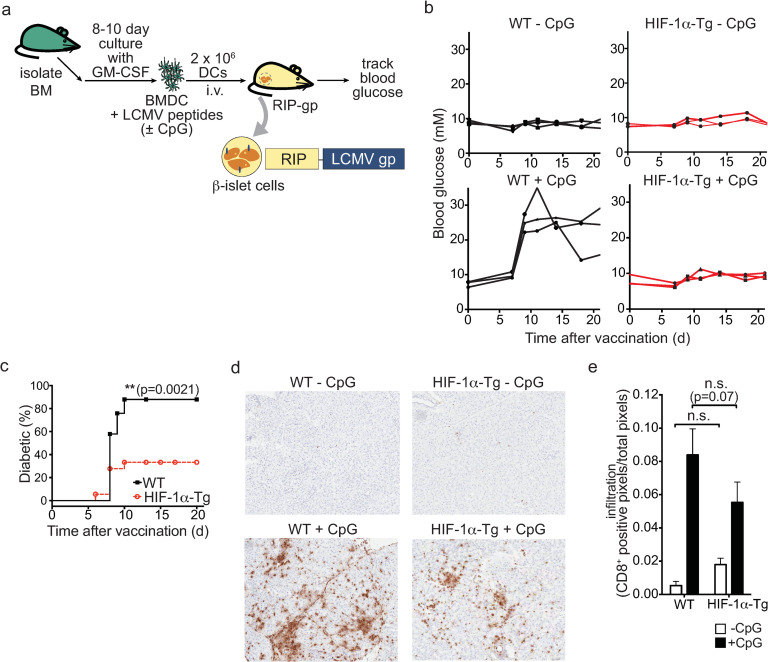
Overexpression of HIF-1α-dPA in BMDCs results in impaired induction of immune pathology.
(a) Experimental overview of the RIP-gp BMDC vaccination model. BMDCs were harvested following 8 days of culture, stimulated with ODN 1826 CpG, pulsed with LCMV gp peptides, and given intravenously into RIP-gp mice. (b) Blood glucose measurements of RIP-gp mice receiving gp-peptide pulsed, unstimulated or CpG-stimulated, WT or HIF-1α-Tg BMDCs (n = 3 mice per group; data representative of three independent experiments). (c) Cumulative diabetes incidence in RIP-gp mice receiving B6 or HIF-1α-Tg, CpG-stimulated, gp peptide-pulsed BMDCs. Data are representative of 20–25 mice per group. (d) Representative CD8+ staining of pancreatic sections 6 days following BMDC administration (10X magnification). (e) Quantification of pancreatic CD8+ infiltration in RIP-gp mice. C57BL/6 or HIF-1α-Tg BMDCs were administered to RIP-gp mice and 6 days later, the pancreas was snap-frozen and processed for histology. CD8+ staining was quantified using positive pixel detection. *P ≤ 0.05; n.s.: not significant. Error bars represent S.D. For (c), a log-rank (Mantel-Cox) test was used to determine significance, and for (e), a two-way ANOVA was performed with Tukey’s test for multiple comparisons.
Further experiments were done to examine the impact of constitutive HIF-1α expression in BMDCs on the induction of immune responses in vivo. We measured the expansion of gp34-tetramer-specific CD8+ T cells in the blood of RIP-gp mice by flow cytometry following WT and HIF-1α-Tg BMDC vaccination (Fig 3A). The frequency of CD8+ gp34-tetramer+ cells was slightly, but not significantly reduced in mice receiving HIF-1α-Tg BMDCs, while HIF-1α-KO BMDCs had similar expansion of gp34-tetramer-specific CD8+ T cells as WT (S3 Fig). To examine whether differences in the functional properties of CD8+ T cells could be found in the pancreas, pancreatic infiltrating lymphocytes (PIL) were isolated and stained for granzyme B expression. Notably, a reduced percentage of PIL expressed granzyme B when RIP-gp mice were given HIF-1α-Tg BMDCs (Fig 3B and 3C), coinciding with the limited induction of islet cell death and reduced frequency of diabetes induction in the RIP-gp model. Together, these data demonstrate that HIF-1α-Tg BMDC vaccination results in suboptimal activation of CD8+ T cells in vivo.
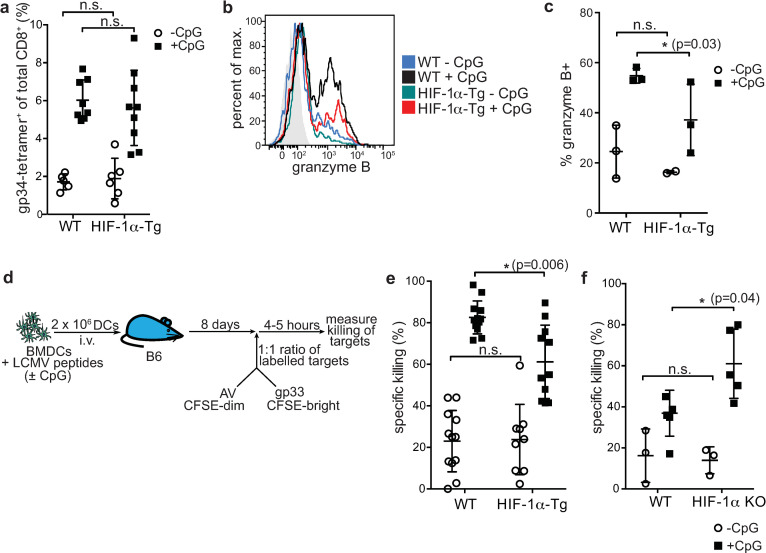
Constitutive expression of HIF-1α-dPA in BMDCs limits cytotoxic CD8+ T cell responses.
(a) RIP-gp mice were vaccinated with gp-peptide pulsed, unstimulated or CpG-stimulated, WT or HIF-1α-Tg BMDCs. Eight days later, blood was collected for tetramer staining of gp-specific CD8+ T cells (gp34-tetramer+). (b) Pancreatic infiltrating lymphocytes (PIL) were isolated from RIP-gp mice 6 days following WT or HIF-1α-Tg BMDC vaccination and stained for granzyme B. Representative staining for CD8+ PIL is shown in the histogram and (c) the percentage of granzyme B+ PIL (of CD8+CD45+ cells) is shown. (d) Overview of the CTL assay used to assess antigen-specific killing of targets in vivo. Peptide-pulsed, CpG stimulated or unstimulated, WT or HIF-1α-Tg BMDCs were infused into naïve B6 host animals. Eight days later, fluorescently labeled, peptide-pulsed target cells were given i.v., and spleens of recipient mice were harvested and analyzed by flow cytometry to assess the antigen-specific, direct killing capacity of CTLs in vaccinated mice. CTL data for mice vaccinated with (e) WT or HIF-1α-Tg BMDCs or (f) WT or HIF-1α KO BMDCs, is shown. For (e) and (f), open circles represent mice given unstimulated BMDCs (-CpG), while shaded squares represent mice given CpG-stimulated BMDCs (+CpG). Data are representative of 2–3 independent experiments, with 2–3 mice per group. For (a) and (e), data were pooled from three independent experiments. *P ≤ 0.05; n.s.: not significant. Statistical testing was performed with two-way ANOVA and Tukey’s test.
To further demonstrate that HIF-1α-Tg BMDCs had a negative impact on their ability to activate CD8+ T cells, we measured the lysis of peptide-pulsed targets in vivo. WT mice were immunized with WT or HIF-1α-Tg BMDCs that had been pulsed with LCMV gp peptides and stimulated with CpG to induce an antigen-specific immune response. Eight days later, a 1:1 mixture of labeled, gp33 peptide-pulsed and adenoviral control peptide-pulsed targets were infused via tail vein injection into the same WT mice. The spleens of these mice were harvested several hours later and analyzed by flow cytometry to assess specific killing of labeled target cells (Fig 3D). We found that HIF-1α-Tg BMDCs induced a suboptimal cytotoxic response compared to WT BMDCs (Fig 3E), and these data are consistent with the limited induction of diabetes observed earlier (Fig 2B and 2C).
To further confirm our findings that HIF-1α limits DC function, we tested HIF-1α KO BMDCs in the RIP-gp model. HIF-1α KO BMDCs were evaluated for their capacity to activate antigen-specific, cytotoxic T cells in vivo (Fig 3D). In contrast to our findings with HIF-1α-Tg BMDCs, HIF-1α KO BMDCs were found to promote an enhanced cytotoxic response compared to WT (Fig 3F), although diabetes incidence was comparable to WT (S3 Fig). Collectively, these observations demonstrate that constitutive expression of HIF-1α impairs the ability of BMDCs to promote the expansion of gp-specific CTLs, which show reduced granzyme B expression and diminished cytotoxic function, while deletion of HIF-1α in BMDCs can augment their immunogenicity.
We next asked whether HIF-1α could be mediating the production of immunosuppressive factors or limiting pro-inflammatory cytokine production that could potentially explain the reduced diabetes incidence observed with HIF-1α-Tg BMDC vaccination in the RIP-gp model. Since we detected increased expression of iNOS in HIF-1α-Tg BMDC cultures (Fig 1E), we analyzed additional targets of HIF-1α from the supernatants of unstimulated and CpG stimulated BMDC cultures. We observed elevated production of nitric oxide (measured as nitrite) and arginase I by HIF-1α-Tg BMDCs following stimulation compared to WT BMDCs (Fig 4A and 4B). Interestingly, although arginase I has been reported to be mainly a target of HIF-2α [30] in primary macrophages, we did not observe significantly altered levels of HIF-2α in HIF-1α-Tg BMDCs compared to WT (Fig 4B). Consistent with these data, arginase I was undetectable in HIF-1α KO BMDCs at both the transcript (S2 Fig) and protein level (Fig 4B), either before or after CpG stimulation, despite similar HIF-2α levels in HIF-1α KO compared to WT, suggesting that HIF-1α may play a predominant role in regulating arginase I expression in BMDCs. We also observed that levels of vascular endothelial growth factor (VEGF)–which is known to have suppressive properties on some immune cell subsets [31–33]–was found to be significantly upregulated in the supernatants of cultures of HIF-1α-Tg BMDCs following CpG stimulation compared to WT BMDCs (Fig 4C). Il10, which has been reported to be a direct transcriptional target of HIF-1α [34], was found to be significantly increased by qPCR (Fig 4D), and secreted IL-10 was also found to be significantly upregulated by stimulated HIF-1α-Tg BMDCs compared to WT (Fig 4E). Conversely, IL-6, IL-12p70, TNFα and TGFβ were not significantly altered with enforced HIF-1α expression in BMDCs at the protein (Fig 4F–4I) or transcript level (S4 Fig). Overall, these data indicate that HIF-1α drives multiple inhibitory mechanisms that contribute to limiting the induction of immunity by DCs.
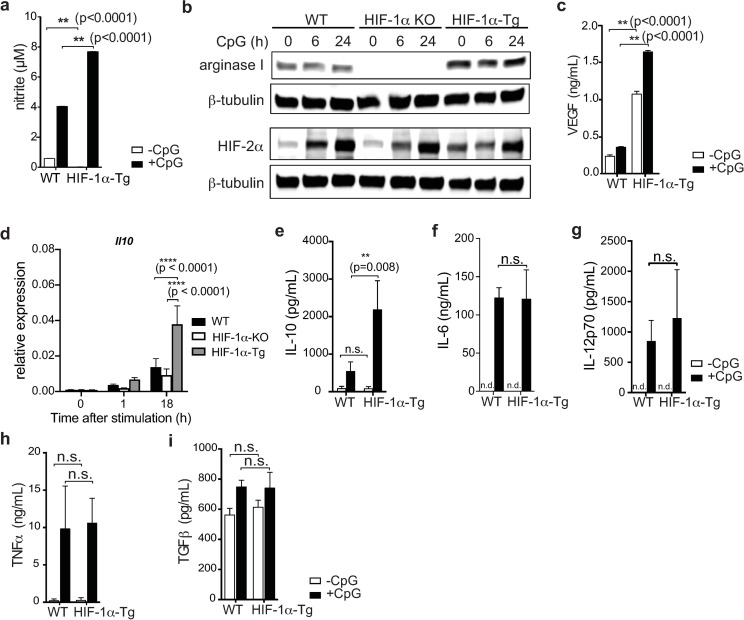
HIF-1α expression in BMDC cultures promotes the production of immunosuppressive cytokines.
(a) Nitric oxide production by WT and HIF-1α-Tg BMDCs was indirectly quantified by measuring nitrite levels (Griess reaction) by ELISA. (b) Arginase I and HIF-2α protein levels in total WT and HIF-1α-Tg BMDC cell lysates. Cell lysates from the same samples were split across different gels and probed separately for arginase I and HIF-2α. β-tubulin was used as a protein loading control. (c) VEGF production by BMDCs was analyzed by ELISA. (d) Il10 expression in BMDCs was analyzed by qPCR before and after CpG-stimulation. (e) IL-10, (f) IL-6, (g) IL-12p70, (h) TNFα, and (i) TGFβ production by BMDCs was analyzed by ELISA. All data are representative of two to three independent experiments. Error bars represent SD of the mean. Statistical testing was performed using two-way ANOVA and Tukey’s test for multiple comparison; n.s.: not significant, n.d.: not detected (below limit of detection).
Our study aimed to examine the effects of HIF-1α in dendritic cells by constitutive expression of a form of HIF-1α that cannot be recognized and targeted for degradation by pVHL. Previous studies examining the effects of HIF-1α on DC function have used conditional knockout approaches [35], culture in hypoxia [17], or small molecules such as cobalt chloride to mimic hypoxic conditions. These and other studies have yielded at times opposing data on the effects of HIF-1α on DCs. Some of these differences could be attributed to the experimental model tested, while HIF-1α-independent effects of hypoxia [36, 37], for example, could also explain some of the conflicting data in the literature.
We generated and evaluated a novel system where HIF-1α was conditionally and constitutively expressed in BMDCs. Our data demonstrate that HIF-1α expression in BMDCs limited their ability to activate a cytotoxic T cell response in vivo. We found that HIF-1α expression did not alter the surface levels of co-stimulatory markers or significantly alter the production of pro-inflammatory cytokines by BMDCs. While previous studies have shown that hypoxia can induce upregulation of CD80, CD86 and MHCII [13, 35] in BMDCs, we did not observe these effects with constitutive expression of HIF-1α. It is likely that hypoxia-driven, but HIF-1α-independent pathways, such as NF-κB [37, 38], were responsible for the phenotypes observed in these studies.
Our findings suggest that constitutive expression of HIF-1α in BMDCs promotes the production of several immune inhibitory mediators. HIF-1α has been shown to directly increase the transcription of the genes for many of these molecules, including Il10 [34], VEGFA [39], NOS2 [40] and ARG1 [41]. Consistent with these data, we observed increased levels of IL-10, VEGF, iNOS, and arginase in HIF-1α-Tg BMDC cultures. Interestingly, Arg1 has been identified to be mainly regulated by HIF-2α [30], while our data suggest that HIF-1α, and not HIF-2α, is the predominant driver of Arg1 expression in BMDCs. The reason(s) for the pre-dominant role of HIF-1α in regulating Arg1 expression in BMDCs is unclear, although the same study showed that HIF-1α can also be involved in regulating Arg1 expression as conditional deletion of HIF-1α in macrophages resulted in reduced expression of Arg1. While HIF-1α and HIF-2α bind to the same hypoxia responsive element (HRE) [42], and can have overlapping targets, the divergence in their regulatory programs has been associated with their interaction with other transcription factors and co-regulators [43]. The precise co-factors that cooperate with HIF-1α to drive Arg1 expression in BMDCs will require further investigation to clarify the context-specific roles of HIF-1α and HIF-2α in regulating Arg1 expression. Overall, our data supports the notion that HIF-1α functions as a crucial transcription factor that drives the production of immunosuppressive molecules. Although we did not identify the specific factor(s) responsible for reduced DC function in our model, it is likely that a combination of factors driven by HIF-1α collectively contributes to inhibiting DC activation of effector T cells.
Consistent with previous reports [14, 44], we also found that HIF-1α was upregulated following TLR stimulation. Several different, but potentially overlapping mechanisms have been described to link increased HIF-1α levels following TLR activation, including the activation of NF-κB [44] and/or ERK signaling [45] pathways, the generation of reactive oxygen species [46], succinate [14], or the sequestration of iron [47] (a co-factor for the PHD family). A later study suggested that in contrast to studies using mouse cells, a p38-MAPK-dependent pathway was important for HIF-1α stabilization in human monocyte-derived DCs following stimulation with LPS [48]. While the dominant mechanism for HIF-1α upregulation downstream of TLR signaling remains to be fully delineated, these data show that increased HIF-1α is a common effect of TLR stimulation of DCs.
Although HIF-1α has been associated with both pro- and anti-inflammatory functions of DCs, we show that conditional deletion of HIF-1α in BMDCs was able to significantly enhance the generation of antigen-specific, cytotoxic T cell responses in vivo. These findings further support work from other groups showing that HIF-1α in DCs limited the expansion of CD8+ T cells and the downstream induction of KLRG1+ CD8+ short-lived effector T cells (SLECs) in a Leishmania infection model [20].
It is important to note that the kinetics of HIF-1α also appears to be cell type-specific. HIF-1α has been shown to be rapidly upregulated upon exposure to hypoxia, but subsequently degraded 4-8h later despite the maintenance of hypoxic conditions [49, 50]. In contrast, HIF-1α expression has been observed to remain upregulated following 6 days of culture of monocyte-derived DCs in hypoxic conditions [17]. These and subsequent studies will benefit from the development of inducible models capable of both temporal and conditional expression of HIF-1α in vivo, and would help to refine our understanding of the functions of HIF-1α in immune cells.
The role of myeloid cells in promoting tumor growth and dampening anti-tumor responses has been well established [51–53]. Our data are consistent with previous reports suggesting that HIF-1α limits dendritic cell function [19, 21]. Taken together, these results suggest that constitutive HIF-1α activation in dendritic cells–for example, following migration within the hypoxic tumor microenvironment–might impair anti-tumor responses by limiting effector T cell cytotoxicity and function. Thus, treatments aiming to modulate HIF-1α activity will ideally consider the immune cell types that would be affected, and will enhance or inhibit HIF-1α function based on the desired therapeutic outcome.
LSL-HIF-1a-dPA mice were obtained from The Jackson Laboratory (JAX stock 009673) and bred to Vav-Cre mice to generate HIF-1α-Tg mice. Hif1afl/fl mice (JAX stock 007561) were bred to Cd11c-Cre-Gfp mice (JAX stock 007567) to generate HIF-1α KO mice (Hif-1afl/fl Cd11c-Cre-Gfp). P14 and RIP-gp mice were previously described [27, 54]. All animal experiments were performed in the Ontario Cancer Institute (OCI) animal facility and with the approval of the OCI Animal Ethics Committee, under Animal User Protocol (AUP) 929.
Bone-marrow derived dendritic cells were cultured from bone marrow harvested from the femurs and tibiae of mice. Additional details can be found in a previously published study from our laboratory [28]. Briefly, BM cells were seeded at 2 x 106 cells per plate in 10cm plates and cultured for 8 days in the presence of GM-CSF. Non-adherent cells were harvested and either stimulated with 10μM ODN1826 CpG (Integrated DNA Technologies), or left unstimulated for 16-20h. Cells were then pulsed with a mixture of gp33-41 (KAVYNFATM), gp276-286 (SGVENPGGYCL) and gp61-80 (GLNGPDIYKGVYQFKSVEFD) peptides at a final concentration of 1μM for 3h, washed three times, and infused intravenously via tail-vein into RIP-gp mice. Blood glucose was monitored using Accu-Chek Performa (Roche) blood glucose meters and Accu-Chek Inform II (Roche) blood glucose test strips.
B6 mice were vaccinated with 2 x 106 peptide-pulsed BMDCs as described above. Eight days after vaccination, a 1:1 ratio of splenocytes pulsed with gp33-41 or a control AV peptide (SGPSNTPPEI), labeled with e450 proliferation dye (eBioscience) at 40μM or 5μM, respectively, were injected via tail vein. Approximately 1 x 107 splenocytes were injected per mouse. Four hours after injection, mice were sacrificed and the spleen harvested and homogenized for flow cytometry analysis. Specific killing was calculated using the following formula: (percentage AV–percentage gp33-41) / (percentage AV) x 100%.
Flow cytometry data was acquired using the BD FACS Canto II (Becton-Dickinson) or BD LSR Fortessa (Becton-Dickinson). Data analysis was performed using Flowjo (Treestar Software). Antibodies were purchased from BD Biosciences, Affymetrix/eBioscience and Biolegend. Antibodies for CCR7 (4B12), CD11b (M1/70), CD11c (N418), CD44 (IM7), CD115 (AFS98), F4/80 (BM8), Gr-1 (RB6-8C5), Ly-6C (HK1.4), Ly-6G (1A8-Ly6G), MHC I (34-1-2S), OX40L (RM134L), PD-1 (J43), PD-L1 (MIH5), PD-L2 (TY25) were purchased from eBioscience. CD25 (PC61), CD40 (3/23), CD80 (16-10A1), CD103 (M290), and annexin V antibodies were from BD Biosciences. 7AAD was also from BD Biosciences. CD86 (GL-1) and MHC II (M5/114.15.2) antibodies were from Biolegend. gp34-tetramer was prepared by sequential addition of streptavidin APC or streptavidin-R-PE (Invitrogen) to biotinylated gp34 monomers (H-2Kb; AVYNFATC) generously provided by the NIH Tetramer Core Facility.
BMDC supernatants were tested for VEGF, IL-6, IL-10, IL-12p70, IL-23, and TNFα by ELISA, with appropriate dilutions. The VEGF ELISA kit was purchased from Peprotech, and the other ELISA kits were purchased from eBioscience (Ready-SET-Go!, Thermo Fisher Scientific). Supernatants were diluted as necessary according to the kit manufacturer’s instructions.
Total RNA was extracted from BMDCs using a RNeasy Plus Mini Kit (Qiagen) or TRIzol reagent (Thermo Fisher Scientific). cDNA was synthesized using a high capacity cDNA reverse transcription kit (Applied Biosystems, Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative, real-time PCR was performed with KAPA SYBR (KAPA Biosystems) and an ABI 7900HT sequence detection system (Applied Biosystems). Actin was used as a housekeeping gene control to normalize target gene expression. Primer sequences can be found in S1 Table.
Protein samples were separated on 4–12% Bis-Tris acrylamide gels (Life Technologies) and transferred to PVDF (Millipore). HIF-1α antibody was purchased from Cayman Chemical (10006421) and Novus Bio (NB100-479). Arginase I (H-52) and NOS2 (M19) antibodies were purchased from Santa Cruz Biotechnology. Rabbit anti-β-actin (A2066) was purchased from Sigma. β-tubulin (AA2) was purchased from Millipore. Secondary antibodies were goat anti-rabbit HRP (Thermo), goat anti-mouse HRP (Thermo), donkey anti-mouse IRDye800CW, donkey anti-goat IRDye680RD and donkey anti-rabbit IRDye680RD (Licor). Blots were developed with Supersignal Femto (Thermo) or Amersham ECL (GE Healthcare Life Sciences) or visualized on an Odyssey CLx imager (Licor).
Statistical testing was performed in Graphpad Prism 7.0e. The threshold for statistical significance was set at alpha = 0.05. Corrections were applied to multiple comparisons testing as indicated in the figure legends.
Masato Sasaki provided the HIF-1α antibody for initial experiments. Tak W. Mak and Vuk Stambolic provided critical feedback for this project.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
 Hypoxia-inducible factor 1 alpha limits dendritic cell stimulation of CD8 T cell immunity
Hypoxia-inducible factor 1 alpha limits dendritic cell stimulation of CD8 T cell immunity
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

