
Competing Interests: The authors have declared that no competing interests exist.
The generation of large metabolomic data sets has created a high demand for software that can fit statistical models to one-metabolite-at-a-time on hundreds of metabolites. We provide the %polynova_2way macro in SAS to identify metabolites differentially expressed in study designs with a two-way factorial treatment and hierarchical design structure. For each metabolite, the macro calculates the least squares means using a linear mixed model with fixed and random effects, runs a 2-way ANOVA, corrects the P-values for the number of metabolites using the false discovery rate or Bonferroni procedure, and calculate the P-value for the least squares mean differences for each metabolite. Finally, the %polynova_2way macro outputs a table in excel format that combines all the results to facilitate the identification of significant metabolites for each factor. The macro code is freely available in the Supporting Information.
Metabolomics is the broad scale analysis of compounds involved in metabolism, primarily utilizing liquid and gas chromatography mass spectrometry, and nuclear magnetic resonance [1]. Matrices analyzed range from plants and cell media to animal and human tissues and fluids. Often capturing hundreds of metabolites from multiple metabolic pathways in one analysis, it is ideal for hypothesis generation. This is particularly true for untargeted metabolomics which typically attempt to analyze as many metabolites as possible to produce peak area or peak height data [2] that is often not calibrated by standards. Typically, the possible chemical identity of metabolites is not known before data are acquired, requiring post data-acquisition identification. This analysis can capture amino acids, sugars, metabolic pathway intermediates, and fatty acids, among many others. Global lipidomics assays are often untargeted and captures a variety of complex lipids including phospholipids, triacylglycerols, and cholesterol esters. Targeted metabolomics analyzes a large number of metabolites but screens for specific classes of metabolites and utilizes calibration standards for each metabolite, in addition to internal standards, to generate quantitative data. Lastly, semi-targeted metabolomics, a hybrid of untargeted and targeted metabolomics, screens for metabolites from several classes whose chemical identity is known prior to data acquisition and produces data in which one calibration curve and internal standard is used to quantify several metabolites.
The generation of these large data sets consisting of hundreds of metabolites has created a high demand for software tools capable of streamlining implementation of sophisticated methods of data analysis for a variety of study designs. Furthermore, the concern for the increased type one error associated with multiple hypothesis testing also requires the utilization of Bonferroni or false discovery rate (FDR) correction [3,4]. Various software packages have been created to enable more efficient statistical analysis of large data sets such as MetaboAnalyst and Metabox [5,6]. As a result, researchers without a programming background can easily conduct multivariate tests on hundreds of metabolites. However, these programs have numerous limitations. More specifically, to our knowledge a software is not available that can conduct analysis of variance (ANOVA) and generate FDR or Bonferroni-adjusted P-values for the selection of metabolites in study designs with a factorial treatment and hierarchical design structure. This is a particular concern since metabolomics is increasingly utilized in complex studies that require advanced statistical modelling to capture all sources of variability. This article details the key aspects and validation of a macro written in SAS software capable of modelling fixed and random effects in multifactorial ANOVA for large metabolomic datasets.
An experimental dataset from a previously published study in our lab [7] was used to illustrate the utilization of the %polynova_2way SAS macro. Data is provided in S1 Data file. Experiments were conducted in accordance with the Institutional Animal Care and Use Committee of California State University (Protocol #1611 approved on 9/13/2017), and the National Research Council Guide for the Care and Use of Laboratory Animals. Animals were euthanized using an intramuscular injection of tiletamine and zolazepam (4 mg ∙ kg-1; Zoetis, Parsippany, NJ), followed by an intracardiac injection of pentobarbital sodium (0.4 mL · kg-1; Schering-Plough, Union, NJ).
The study was designed to investigate the effects of a high-fructose high-fat diet and probiotic supplementation in the pathogenesis and progression of pediatric non-alcoholic fatty liver disease. Twenty-eight 13-d-old Iberian pigs were housed in pairs in 1.5 × 1.5 m pens, and randomly distributed to receive 1 of the 4 liquid diets for 10 consecutive weeks: 1) control (CON-N; n = 4 pens), 2) high-fructose high-fat (HFF-N; n = 3 pens), 3) CON + probiotics (CON-P; n = 4 pens), and 4) HFF-N + probiotics (HFF-P; n = 3 pens). All animals were euthanized on d 70 of the study, and tissue from left medial segment in the liver was collected and frozen in liquid nitrogen for metabolomic analysis.
Metabolomics assays were performed in 26 liver samples (8 CON-N, 8 CON-P, 5 HFF-N, 5 HFF-P) using protein precipitation extraction with ultra-performance liquid chromatography-tandem mass spectrometry. Metabolite intensities were normalized to that of internal standards added during the extraction and to sample weight to account for small variations in starting tissue. Data was expressed as peak areas under the curve. A total of 218 metabolites were detected in liver tissue, with a few missing values produced due to metabolite peaks not being detected. These measurements were set to missing by placing a dot (.) in the CSV file.
Before a model fitting to one-metabolite-at-a-time can be implemented with our %polynova_2way macro, data must be analyzed with multivariate statistics using software such as R prcomp() function [8], MetaboAnalyst or Primer-E (v.7; Primer-E Ltd., Plymouth, UK), as previously described [7]. Briefly, liver data was imported into Primer-E, log transformed into a normal distribution approximation and analyzed with a Euclidean distance matrix. A nonparametric permutational analysis of variance (PERMANOVA; Primer-E) with diet × probiotic as fixed effects, and pen nested in diet × probiotic as random effect, was used for testing the null hypothesis of no difference between groups under a reduced model, 9,999 permutations, and type III (partial) sum of squares. PERMANOVA compares groups (i.e. diets) of multivariate sample units (i.e. pigs) by constructing ANOVA-like test statistics from a matrix of resemblances (distances, dissimilarities, similarities) calculated among the sample units, and obtains P-values using random permutations of observations among the groups. Results are presented in Table 1.

| Item | df | SS | MS | Pseudo-F | P(perm) | Unique perms |
|---|---|---|---|---|---|---|
| Probiotic | 1 | 2.598 | 2.598 | 0.489 | 0.905 | 9921 |
| Diet | 1 | 192.3 | 192.3 | 36.18 | 0.0002 | 9891 |
| Probiotic × Diet | 1 | 4.114 | 4.114 | 0.774 | 0.644 | 9931 |
| Pen (Probiotic × Diet) | 10 | 53.64 | 5.364 | |||
| Residual | 12 | 57.42 | 4.785 | |||
| Total | 25 | 322.7 |
Metabolomics data was further assessed by Principal Component Analysis (PCA) to visualize group discrimination in a 2-dimensional scores plot (Fig 1).

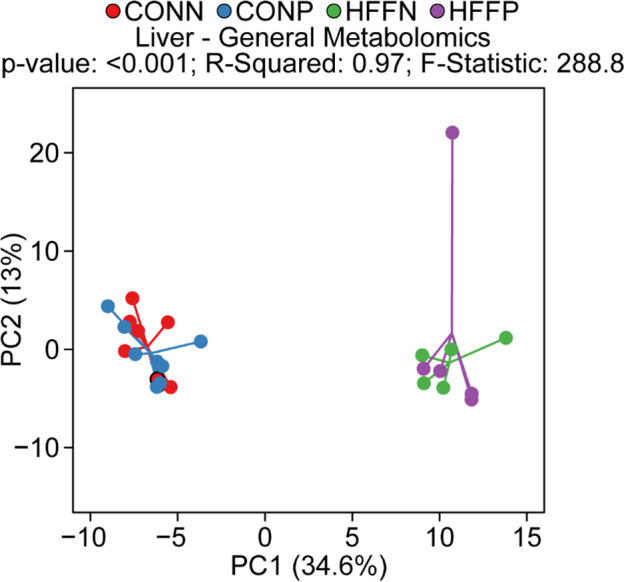
PCA analysis to visualize group discrimination in a 2-dimensional scores plot.
Two-dimensional visualization of PCA scores are projected from their group centroid along components 1 and 2. P-value, R-squared, and F-statistic are derived from ANOVA assessed on first principal component. Each point represents an individual pig and color of point denotes diet. Consistent with PERMANOVA analysis, PCA showed a separation of HFF and CON samples, but not a probiotic effect.
PCA analyses were conducted in the R Statistical Language using the prcomp() function [8]. Cube-root transformed concentrations or relative abundance data were scaled to unit variance prior to PCA assessment. PCA scores from components 1 and 2 were plotted and overlaid with diet and probiotic classifiers. Component 1 was assessed for group differences using ANOVA and we reported the total R2, F-statistic, and P-value from these models.
Our %polynova_2way macro, written in SAS software [9], allows for the identification of metabolites differentially expressed between treatment groups in complex study designs that involve treatment interactions and random effects. The SAS code is provided in S1 Code file. For each metabolite, the macro calculates the least squares means using a linear mixed model with fixed and random effects, runs a two-way ANOVA, adjusts the ANOVA P-values for the number of metabolites using the FDR [3] or Bonferroni [4] procedure, calculate the P-values of the least squares mean differences, and adjust these P-values for the number of pairwise comparisons using Tukey-Kramer or Bonferroni procedures. Finally, the macro combines the results into a single table (easily viewable in Microsoft Excel) that allows the researcher to easily sort through the metabolites and identify those significantly different among treatments (Fig 2).
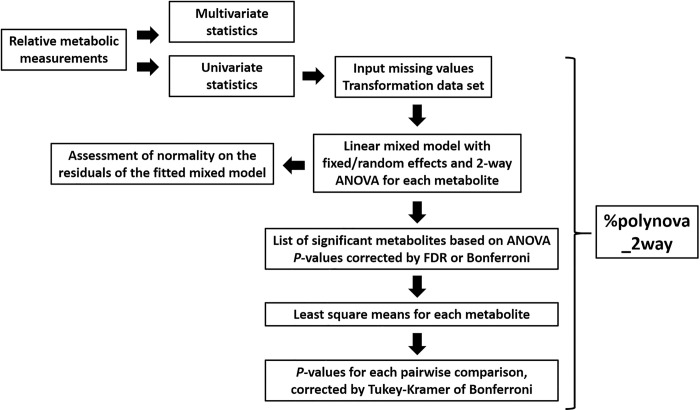
Overview of analytic workflow of %polynova_2way SAS macro.
Input is the table of relative metabolic measurements. The %polynova_2way macro outputs a table in excel format that combines all the results to facilitate the identification of significant metabolites.
The %polynova_2way macro is comprised of multiple sub-steps for fully processing and modeling metabolomics data: 1) The raw CSV data file is imported into SAS using PROC IMPORT. The macro assumes that the raw data file is in wide format with a separate column for each treatment and metabolite measurement and a separate row for each individual in the study (See S1 Data file). 2) The data are transposed into a long format so that later procedures can exploit the BY statement to analyze all of the data in a single PROC step. 3) The macro transforms the data to investigate model assumptions for each individual metabolite (see Results and Discussion). 4) The mixed model is fitted using PROC MIXED (output of the model, including least squares means, is saved in a corresponding dataset). 5) Tests for normality are performed on the residuals of the fitted models and corresponding P-values are saved. 6) P-values from the previously run PROC MIXED are FDR- or Bonferroni-adjusted for the number of metabolites using PROC MULTTEST. 7) Least squares mean estimates and confidence intervals back-transformed to the scale of the original data are computed and saved in a corresponding dataset. 8) P-values from least squares mean differences are corrected for the number of pairwise comparisons by Tukey-Kramer or Bonferroni and saved in a corresponding dataset. The remainder of this macro re-organizes and consolidates all of these results into a single, concise table of information. A separate, secondary macro (%export_mixed_res) exports the results of %polynova_2way in one of two ways: 1) one large, consolidated CSV file, or 2) a separate CSV file for each component of the results listed above.
In its current state the macro is robust and requires only minimal changes based on the dataset specified. As mentioned, the macro assumes that the data are in wide format (step one above). The %polynova_2way macro can accommodate many different sizes of dataset without adjustment. The user must provide the input and output parameters, and macro variable values as specified in Table 2. All parameters of the %polynova_2way macro are required. There are four variables defined above the macro which specify: 1) path: desired location of SAS dataset created by the macro (a single, SAS dataset version of the original CSV file), 2) respath: desired location of the resulting output, 3) normgraphpath: desired location of the normality diagnostics, including normal quantile-quantile plots, and 4) fitgraphpath: desired location of the fitted values versus residuals plots. The class_statement, model_vars, and random_statement parameters are all used in the respective PROC MIXED components: class statement, model statement, and random statement. These parameters give the user some flexibility to specify different types of models that PROC MIXED can accommodate. However, the remainder of the macro assumes that the user is fitting a model with two main effects plus an interaction that could include random effects. For more detail we refer the reader to the SAS documentation for PROC MIXED [9].
| Parameter | Description |
|---|---|
| Input | |
| path | Path to desired location of SAS dataset version of original CSV data file created by the macro. Do not include file name. e.g. C:\Users\manjarin\PLOSONE\dataset |
| respath | Path to desired results location, not including file name. e.g. C:\Users\manjarin\PLOSONE\dataset |
| normgraphpath | Path to normal quantile-quantile plots if requested. e.g. C:\Users\manjarin\PLOSONE\graphs |
| fitgraphpath | Path to fitted value plots if requested. e.g. C:\Users\manjarin\PLOSONE\graphs |
| raw_dsetin | Name of input dataset; Assumes this is path to CSV file, including file name C:\Users\manjarin\PLOSONE\dataset\S1_Data.csv |
| Macro variables (%macro polynova_2way) | |
| normtrans | Transformation for normality 0 (None), 1 (Log; default), 2 (Square root) |
| pval_adjust | Correction of ANOVA P-values by number of metabolites 1(FDR; default), 2(Bonferroni) |
| excludevars | Names of variables not to use as response, but used in model e.g. Trx1 Trx2 pig pen |
| ignorevars | Names of variables not used in model at all e.g. Sex |
| treatment_var1 | Name of the first treatment variable in your dataset e.g. Trx1 |
| treatment_var2 | Name of the second treatment variable in your dataset e.g. Trx2 |
| class_statement | Class variables in the PROC MIXED e.g. Trx1 Trx2 pen |
| model_vars | Fixed effects and interaction in the PROC MIXED e.g. Trx1|Trx2 |
| random_statement | Random effects in PROC MIXED e.g. pen(Trx1*Trx2) |
| pairwise_adjust | Correction of P-values of least squares mean differences by number of pair-wise comparisons. For comparisons of marginal means (main effects), use Tukey-Kramer. For comparisons of cell means (interaction), use Bonferroni (see Results and Discussion section for additional information) Tukey-Kramer (default), Bonferroni |
| Output labels (%export_mixed_res) | |
| name | Name of output CSV dataset file without “.csv” e.g. Liver_PLOS |
| trans | Name of transformation applied e.g. Log |
| adjust | ANOVA P-value correction e.g. FDR |
| singlefile | TRUE specifies all results should be consolidated and exported in a single CSV file TRUE (default) or FALSE |
Use of the macro does require a few precautions. First, SAS maintains a number of restrictions on variable names (metabolite names in the applications presented here). Before applying the macro to a dataset all of the variable names should conform to these restrictions, most importantly that they are less than 32 characters long and do not contain special characters other than underscores. Second, file names and file paths specified as parameters of the macro cannot contain spaces. Finally, the results are currently sorted by the first treatment variable’s P-values. So, the user should keep this in mind in determining which column they want to make the first treatment variable.
We demonstrate the use of the %polynova_2way macro in the analysis of metabolomics data generated in our pig study to investigate the role of HFF diets and probiotics in pediatric NAFLD. The dataset used in this example included pig identification number, pen, sex, Trx1 (probiotic) and Trx2 (diet), followed by a list of all the metabolites analyzed in liver tissue. In particular, the statistical model being used here is
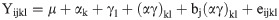
proc mixed data = metabolitedata;
by Response;
class Trx1 Trx2 pen;
model Value = Trx1|Trx2/ddfm = KENWARDROGER outpm = resids residual;
random pen(Trx1*Trx2);
lsmeans Trx2|Trx2/adjust = tukey cl;
run;
The SAS output consists of a single horizontal table in an excel file that contains results for each metabolite organized in rows, and the parameters from the SAS output in columns. To fit the table in the result section of the manuscript, it has been subdivided in 4 smaller tables presented below (Tables 3–6). Information included in the SAS output presented in Table 3 includes: 1) response variable, 2) results from the Shapiro-Wilk and Kolmogorov–Smirnov tests of normality conducted on residuals, 3) results of the 2-way ANOVA expressed as P-values and FDR- or Bonferroni corrected P-values for Trx1, Trx2 and their interaction (inter).
| Response | Transformation | Shapiro_Wilk | Kolmogorov | Pvalue_Trx1 | FDR Pvalue_Trx1 | Pvalue_Trx2 | FDR Pvalue_Trx2 | Pvalue_Inter | FDR Pvalue_Inter |
|---|---|---|---|---|---|---|---|---|---|
| pyridoxate | log | 0.54 | 0.15 | 0.66 | 0.99 | 0.001 | 0.001 | 0.15 | 0.95 |
| acetylchol | log | 0.55 | 0.15 | 0.94 | 0.98 | 0.008 | 0.01 | 0.87 | 0.95 |
| adenosine | log | 0.72 | 0.15 | 0.05 | 0.98 | 0.05 | 0.05 | 0.30 | 0.95 |
| anthranilic | log | 0.65 | 0.15 | 0.59 | 0.98 | 0.01 | 0.01 | 0.90 | 0.89 |
| Response | LS_N | CI_Lower_N | CI_Upper_N | LS_P | CI_Lower_P | CI_Upper_P |
|---|---|---|---|---|---|---|
| pyridoxate | 0.22 | 0.18 | 0.27 | 0.21 | 0.17 | 0.26 |
| acetylchol | 68154 | 52212 | 88965 | 69049 | 52897 | 90133 |
| adenosine | 2.31×108 | 2.10×108 | 2.53×108 | 2.65×108 | 2.40×108 | 2.92×108 |
| anthranilic | 29050 | 26594 | 31732 | 29962 | 27430 | 32729 |
| Response | P_valueD Trx1 N_P | P_valueD Trx2 CON_HFF | P_valueD Trx1_Trx2 N_CON P_CON | P_valueD Trx1_Trx2 N_CON N_HFF | P_valueD Trx1_Trx2 N_CON _PHFF | P_valueD Trx1_Trx2 N_HFF P_CON | P_valueD Trx1_Trx2 P_CON P_HFF | P_valueD Trx1_Trx2 N_HFF P_HFF |
|---|---|---|---|---|---|---|---|---|
| pyridoxate | 0.67 | 0.0007 | 0.41 | 0.09 | 0.005 | 0.02 | 0.0008 | 0.23 |
| acetylchol | 0.94 | 0.008 | 0.94 | 0.04 | 0.06 | 0.05 | 0.07 | 0.88 |
| adenosine | 0.04 | 0.05 | 0.35 | 0.04 | 0.96 | 0.009 | 0.47 | 0.06 |
| anthranilic | 0.59 | 0.01 | 0.74 | 0.04 | 0.09 | 0.02 | 0.05 | 0.67 |
| Response | TukeyD Trx1 NP | TukeyD Trx2 CON HFF | TukeyD Trx1_Trx2 N_CON P_CON | TukeyD Trx1_Trx2 N_CON N_HFF | TukeyD Trx1_Trx2 N_CON _P_HFF | TukeyD Trx1_Trx2 N_HFF P_CON | TukeyD Trx1_Trx2 P_CON P_HFF | TukeyD Trx1_Trx2 N_HFF P_HFF |
|---|---|---|---|---|---|---|---|---|
| pyridoxate | 0.67 | 0.001 | 0.83 | 0.32 | 0.02 | 0.09 | 0.004 | 0.61 |
| acetylchol | 0.94 | 0.008 | 1.00 | 0.16 | 0.22 | 0.18 | 0.24 | 0.99 |
| adenosine | 0.04 | 0.05 | 0.75 | 0.13 | 1.00 | 0.03 | 0.87 | 0.22 |
| anthranilic | 0.60 | 0.009 | 0.99 | 0.14 | 0.28 | 0.09 | 0.19 | 0.97 |
In addition to Shapiro-Wilk and Kolmogorov-Smirnov tests, normality diagnostics including quantile-quantile and fitted values vs. residuals plots are also generated in an additional pdf file. The decision to implement a transformation of the data or not needs to be made on a metabolite-by-metabolite basis, especially since mixed models will be fitted to one-metabolite-at-a-time. In fact, if transformation is applied indiscriminately to metabolites for which it is not needed, the transformation is likely to distort distributional assumptions and create problems where none were present. Therefore, the normality diagnostic output in the macro must be used along with the function normtrans = 0 (not-transformed), 1 (log-transformed), 2 (square-root transformed) to determine which metabolites need to be transformed, and which transformation need to be implemented, before examining the PROC MIXED output. The %polynova_2way macro has 2 limitations. The macro cannot transform individual metabolites within the dataset, so once non-normally distributed metabolites have been identified, the user will have to individually transform them in the csv file, and then rerun the macro with the function normtrans = 0. In addition, the macro can only implement log- and square root-transformations of the data. If different transformations are needed, the user will have to investigate them with methods such as Box-Cox. A SAS macro for Box-Cox that utilizes PROC MIXED has been published by Piepho [10].
An important output in Table 3 are the P-values and corrected P-values of the ANOVA for each metabolite, which allows the researcher: 1) to select significant metabolites for each factor and their interaction, and 2) to correct the P-values for the total number of metabolites using the FDR or Bonferroni procedure. For example, in our liver dataset the multivariate analysis (Table 1) identified a significant effect for diet, but not for probiotics or diet × probiotics, so we used Trx2 FDR-corrected P-values to select significant metabolites between CON and HFF diets.
To describe the expected magnitude of treatment effects, the SAS output also includes the least square means estimates (marginal means) and confidence intervals (Table 4). If the data were transformed for analysis, least squares means are back-transformed to the scale of the original data for reporting purposes.
Tables 5 and 6 consist of raw P-values and corrected P-values of the least squares mean differences for each metabolite, which allows the researcher: 1) to investigate the level of significance for each pairwise comparison, and 2) to correct the P-values for the number of pairwise comparisons by using Tukey-Kramer or Bonferroni procedures. In our liver dataset, given that we are only interested in the effect of diet, we used Trx2 CON_HFF Tukey-adjusted P-values to investigate differences between CON and HFF diets. Of note, Tukey-Kramer’s adjustment for multiplicity is appropriate to control Type I error when main effects are significant and the pairwise comparisons of interest are between marginal treatment means (i.e. N vs P, or CON vs HFF). This is indeed the case for the example explored herein. However, when interactions are significant and pairwise comparisons of interest need to be made between treatment cell means (i.e. simple effects), Tukey-Kramer’s adjustment quickly starts to overpenalize due to multiple unnecessary tests. Instead, relevant simple effects to follow significant interactions should be adjusted using a Bonferroni adjustment based on the number of comparisons of interest [11].
Strengths of the %polynova_2way SAS macro include its ability to statistically analyze group differences in metabolites in study designs with a factorial treatment structure and hierarchical design structure, and subsequently package an assortment of results into a single Excel. A limitation of the macro is that before it can be implemented, the user must investigate the data using multivariate statistics that needs to be run in additional software such as Primer-e, MetaboAnalyst or R language.
In conclusion, this paper presents a flexible SAS macro for analysis of metabolomic studies with two-way factorial treatment structures and experimental design leading to structured data. The macro represents a powerful tool that allows researchers to incorporate both fixed and random effects in the statistical models, implement transformation of the data to improve normality, select significant metabolites using FDR or Bonferroni corrections, and adjust P-values for multiple pairwise comparisons. The macro described herein increases the capacity of metabolomics researchers to appropriately analyze complex study designs by streamlining the implementation of sophisticated methods for large data sets. Because we acknowledge the focus on two-way analyses could be limiting, we have included code for a second macro, %polynova_1way which performs similar one-way analyses.
We thank Dr. Brian Piccolo for his assistance with the multivariate analysis.
1
2
3
4
5
6
7
8
9
10
11
 %polynova_2way: A SAS macro for implementation of mixed models for metabolomics data
%polynova_2way: A SAS macro for implementation of mixed models for metabolomics data
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

