
 ,
Hayley Tomes,
Jana Hagen,
Katherine Smith,
Chummy Sikasunge,
Dorit Hockman,
Murray E. Selkirk,
Joseph Valentino Raimondo
,
Hayley Tomes,
Jana Hagen,
Katherine Smith,
Chummy Sikasunge,
Dorit Hockman,
Murray E. Selkirk,
Joseph Valentino Raimondo
The authors have declared that no competing interests exist.
Larvae of the cestodes Taenia solium and Taenia crassiceps infect the central nervous system of humans. Taenia solium larvae in the brain cause neurocysticercosis, the leading cause of adult-acquired epilepsy worldwide. Relatively little is understood about how cestode-derived products modulate host neural and immune signalling. Acetylcholinesterases, a class of enzyme that breaks down acetylcholine, are produced by a host of parasitic worms to aid their survival in the host. Acetylcholine is an important signalling molecule in both the human nervous and immune systems, with powerful modulatory effects on the excitability of cortical networks. Therefore, it is important to establish whether cestode derived acetylcholinesterases may alter host neuronal cholinergic signalling. Here we make use of multiple techniques to profile acetylcholinesterase activity in different extracts of both Taenia crassiceps and Taenia solium larvae. We find that the larvae of both species contain substantial acetylcholinesterase activity. However, acetylcholinesterase activity is lower in Taenia solium as compared to Taenia crassiceps larvae. Further, whilst we observed acetylcholinesterase activity in all fractions of Taenia crassiceps larvae, including on the membrane surface and in the excreted/secreted extracts, we could not identify acetylcholinesterases on the membrane surface or in the excreted/secreted extracts of Taenia solium larvae. Bioinformatic analysis revealed conservation of the functional protein domains in the Taenia solium acetylcholinesterases, when compared to the homologous human sequence. Finally, using whole-cell patch clamp recordings in rat hippocampal brain slice cultures, we demonstrate that Taenia larval derived acetylcholinesterases can break down acetylcholine at a concentration which induces changes in neuronal signalling. Together, these findings highlight the possibility that Taenia larval acetylcholinesterases can interfere with cholinergic signalling in the host, potentially contributing to pathogenesis in neurocysticercosis.
Infection of the human nervous system with larvae of the parasite Taenia solium is a significant cause of acquired epilepsy worldwide. Despite this, the precise cellular and molecular mechanisms underlying epileptogenesis in neurocysticercosis remain unclear. Acetylcholinesterases are a family of enzymes widely produced by helminthic parasites. These enzymes facilitate the breakdown of acetylcholine, which is also a major neurotransmitter in the human nervous system. If T. solium larvae produce acetylcholinesterases, this could potentially disrupt host cholinergic signalling, which may in turn contribute to seizures and epilepsy. We therefore set out to investigate the presence and activity of acetylcholinesterases in T. solium larvae, as well as in Taenia crassiceps larvae, a species commonly used as a model parasite in neurocysticercosis research. We found that both T. crassiceps and T. solium larvae produce acetylcholinesterases with substantial activity and that the functional protein domains in the Taenia solium acetylcholinesterases have great similarity to those of human acetylcholinesterases. We further demonstrate that the acetylcholinesterase activity in the products of these parasites is sufficient to break down acetylcholine at a concentration which induces changes in neuronal signalling in an ex vivo brain slice model. This study provides evidence that Taenia larvae produce acetylcholinesterases and that these can potentially interfere with cholinergic signalling in the host and contribute to pathogenesis in neurocysticercosis.
Neurocysticercosis is a human disease which arises when larvae of the cestode Taenia solium (T. Solium) infect the central nervous system [1]. The most common symptom of this infection is the development of epileptic seizures, which occur in 70–90% of symptomatic neurocysticercosis cases [2]. As a result, neurocysticercosis is a major cause of adult-acquired epilepsy worldwide. Neurocysticercosis impacts heavily on the quality of life of those infected, and is also a significant drain on the medical and economic resources of endemic countries [3–5]. Despite the global impact of neurocysticercosis, precisely how cerebral infection with T. solium relates to the development of seizures remains unclear.
It has been well documented that many parasitic worms of the alimentary tract produce substances that aid them in modulating host responses in ways that benefit the parasite [6–8]. Acetylcholinesterases (AChEs), which catalyse the breakdown of acetylcholine, are one family of enzymes that have been implicated in the modulation of host responses. Helminths widely express membrane-bound forms of AChEs, which are classically associated with the facilitation of rapid acetylcholine signalling to parasite muscle, sensory, and neural structures [9,10]. Some also produce surface-presenting membrane-bound AChEs [11–14], or can actively excrete/secrete AChEs, which may modulate acetylcholine dependent components of the host immune response, play a role in detoxification of ingested cholinesterase inhibitors, or inhibit smooth muscle contraction and mucus and fluid secretion associated with clearance of intestinal parasites [10,15–17].
Acetylcholine is also a major neurotransmitter in the human brain, with powerful effects on the excitability of cortical circuits [18,19]. It is a critical component of multiple brain systems that are responsible for functions such as attention, learning, memory, sleep and motor activity [20,21]. Disruption of cholinergic signalling is well known to lead to seizures. For instance, mutations of the nicotinic acetylcholine receptor (in the genes coding for the β4 or α2 subunits) underlie a heritable form of epilepsy called autosomal dominant nocturnal frontal lobe epilepsy [22]. The mutant receptors are more sensitive to acetylcholine than normal receptors, and may generate seizures by promoting and synchronizing spontaneous oscillations in thalamo-cortical circuits [22]. Further, pilocarpine (an acetylcholine muscarinic receptor agonist) is a well described proconvulsant agent [23] and the blockade of endogenous brain AChEs by organophosphate pesticides or poisons can also cause seizures [24,25].
Since T. solium larvae invade the central nervous system in neurocysticercosis, it is important to determine potential AChE activity expressed by these larvae, as such activity could conceivably interfere with endogenous cholinergic signalling in the brain by breaking acetylcholine down into neurologically inactive products [26]. Taenia crassiceps (T. crassiceps) is a related cestode, which has also been known to invade the human nervous system, and is widely utilised as a model parasite for T. solium in animal models of cysticercosis and neurocysticercosis [27–29]. It is therefore also important to ascertain how AChE activity might compare between the larvae of these two Taenia species.
AChEs have been reported in the adult forms of several members of the broader Taeniidae family [30–32] as well as in larval stages [9,11,13,33,34]. The AChEs are often associated with the neural structures and parasite tegument of Taeniids, and there is also some suggestion that some Taeniid larvae may release AChEs into the host environment [33]. Studies describing cholinesterases in T. crassiceps larvae are scarce, with one report of AChEs in the bladder wall of T. crassiceps [35], and one other study which refers to the presence of “unidentified esterases” in the cystic fluid of T. crassiceps [36].
The genome of T. solium has recently been sequenced [37] and bioinformatics have revealed three T. solium acetylcholinesterase homologs [16]. Whether the acetylcholinesterases are expressed during the larval stage of T. solium, and if so, what their activities and locations are, is not yet clear. A histological study by Vasantha et al. [38] in T. solium larvae demonstrated AChE staining in neural structures of the larvae. No obvious staining of AChEs on the surface of the larvae is described, apart from positive staining in a few surface nerve endings. We further found one other report of cholinesterase activity in T. solium larvae, with activity predominantly present in the isolated cyst bladder ([39] cited in [40]).
Therefore, there is an important need for a detailed characterization of AChE activity in the larvae of different Taenia species, as well as an investigation into whether larval derived AChEs could conceivably disrupt host neuronal cholinergic signalling. Here, we used multiple techniques to explore AChEs activity in different extracts of both T. crassiceps and T. solium larvae. We find that both the larvae of T. crassiceps and T. solium contain significant AChE activity, but it is broadly lower in T. solium as compared to T. crassiceps larvae. In addition, whilst AChEs were present in all fractions of T. crassiceps larvae, including the membrane surface and excreted/secreted extracts, we could not identify AChEs on the membrane surface or within the excreted/secreted extracts of T. solium larvae. Using bioinformatic approaches we show that functional protein domains in the Taenia solium acetylcholinesterases are conserved, when compared to the homologous human sequence. Finally, using whole-cell patch clamp recordings in rodent hippocampal brain slice cultures we demonstrate that Taenia larval derived AChEs can break down acetylcholine at a concentration which induces changes in neuronal signalling.
All animal handling, care and procedures were carried out in accordance with South African national guidelines (South African National Standard: The care and use of animals for scientific purposes, 2008) and with approval from the University of Cape Town Animal Ethics Committee (Protocol No: AEC 019/025, AEC 014/035).
The protein sequences for T. solium and T. saginata cholinesterase homologues, identified in Tedla et al. [16], were downloaded from https://parasite.wormbase.org/ (Accession numbers: TsM_000001700 [Tso1], TsM_000234300 [Tso2], TsM_001220100 [Tso3], TSAs00071g07627m00001 [Tsa]). The human protein sequence was downloaded from NCBI (Accession number NP000656 [Hs]). Protein sequences were aligned using ClustalW in SnapGene (v5.5.5) and the presented alignment prepared with BOXSHADE (https://embnet.vital-it.ch/software/BOX_form.html). Annotations of protein domains were based on those in Tedla et al. [16]. The percent identity matrix for the analysed protein sequences was generated using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Larvae (ORF strain) were donated by Dr Siddhartha Mahanty (University of Melbourne, Melbourne, Australia) and propagated in vivo by serial intraperitoneal infection of 5-8-week-old female C57BL/6 mice. Every 3 months parasites were harvested by peritoneal lavage and washed 6 times in phosphate buffered saline (PBS, 1X, pH 7.4) before further processing.
Larvae were frozen at -80°C immediately after harvesting. Upon thawing, larvae were suspended in a volume of PBS threefold that of the larvae. A protease inhibitor cocktail was added to this suspension (1% vol/vol, Sigma-Aldrich). The larvae were then homogenised on ice using a glass tissue grinder. The resulting mixture was centrifuged at 3100 g for 20 minutes at 4°C. The liquid supernatant (excluding the low density white floating layer) was collected and sterile filtered through a 0.22 μm size filter (Millex-GV syringe filter, Merck). This supernatant was then aliquoted and stored at -80°C until use. This preparation is referred to as “T. crassiceps whole cyst homogenate”.
After harvesting, washed larvae (+/- 10ml) were placed onto a piece of filter paper (which had been saturated with 1X PBS) in a metal sieve. Cysts were then ruptured using a weighing spatula. The fluid from the ruptured cysts that passed through the filter paper was collected in a beaker and was centrifuged at 3100 g for 20 minutes at 4°C, and the supernatant was collected, aliquoted and stored at -80°C until use. This extract is referred to as “T. crassiceps cyst vesicular fluid”. The fraction of the cysts that remained on the filter paper were scraped off with the weighing spatula and suspended in an equal volume of PBS containing a protease inhibitor cocktail (1% vol/vol, Sigma-Aldrich). This mixture was freeze-thawed once at -80°C, homogenised on ice using a glass tissue grinder, and centrifuged at 3100 g for 20 minutes at 4°C. The liquid supernatant was collected, aliquoted, and stored at -80°C until use. This extract is referred to as “T. crassiceps cyst membrane”.
After harvesting, washed larvae (+/- 10 ml) were placed in a 50 ml culture flask with 10 ml culture medium (Earle’s Balanced Salt Solution with 5.3 g/L glucose, 1X Glutamax, 50 U/ml penicillin, 50 μg/ml streptomycin, 100 μg/ml gentamicin sulphate and 11.4 U/ml nystatin). Larvae were maintained at 37°C in 5% CO2. After 48 hrs the medium was discarded and replaced with 10 ml fresh media. At 20 days in vitro (at which point larvae still displayed motility) the culture media was collected, aliquoted, and stored at -80°C until use. For electrophysiology experiments the excretory/secretory extracts were dialysed/buffer exchanged with PBS using an Amicon stirred cell (Merck) with a 3 kDa molecular weight cut-off membrane, in order to remove small molecules that could potentially induce electrophysiological responses that would interfere with the acetylcholine effect (such as glutamate—see [41].
Larvae of T. solium were harvested from the muscles of a heavily infected, freshly slaughtered pig in Lusaka, Zambia. Larvae were removed from the muscle by vigorous shaking and collected in petri dishes containing sterile PBS (1X, pH 7.4).
After extensive washing with sterile PBS (1X, pH 7.4), larvae were suspended in a volume of PBS threefold that of the larvae, containing phenylmethyl-sulphonyl fluoride (5 mM) and leupeptin (2.5 μM). Larvae were then homogenised using a sterile handheld homogenizer at 4°C. The resulting homogenate was sonicated (4 x 60 s at 20 kHz, 1 mA, with 30 s intervals), gently stirred with a magnetic stirrer (2 hrs at 4°C), and centrifuged at 15 000 g for 60 min at 4°C. The liquid supernatant (excluding the low density white floating layer) was collected and sterile filtered through 0.45 μm size filters (Millex-GV syringe filter, Merck). This supernatant was then collected, aliquoted and stored at -80°C until use. This preparation is referred to as “T. solium whole cyst homogenate”.
After extensive washing with sterile PBS, larvae were placed in a petri dish and individually ruptured with a sterile needle. The resulting fluid in the petri dish was collected and centrifuged at 15 000 g for 60 min at 4°C. The supernatant was then sonicated (4 x 60 s at 20 kHz, 1 mA, with 30 s intervals), phenylmethyl-sulphonyl fluoride (5 mM) and leupeptin (2.5 μM) were added, and the solution was centrifuged a second time at 15,000 g for 60 min at 4°C. The supernatant was collected, aliquoted and stored at -80°C until use. This extract is referred to as “T. solium cyst vesicular fluid”. The remaining parts of the larvae were again extensively washed with PBS and then suspended in an equal volume of PBS containing phenylmethyl-sulphonyl fluoride (5 mM) and leupeptin (2.5 μM). This suspension was again homogenised using a sterile handheld homogenizer at 4°C. The resulting homogenate was sonicated (4 x 60 s at 20 kHz, 1 mA, with 30 s intervals), gently stirred with a magnetic stirrer (2h at 4°C), and centrifuged at 15,000 g for 60 min at 4°C. The liquid supernatant (excluding the low density white floating layer) was collected and sterile filtered through 0.45 μm size filters (Millex-GV syringe filter, Merck). This supernatant was then aliquoted and stored at -80°C until use. This extract is referred to as “T. solium cyst membrane and scolex”.
After harvesting, washed larvae were placed into 6 well plates (+/- 15 per well) with 2 ml culture medium (RPMI 1640 with 10 mM HEPES buffer, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B and 2 mM L-glutamine). Every 24 h, 1 ml of culture medium was collected from each well and replaced with fresh culture medium. Medium from all wells was pooled each day, aliquoted and stored at -80°C. Media collected on days 1, 2, and 3 in vitro were pooled, and are referred to as “T. solium excretory/secretory extracts”.
All T. crassiceps and T. solium larval extracts were assessed for protein concentration using a BCA or Bradford protein assay kit (Sigma-Aldrich), respectively.
AChE activity was determined by the method of Ellman et al. [42] at room temperature with 1 mM acetylthiocholine iodide as substrate in the presence of 1 mM 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB) in 100 mM sodium phosphate (pH 7.0). The reaction was monitored by measuring the absorbance at 412 nm, and hydrolysis of acetylthiocholine iodide calculated from the extinction coefficient of DTNB [42]. Activity was expressed as nanomoles of acetylthiocholine hydrolysed per minute per milligram of total protein in each larval extract (nmol min-1 mg-1). To test the sensitivity of Taenia AChEs to different inhibitors, extracts were preincubated with different concentrations of 1,5-bis(4-allyldimethylammoniumphenyl)pentan-3-one dibromide (BW 284c51), tetraisopropyl pyrophosphoramide (iso-OMPA) or eserine salicylate for 20 min at room temperature in Ellman buffer, prior to the addition of 1 mM acetylthiocholine iodide and enzyme activity determination. Each reaction was assayed a minimum of three times. Where AChE activity was reduced to undetectable levels by inhibitors, a residual activity of 0% was allocated on inhibition curves.
Extracts were electrophoresed in Tris-glycine buffer, pH 8.3, through 7.5% polyacrylamide gels in the absence of denaturing and reducing agents. Electrophoresis was performed at 150 V for 3 hrs on ice. Protein staining (Coomassie) was performed on one set of PAGE gels, and specific staining for AChE activity was performed overnight as described by Selkirk and Hussein [43] adapted from the method of Karnovsky and Roots [44]. The maximum volume of each protein extract was loaded (20 μl), to ensure maximal staining. Protein concentrations of the different extracts varied (T. crassiceps: whole cyst homogenate = 1.9 mg/ml, cyst membrane = 3.4 mg/ml, cyst vesicular fluid = 3.0 mg/ml and excretory/secretory extracts = 1.32 mg/ml; T. solium: all extracts = 1.5 mg/ml). AChE stains were performed at least three times to ensure reproducibility.
To localise Taenia AChEs, Taenia larvae were submerged in 10% formalin for 60 min, to fix the tissue. Some of the larvae were then stained overnight for AChE activity as described by Selkirk and Hussein [43], and mounted onto slides as whole mounts. A subset of the fixed larvae was embedded in cryo-embedding medium, frozen overnight at -80°C, and cryo-sectioned the following day at 50 μm. The sections were then similarly stained overnight for AChE activity, placed on positively charged slides and dehydrated in graded alcohols before mounting. To assess non-specific staining, in a subset of the whole mount and cryo-section specimens, acetylthiocholine iodide (the substrate) was omitted during the AChE staining procedure. Specimens were imaged using an upright light microscope.
Organotypic brain slices were prepared using 6-8-day-old Wistar rats following the protocol originally described by Stoppini et al. [45]. Briefly, brains were extracted and swiftly placed in cold (4°C) dissection media consisting of Earle’s Balanced Salt Solution (Sigma-Aldrich) supplemented with D-glucose (6.1 g/L) and HEPES (6.6 g/L). The hemispheres were separated, and individual hippocampi were removed and immediately cut into 350 μm slices using a Mcllwain tissue chopper (Mickle). Cold dissection media was used to separate and rinse the slices before placing them onto Millicell-CM membranes (Sigma-Aldrich). Slices were maintained in culture medium consisting of 25% (vol/vol) Earle’s balanced salt solution; 49% (vol/vol) minimum essential medium (Sigma-Aldrich); 25% (vol/vol) heat-inactivated horse serum (Sigma-Aldrich); 1% (vol/vol) B27 (Invitrogen, Life Technologies) and 6.2 g/l D-glucose (Sigma-Aldrich). Slices were incubated in a 5% carbon dioxide (CO2), humidified incubator at 37°C. Recordings were made after 6–14 days in culture.
Brain slices were transferred to a submerged recording chamber on a patch clamp rig, which was maintained at a temperature between 28 and 34°C, and were continuously superfused with standard artificial cerebrospinal fluid (120 mM NaCl, 3mM KCl, 2 mM MgCl2, 2 mM CaCl2, 1.2 mM NaH2PO4, 23 mM NaHCO3 and 11 mM D-Glucose in deionised water with pH adjusted to between 7.35–7.40 using 0.1 mM NaOH) bubbled with carbogen gas (95% O2: 5% CO2) using peristaltic pumps (Watson-Marlow). Micropipettes were prepared (tip resistance between 3 and 7 MΩ) from borosilicate glass capillaries (outer diameter 1.2 mm, inner diameter 0.69 mm) (Harvard Apparatus Ltd) using a horizontal puller (Sutter). Micropipettes utilised for whole cell patch clamping were filled with an artificial cell internal solution (126 mM K-gluconate, 4 mM KCl, 10 mM HEPES, 4 mM Na2ATP, 0.3 mM NaGTP and 10 mM Na2-phosphocreatine) before being placed over the recording electrode.
Neurons in the CA3 region of the hippocampus were visualized using an upright microscope with a 20X water immersion objective. Surface cells with a typical pyramidal cell body morphology were selected for whole cell patching. To explore the ability of Taenia acetylcholinesterase activity to alter neuronal acetylcholine signalling, two additional “puffer” micropipettes were lowered to the cell surface once a neuron had been patched. One of these micropipettes contained a solution of 200 μm acetylcholine with 1.3 mg/ml T. crassiceps excretory/secretory extracts, while the second contained a solution of 200 μm acetylcholine with 1.3 mg/ml T. crassiceps excretory/secretory extracts that had been heated to 56°C for 30 min to inactivate enzymes. Current was injected to hold the membrane potential of cells close to their action potential firing threshold. Five 30 ms puffs (~20 psi) of one of the solutions was then applied to the cell’s surface using an OpenSpritzer [46] and the neuron’s response recorded for 26 s before a 94 s “recovery” period was allowed. Thereafter an identical puff train of the other solution was applied, and the neuron’s response again recorded for 26 s. After another 94 s recovery period, the cycle was repeated.
Post-recording analysis consisted of counting the number of action potentials induced by the application of each solution within a 5 s period of the onset of the puff train. Only traces with a baseline membrane potential prior to the puff application of between -60 mV and -45 mV were included. Each data point in the puffing experiments represents the average of between 2 and 5 repeats of the puff cycle. Matlab (MathWorks) was utilised for trace analysis.
Data was visualised and analysed using Matlab, Microsoft Excel and GraphPad Prism. Each dataset was subjected to a Shapiro-Wilk test to determine whether it was normally distributed. Most datasets proved to be non-normal and as such non-parametric analyses were utilised throughout. These included: Kruskal-Wallis analyses with Dunn’s multiple comparison post-hoc tests and Mann-Whitney tests. The confidence interval for all tests was set at 95%.
In order to quantify AChE activity in T. crassiceps larvae, Ellman’s assays were employed, using acetylthiocholine as a substrate [42]. These assays revealed that all T. crassiceps larval extracts had significant AChE activity (Table 1and Fig 1A). A Kruskal Wallis one-way ANOVA with post hoc Dunn’s Multiple Comparison tests revealed that the only statistically significant difference between the median activities of the different T. crassiceps larval extracts was between that of the cyst vesicular fluid and that of the excretory/secretory extracts (P ≤ 0.01, Fig 1A).

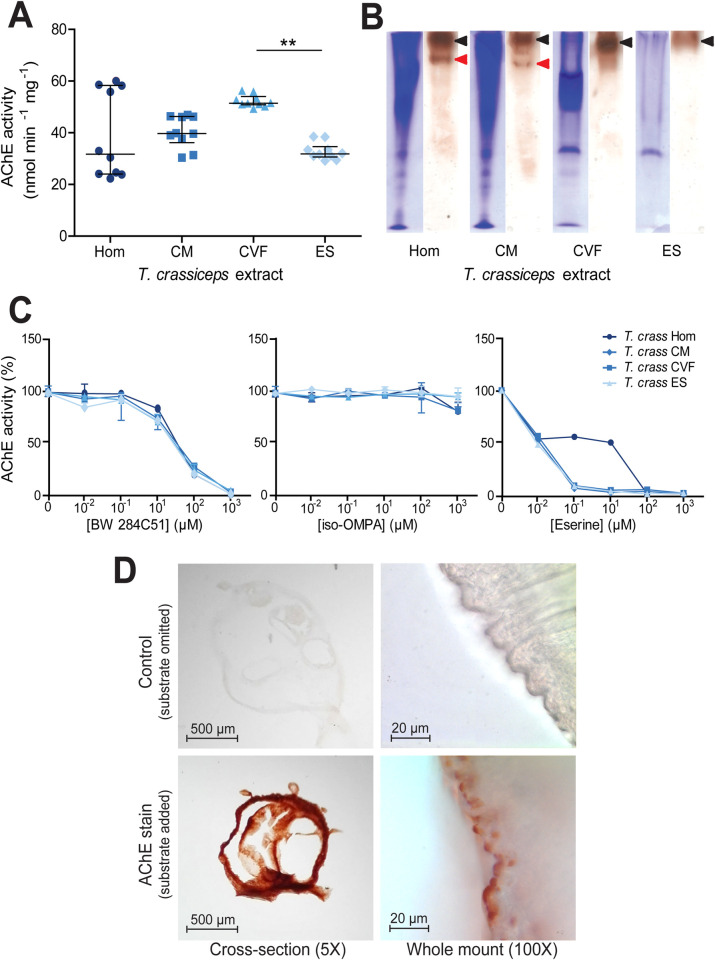
Identification and characterisation of acetylcholinesterases in Taenia crassiceps larval extracts.
A) Quantification of acetylcholinesterase (AChE) activity in different Taenia crassiceps larval extracts. AChE activity was quantified using the method of Ellman et al. [42] with 1 mM acetylthiocholine iodide as substrate in the presence of 1 mM 5,5’-dithiobis(2-nitrobenzoic acid) in 100 mM sodium phosphate, pH 7.0, at room temperature. The extracts assessed were: whole cyst homogenate (Hom); cyst membrane (CM), cyst vesicular fluid (CVF) and larval excretory/secretory extracts (ES). Values with median ± IQR, N = 10 for all extracts assayed, **p ≤ 0.01, Kruskal-Wallis test with Dunn’s multiple comparison post-hoc tests B) Non-denaturing polyacrylamide gel electrophoresis of Taenia crassiceps extracts. Extracts were electrophoresed in Tris-glycine buffer, pH 8.3, through 7.5% polyacrylamide gels in the absence of denaturing and reducing agents. Coomassie staining was performed on one set of gels (left tracks), and staining for AChE activity [44] was performed on another set of gels for 16 hrs after incubation with the substrate (right tracks). The maximum volume of each extract was loaded (20 μl), to ensure maximal staining. Protein concentrations of the different extracts varied: Hom = 1.9 mg ml-1, CM = 3.4 mg ml-1, CVF = 3.0 mg ml-1 and ES = 1.32 mg ml-1. C) Inhibitor sensitivity of Taenia crassiceps AChEs. Taenia crassiceps extracts were preincubated with BW 284C51, iso-OMPA or eserine salicylate (Eserine) for 20 min at room temperature in Ellman buffer, prior to the addition of 1 mM acetylthiocholine iodide and enzyme activity determination. Median ± Range, N = 10 for all extracts in absence of inhibitors, N = 3 for all extracts at all inhibitor concentrations. D) Localisation of larval AChEs. Cryo-sections (N = 19 for control and N = 19 for AChE stain) and whole mounts (N = 15 for control and N = 15 for AChE stain) of Taenia crassiceps larvae were subjected to AChE staining [44] for 16 hrs prior to dehydration and mounting. Images in top panels show time-matched controls where acetylthiocholine iodide was omitted from the staining solution. Cross sections were imaged at 5X magnification and whole mounts were imaged at 100X magnification.

| Larval species | Larval extract | # of assays | Median AChE activity (nmol min-1 mg-1) |
|---|---|---|---|
| Whole cyst homogenate | 10 | 31.7 (IQR 24.0–58.3) | |
| Taenia crassiceps | Cyst membrane | 10 | 39.7 (IQR 36.1–46.3) |
| Cyst vesicular fluid | 10 | 51.5 (IQR 50.9–54.1) | |
| Excretory/secretory extracts | 10 | 31.8 (IQR 50.9–54.1) | |
| Whole cyst homogenate | 5 | 4.1 (IQR 4.1–5.1) | |
| Taenia solium | Cyst membrane & scolex | 5 | 14.0 (IQR 13.8–15.3) |
| Cyst vesicular fluid | 5 | 4.7 (IQR 3.37–5.03) | |
| Excretory/secretory extracts | 4 | Undetectable |
To visually confirm T. crassiceps AChE activity, and to assess whether there may be more than one AChE isoform in T. crassiceps larval extracts, non-denaturing PAGE gels were run, and stained for AChE. A second set of non-denaturing PAGE gels were run simultaneously and Coomassie stained. These demonstrated that the different T. crassiceps larval extracts contained different protein compositions (left tracks in Fig 1B). The AChE stained gels (right track for each larval extract in Fig 1B) showed distinct dark bands (indicated by black arrowheads) in the tracks of all the T. crassiceps larval extracts, thereby confirming that all the T. crassiceps larval extracts show AChE activity. In the whole cyst homogenate and the cyst membrane tracks of the AChE stained gels, there was an additional smaller band (indicated by the red arrowheads in Fig 1B). These results suggest that T. crassiceps larvae express more than one isoform of AChE.
The sensitivity of AChE activity in T. crassiceps larval extracts to different inhibitors was tested by preincubating them for 20 min with different concentrations of BW 284c51 (a selective AChE inhibitor), iso-OMPA (a selective butyryl cholinesterase inhibitor) or eserine salicylate (a nonselective cholinesterase inhibitor) before assaying AChE activity [42]. All T. crassiceps extracts showed a similar dose-dependent inhibitory response to BW 284c51, with AChE activity in all extracts being almost completely inhibited by the presence of 1000 μM BW 284c51 (Fig 1C and S1 Table). Conversely, the AChE activity of T. crassiceps extracts was not greatly inhibited by iso-OMPA, with only very small reductions in activity being observed even at high (1 mM) inhibitor concentration (Fig 1C and S1 Table). AChE activity in T. crassiceps cyst membrane, cyst vesicular fluid and excretory/secretory extracts was highly sensitive to eserine inhibition, with strong inhibition apparent at low (1 μM) eserine concentration (Fig 1C and S1 Table). The AChE activity of T. crassiceps whole cyst homogenate was less sensitive to eserine inhibition, only displaying strong inhibition at a much higher eserine concentration (100 μM) (Fig 1C and S1 Table). These inhibition patterns suggest that the cholinesterase produced by T. crassiceps larvae can be classified as true AChEs, as opposed to pseudocholinesterases [47].
To spatially localise AChEs within the larvae, both cross-sections of larvae and whole larvae were subjected to AChE staining [44], (Fig 1D). To evaluate non-specific staining, a second set of larval cross-sections and whole larvae were subjected to the same staining procedure, with the exception that the substrate (acetylthiocholine iodide) was omitted. Samples where the substrate was omitted (top panels, Fig 1D) showed minimal staining. In contrast, cross sections stained for AChE activity displayed dense, uniform staining, indicating that AChEs are localised ubiquitously throughout the tegument membrane (bottom-left panel, Fig 1D). Whole larvae stained for AChE revealed that staining localised to numerous small protrusions on the surface of the cyst tegument membrane (bottom-right panel, Fig 1D).
Next, we focussed on the major pathogenic cestode of humans; T. solium. Ellman’s assays revealed that T. solium whole cyst homogenate, cyst membrane and scolex, and cyst vesicular fluid had detectable AChE activity, whilst the excretory/secretory extracts consistently displayed no detectable AChE activity (Table 1 and Fig 2A). A Kruskal Wallis one-way ANOVA with post hoc Dunn’s Multiple Comparison tests revealed that the cyst membrane and scolex had a statistically significantly higher activity than that of the whole cyst homogenate and the cyst vesicular fluid, whilst the median activity of two latter extracts did not differ significantly from one another (P ≤ 0.01, Fig 2A).
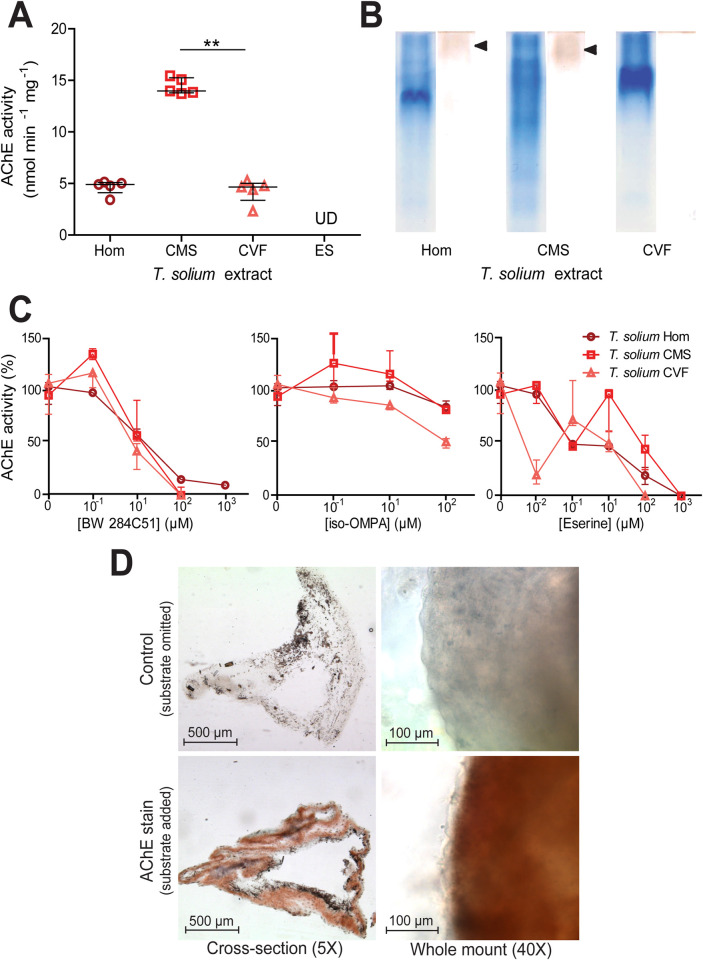
Identification and characterisation of acetylcholinesterases in Taenia solium extracts.
A) Quantification of acetylcholinesterase (AChE) activity in different Taenia solium extracts as in Fig 1. The extracts assessed were: whole cyst homogenate (Hom); cyst membrane and scolex (CMS), cyst vesicular fluid (CVF) and larval excretory/secretory extracts (ES). Values with median ± IQR, N = 5 for all extracts assayed, **p ≤ 0.01, Kruskal-Wallis test with Dunn’s multiple comparison post-hoc tests. UD = undetectable. B) Non-denaturing polyacrylamide gel electrophoresis of different Taenia solium extracts. Coomassie staining was performed on one set of PAGE gels (left tracks), and staining for AChE activity [44] was performed on another set of gels (right tracks). For each extract, 30 μg of total protein was loaded. C) Inhibitor sensitivity of Taenia solium AChEs. Taenia solium extracts were preincubated with BW 284C51, iso-OMPA or eserine salicylate (Eserine) for 20 min at room temperature in Ellman buffer, prior to the addition of 1 mM acetylthiocholine iodide and enzyme activity determination. Median ± Range, N = 5 for all extracts in absence of inhibitors and at 10 μM inhibitor concentration, N = 3 for all extracts at all other inhibitor concentrations. D) Localisation of larval AChEs. Cryo-sections (N = 20 for control and N = 20 for AChE stain) and whole mounts (N = 4 for control and N = 4 for AChE stain) of Taenia solium larvae were subjected to AChE staining [44]. Images in top row show time-matched controls where acetylthiocholine iodide was omitted from the staining solution. Cross sections were imaged at 5X magnification and whole mounts were imaged at 40X magnification.
To visually confirm AChE activity in T. solium, and to assess whether there may be more than one AChE isoform in larval extracts, non-denaturing PAGE gels were resolved and stained for AChE (Fig 2B). Again, a second set of non-denaturing PAGE gels were run simultaneously and Coomassie stained. The Coomassie stain showed that each extract has a distinct protein profile composition (left tracks in Fig 2B). The AChE stained gels (right track for each larval extract in Fig 1B) revealed bands in the whole cyst homogenate and in the cyst membrane and scolex preparations (indicated by black arrowheads), but no apparent band in the cyst vesicular fluid track. The absence of bands in the cyst vesicular fluid tract, and the relatively faint bands in tracks of the other extracts can be attributed to the fact that the amount and concentrations of the extracts loaded contain a drastically lower total AChE activity than is usually recommended for this technique [43]. In future investigations this could be solved by concentration or purification of the AChEs in T. solium extracts.
The sensitivity of AChE activity in T. solium larval extracts to different inhibitors was tested by preincubating them for 20 min with different concentrations of BW 284c51, iso-OMPA or eserine salicylate, before assaying AChE activity [42]. All T. solium extracts showed a similar dose-dependent inhibitory response to BW 284c51, although the T. solium whole cyst homogenate appears somewhat less sensitive to inhibition than the cyst membrane and scolex and cyst vesicular fluid (Fig 2C and S2 Table). T. solium extracts showed low sensitivity to inhibition by iso-OMPA (Fig 2C and S2 Table). T. solium whole cyst homogenate, cyst membrane and scolex, and cyst vesicular fluid showed very variable sensitivities to inhibition by increasing concentrations of eserine, but were ultimately all strongly inhibited at an eserine concentration of 1 mM or less (Fig 2C and S2 Table). These inhibition patterns suggest that the cholinesterases produced by T. solium larvae can be classified as true AChEs, as opposed to pseudocholinesterases [47].
Next, we set out to spatially localise AChEs within the larvae. To do so both cross-sections of larvae and whole larvae were subjected to the same AChE staining as was applied to T. crassiceps larvae. Control samples where the substrate was omitted to evaluate non-specific staining (top panels, Fig 2D), showed some patchy black background staining. AChE stained cross-sections showed uniform, although not very dense, AChE staining throughout the tegument membrane (in addition to the black background staining) (bottom-left panel, Fig 2D). High magnification images of the surface of the tegument membrane in whole-mounted AChE-stained larvae revealed that, unlike in T. crassiceps, AChEs in the tegument membrane of T. solium are not surface-presenting (bottom-right panel, Fig 2D).
Given that the genome for T. solium and the related species Taenia saginata (T. saginata) are available (the genome for T. crassiceps has not yet been sequenced), we performed amino acid sequence alignments to determine the putative structural resemblance between host (Homo sapiens) and parasite acetylcholinesterase enzymes. The analysis of full amino acid sequence alignments for T. solium, T. saginata and Homo sapiens (H. sapiens) acetylcholinesterases (Fig 3) revealed the percent identity between the acetylcholinesterase of T. saginata (TSa_s00071g07627m00001) and one of the T. solium acetylcholinesterase sequences (Ts_000234300) to be 97%, whilst there was only a 33% and 38% identity between the acetylcholinesterase of T. saginata and the other two T. solium acetylcholinesterase sequences (Ts_000001700 and Ts_001220100, respectively). The percent identity between the three different T. solium acetylcholinesterase sequences ranged from 33% to 38%, and the percent identity of these to the of H. sapiens acetylcholinesterase (Hs_NP_000656) similarly ranged from 36% to 39%. Importantly, the “catalytic triad”, which has been described to form the active site for ester hydrolysis within the enzyme, is fully conserved between H. sapiens and two of the three T. solium acetylcholinesterase amino acid sequences (TsM 000001700 [Tso1], and TsM 001220100 [Tso3], yellow boxes in Fig 3), while three of the four sites that form the “acyl binding pocket”, a region of the enzyme that confers substrate specificity, are fully conserved across all the analysed sequences (green boxes in Fig 3) [16,48]. These findings support our earlier findings that T. solium contains cholinesterases and that these are true acetylcholinesterases, as opposed to pseudocholinesterases.
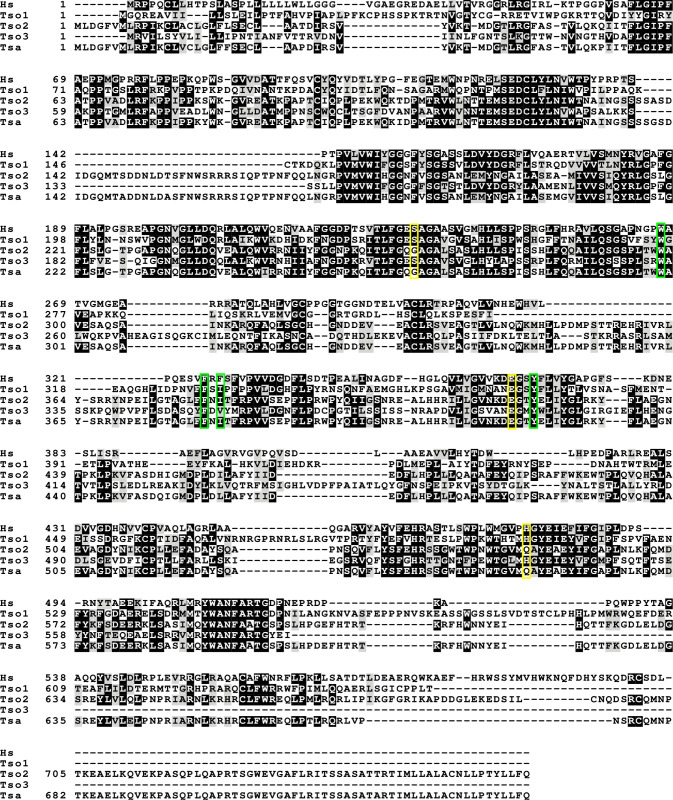
Full amino acid sequence alignment for Taenia solium, Taenia saginata and Homo sapiens acetylcholinesterase proteins.
(Accession numbers: NP000656 [Hs], TsM_000001700 [Tso1], TsM_000234300 [Tso2], TsM_001220100 [Tso3], TSAs00071g07627m00001 [Tsa]). The catalytic triad is indicated in yellow and the acyl binding pocket is indicated in green.
Comparison of AChE activity in the extracts of T. crassiceps versus the comparable T. solium extracts show that T. solium extracts consistently have significantly lower AChE activities than those of T. crassiceps (P ≤ 0.001, Mann Whitney tests, Table 1). Further, a different pattern of AChE distribution within the cyst is observed between the two species–T. solium AChEs appear to be predominantly located in the cyst membrane and scolex, whilst T. crassiceps AChEs appear abundant in both the cyst membrane and in the cyst vesicular fluid and are additionally excreted/secreted (Table 1). T. crassiceps also displays surface presenting AChEs, whilst T. solium do not, as revealed by AChE staining of whole larvae (Figs 1D and 2D). It is also noteworthy that T. crassiceps and T. solium larval extracts display different inhibitor sensitivities to BW 284c51, iso-OMPA and particularly to eserine (Fig 1C versus Fig 2C). This suggests that the two parasites produce different forms of AChE, although further investigation would be required to confirm this.
In order to investigate what implications the presence of Taenia larval AChEs may have in the context of neurocysticercosis, 200 μM acetylcholine (known to induce depolarisation in hippocampal pyramidal neurons [49]) was applied to neurons in hippocampal organotypic cultures, together with either heat-inactivated T. crassiceps excretory/secretory products, or active T. crassiceps excretory/secretory products. The response of the membrane potential of the neurons was measured using whole-cell patch-clamp recordings (see Materials and Methods, and Fig 4A and 4B). When neurons were held at a voltage close to their action potential threshold and picolitre volumes of 200 μM acetylcholine with heat-inactivated excretory/secretory products were puffed toward the soma of the neurons, they depolarised and fired action potentials (APs) (median = 3.6 APs, IQR = 1.0–11.2 APs, N = 16, Fig 4B and 4C). However, when 200 μM acetylcholine with active excretory/secretory products were applied to the same neurons just 2 min prior to/after this, the neurons did not show the same response, often firing no action potentials despite still being held at a voltage close to their action potential threshold (median = 0.2 APs, IQR = 0.0–1.7 APs, N = 16, P = 0.0017, Wilcoxon signed-rank test, Fig 4C). Whilst this formally only demonstrates that a heat-labile component of the extract has an inhibitory effect, this is highly likely due to AChEs in the T. crassiceps larval excretory/secretory products. These experiments therefore indicate that T. crassiceps larval excretory/secretory products have sufficient AChE activity to break down acetylcholine at a concentration which induces changes in neuronal signalling in this ex vivo brain slice model. This would hold true for T. solium cyst membrane and scolex AChEs, given twice the amount of time, or the use of an extract of double the concentration, as these break down acetylcholine at roughly half the rate of T. crassiceps larval excretory/secretory extracts (Table 1).
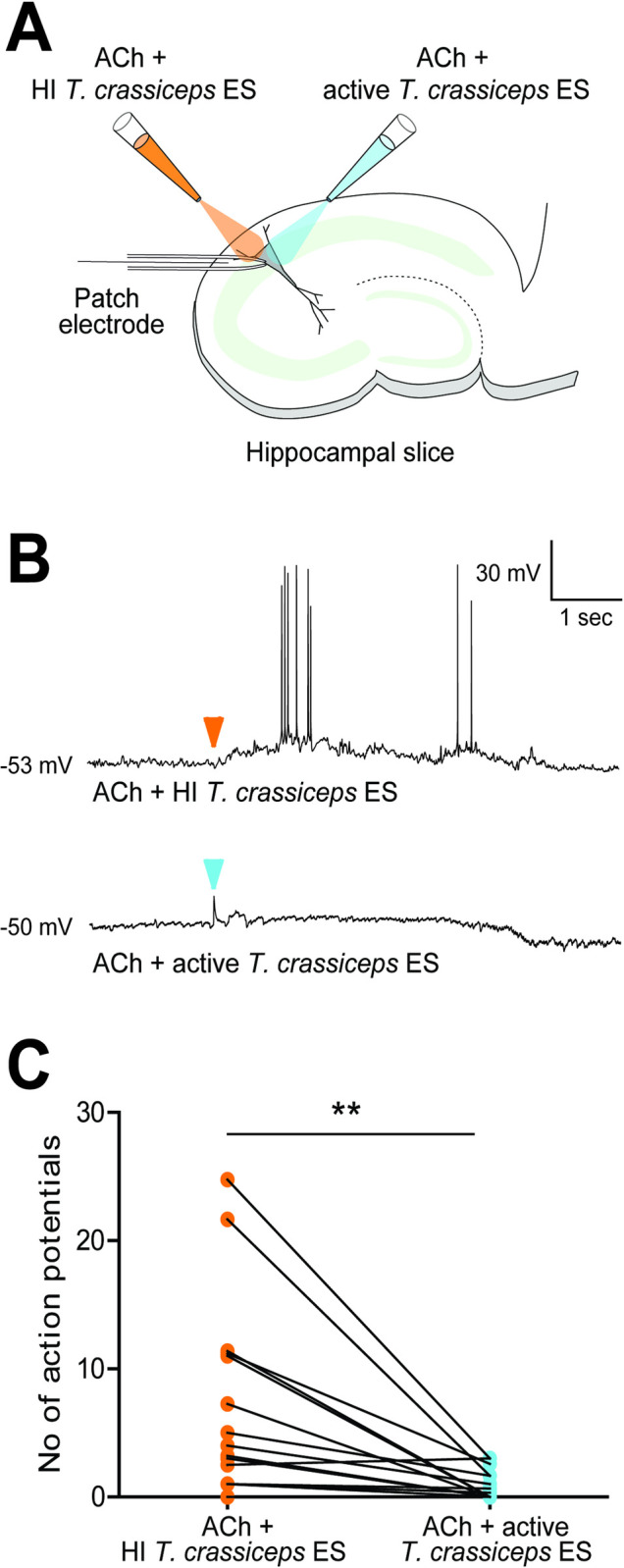
Taenia acetylcholinesterases can break down acetylcholine at a concentration which induces changes in neuronal signalling.
A) Schematic depicting the experimental setup where whole-cell patch-clamp recordings were made from rat CA3 pyramidal neurons in organotypic hippocampal brain slice cultures. Whilst recording the electrical activity from the neurons, two glass pipettes delivered picolitre volumes of acetylcholine (ACh) (200 μM) with heat-inactivated (at 56°C for 30 min) Taenia crassiceps excretory/secretory extracts (1.3 mg ml-1), or of ACh (200 μM) with unheated/active Taenia crassiceps excretory/secretory extracts (1.3 mg ml-1). B) The membrane potential responses of a pyramidal neuron when a solution of ACh with heat-inactivated (HI) Taenia crassiceps excretory/secretory extracts (ES) (top trace) or of ACh with active Taenia crassiceps ES (bottom trace) was puffed onto the cell body (arrowheads indicate moment of application). C) Population data (median ± IQR) where each point represents the mean number of action potentials evoked in 5 s after neurons were exposed to 5 x 30 ms puffs (2–5 repeat cycles) of either a solution of ACh with HI Taenia crassiceps ES (N = 13) or of ACh with active Taenia crassiceps ES (N = 13). **p ≤ 0.01, Wilcoxon signed-rank test.
Here we have used multiple methods to characterise the amount and spatial localization of AChE activity in larvae of the cestodes T. crassiceps and T. solium. Previous studies have identified AChE activities in the larvae of multiple species of the broader Taeniidae family including Echinococcus granulosus (dog tapeworm) [30,33,34] and Taenia pisiformis (rabbit tapeworm) [13]. To our knowledge our data represent the first definitive measurements of AChE activity from larvae of T. crassiceps. The amount of AChE activity we report in T. crassiceps whole cyst homogenate is similar to that previously reported for the larval homogenate of T. pisiformis (T. crassiceps whole cyst homogenate median activity = 31.69 nmol min-1 mg-1, T. pisiformis homogenate mean activity = 24.8 nmol min-1 mg-1) [13]. Interestingly we found that whilst T. solium larvae also exhibit substantial AChE activity, this activity is broadly less than in extracts from T. crassiceps. Furthermore, the spatial profile of larval AChE activity is different between these two species. Whilst AChEs were present in all fractions of T. crassiceps larvae, and presented on the tegument membrane surface, we could not identify AChEs on the tegument membrane surface or within the excreted/secreted extracts of T. solium larvae. The lack of surface staining in T. solium larvae is in accordance with a previous study by Vasantha et al. (1992), which reports AChEs in T. solium larvae to be associated with a sub-tegumental network of nerves in the strobila and bladder wall.
Our findings have important implications in the context of T. crassiceps being utilised as a model organism for neurocysticercosis research, where both larval extracts and whole early-stage cysts of T. crassiceps have been employed [50–53]. Our data illustrate that there exist significant differences in the cholinergic biology of T. solium and species that have been utilised as model parasite for T. solium, such as T. crassiceps. Once the genome for T. crassiceps has been sequenced, future work will also be able to determine potential differences in AChE amino-acid sequences between cestode species. Notably, the reported AChE activity of tetrathyridia of Mesocestoides corti, another popular model parasite for neurocysticercosis research, is far greater than that which we report for both T. crassiceps and T. solium [54]. These differences should be considered when selecting an appropriate model parasite for NCC research.
Our observation that T. solium larvae do not excrete/secrete AChEs is interesting, as many helminth species have been observed to secrete these enzymes in substantial amounts, with proposed benefits to parasite survival, such as protection against ingested AChE inhibitors and modulation of the host immune response [7,10,16]. Recently, a study by Vaux et al. (2016) demonstrated that in vivo exposure to secreted AChE from Nippostrongylus brasiliensis promoted classical activation of macrophages (as opposed to alternative activation), a state which is permissive to the survival of parasitic nematodes. In contrast, classically activated macrophages appear to be deleterious to the survival of Taeniid larvae, occurring in the resistant Th1 acute phase of infection, whilst their phenotype is shifted to an alternatively activated state during chronic Taeniid infection [8,55]. This could potentially explain why it may not be beneficial for Taeniids to secrete large amounts of AChE.
Our bioinformatics analysis demonstrated that there was modest overall sequence identity between T. solium and H. sapiens acetylcholinesterase sequences, as has been previously reported for Schistosoma mansoni [16]. However, the fact that protein functional domains appear to be conserved between T. Solium and H. sapiens lends support to our experimental findings that Taenia larvae contain active AChEs. In addition, using whole-cell patch clamp recordings in rodent hippocampal brain slice cultures, we show that Taenia larval-derived AChEs have sufficient activity to break down acetylcholine at a concentration which induces changes in neuronal signalling. These experiments demonstrate that although the activities of Taenia larval-derived AChEs may not seem substantial when compared to that of other parasitic worms, such as Nippostrongylus brasiliensis (which has a reported excretory/secretory extract AChE activity of 5.3 μmol min-1 mg-1 [56]), they may well be substantial enough to exert an effect in the sensitive brain environment. It should also be taken into consideration that in clinical presentations of NCC, T. solium larvae interface directly with brain tissue, which means that the membrane-bound AChEs in T. solium larvae identified in this study could potentially have a direct and spatially concentrated impact on brain tissue when the cyst starts to degrade and lose membrane integrity. Although we have demonstrated in this study that larval-derived AChEs have sufficient activity to break down acetylcholine at a concentration which induces changes in neuronal signalling in an ex vivo brain slice model, further electrophysiological investigations, preferably using purified T. solium AChE applied in an in vivo model, are required to determine whether the activity of larval-derived AChEs can, in fact, functionally alter neuronal signalling.
The cause of seizures and epilepsy secondary to neurocysticercosis has been a matter of great controversy, with some researchers questioning whether neurocysticercosis is truly causative of epilepsy, and with different studies sometimes reporting contrasting results [57–59]. One thing that has become increasingly clear in recent years, however, is that inflammation is closely linked to the generation of seizures and epilepsy, both generally, and within the context of neurocysticercosis [60,61]. In the healthy brain, microglia and astrocytes regulate inflammatory signalling in the brain via, amongst others, acetylcholine receptor dependent signalling [62]. Activation of acetylcholine receptors has been shown to strikingly impair acute phase inflammation [63,64]. One might hypothesise, then, that exposure of the brain to T. solium membrane AChEs during cyst degeneration could exacerbate perilesional inflammation if the larval AChEs break down acetylcholine molecules involved in anti-inflammatory signalling. Based on this supposition, future investigations involving the addition of AChE inhibitors to NCC model systems that present with severe perilesional inflammation, could be of potential value.
In summary, our findings describe distinct profiles of acetylcholinesterase activity in T. crassiceps and T. solium larvae. In light of this, we encourage neurocysticercosis researchers to take into consideration that differences do exist between T. solium and related cestodes such as T. crassiceps. We also highlight the possibility of larval-derived acetylcholinesterases interfering with host neural and immune signalling in the brain.
T. crassiceps larvae were generously donated to us by Dr Siddhartha Mahanty (University of Melbourne, Melbourne, Australia). The authors would also like to thank the CYSTINET-Africa consortium for the ongoing support of CPdC, UFP and CS as members.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
 Taenia larvae possess distinct acetylcholinesterase profiles with implications for host cholinergic signalling
Taenia larvae possess distinct acetylcholinesterase profiles with implications for host cholinergic signalling
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
