 Oligomeric quaternary structure of Escherichia coli and Mycobacterium smegmatis Lhr helicases is nucleated by a novel C-terminal domain composed of five winged-helix modules
Oligomeric quaternary structure of Escherichia coli and Mycobacterium smegmatis Lhr helicases is nucleated by a novel C-terminal domain composed of five winged-helix modules
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

- Altmetric
Mycobacterium smegmatis Lhr (MsmLhr; 1507-aa) is the founder of a novel clade of bacterial helicases. MsmLhr consists of an N-terminal helicase domain (aa 1–856) with a distinctive tertiary structure (Lhr-Core) and a C-terminal domain (Lhr-CTD) of unknown structure. Here, we report that Escherichia coli Lhr (EcoLhr; 1538-aa) is an ATPase, translocase and ATP-dependent helicase. Like MsmLhr, EcoLhr translocates 3′ to 5′ on ssDNA and unwinds secondary structures en route, with RNA:DNA hybrid being preferred versus DNA:DNA duplex. The ATPase and translocase activities of EcoLhr inhere to its 877-aa Core domain. Full-length EcoLhr and MsmLhr have homo-oligomeric quaternary structures in solution, whereas their respective Core domains are monomers. The MsmLhr CTD per se is a homo-oligomer in solution. We employed cryo-EM to solve the structure of the CTD of full-length MsmLhr. The CTD protomer is composed of a series of five winged-helix (WH) modules and a β-barrel module. The CTD adopts a unique homo-tetrameric quaternary structure. A Lhr-CTD subdomain, comprising three tandem WH modules and the β-barrel, is structurally homologous to AlkZ, a bacterial DNA glycosylase that recognizes and excises inter-strand DNA crosslinks. This homology is noteworthy given that Lhr is induced in mycobacteria exposed to the inter-strand crosslinker mitomycin C.
INTRODUCTION
Helicases are omnipresent and have wide-ranging functions in DNA and RNA metabolism. Helicases use the chemical energy of NTP hydrolysis to either affect mechanical changes in the secondary structure of nucleic acids or remodel the structures of protein–nucleic acid complexes. Helicases have been classified into superfamilies, families and subfamilies according to their distinctive primary, tertiary and quaternary structures, and their biochemical specificities, i.e., their NTP preference, nucleic acid preference and directionality of translocation or unwinding (1). Helicases are versatile in their use of flanking auxiliary domains to affect substrate specificity or tack on catalytic functionality (1).
The bacterial DNA helicase Lhr is transcriptionally upregulated in mycobacteria by the LexA/RecA-independent PafBC pathway in response to DNA damage by the inter-strand crosslinker mitomycin C (2–4). Mycobacterium smegmatis (Msm) Lhr exemplifies a new clade of superfamily 2 (SF2) DNA helicase, by virtue of its signature domain structure and its distinctive DNA interface (5,6). MsmLhr is a 1507-amino acid (aa) nucleic acid-dependent ATPase/dATPase that uses ATP hydrolysis to drive unidirectional 3′ to 5′ translocation along single-strand (ss) DNA (the tracking strand) and to unwind duplexes en route. The strand displaced by the translocating MsmLhr helicase can be either RNA or DNA. The ATPase, DNA translocase and RNA:DNA helicase activities of mycobacterial Lhr are encompassed within the N-terminal 856-aa segment (5). This autonomous helicase, referred to as Lhr-Core, consists of two N-terminal RecA-like modules (aa 8–230 and 231–435, respectively), a winged-helix (WH) domain (aa 436–529) and a unique core C-domain (aa 530–856) that adopt a novel fold (6) (Figure 1). All four protein domains of MsmLhr-Core contribute to an extensive interface with the DNA tracking strand (6). Lhr-Core homologs of similar size (800–900 aa) are present in the proteomes of many diverse bacterial and archaeal taxa (5,8).


Homology of EcoLhr and MsmLhr. The top panel depicts in cartoon form the domain organization of the full-length bacterial Lhr protein, as revealed by the crystal structure of the Core helicase unit of MsmLhr. The large C-terminal domain (CTD) of full-length Lhr, as yet structurally uncharacterized (denoted by ???), is colored gray. In the bottom panel, the amino acid sequence of the 1538-aa EcoLhr polypeptide is aligned to that of the homologous 1507-aa MsmLhr protein. Positions of side chain identity/similarity are highlighted in yellow. Gaps in the alignments are denoted by dashes. The SF2 NTPase motifs I, II, III and VI are labeled and highlighted by bars above the sequences. The EcoLhr motif II Asp179 and Glu180 residues that were mutated to alanine in the present study are denoted by black arrowheads below the alignment. The C-terminal margins of MsmLhr-Core (aa 1–856) and EcoLhr-Core (aa 1–877) proteins are denoted by arrows. A conserved tryptophan side chain essential for translocation in MsmLhr is denoted by a red triangle and red box. The amino acids in the MsmLhr CTD that contribute to the subunit interfaces in the CTD tetramer are denoted by black dots above the alignment.
Full-length Lhr homologs are found in a narrower spectrum of bacteria from eight phyla, being especially prevalent in Actinobacteria and Proteobacteria (5). Attention was drawn to the Escherichia coli Lhr protein by the Deutscher lab (7), who sequenced the lhr gene, and found that it specified what was then, at 1538-aa, the largest known E. coli polypeptide. In search of evidence for its predicted biochemical activity, Reuven et al. (7) over-produced EcoLhr under the control of an inducible T7 promoter but did not detect any increase in nucleic acid-dependent ATPase activity in extracts of induced bacteria versus control extracts lacking the expression plasmid. Thus, it was not clear whether EcoLhr is a bona fide ATPase or helicase, notwithstanding its extensive primary structure homology to MsmLhr (Figure 1), which includes conservation of SF2 helicase motifs and specific amino acids within MsmLhr-Core that are critical for its translocase or helicase activity (6). The large C-terminal halves of MsmLhr (aa 857–1507) and EcoLhr (aa 878–1538) that flank the Core are conserved but have no instructive motifs or apparent primary structure similarities to any proteins outside of the Lhr helicase clade (5).
In the present study we address three outstanding questions: (i) is E. coli Lhr an ATPase or helicase and, if so, what is its biochemical specificity; (ii) how does the large C-terminal half of EcoLhr and MsmLhr impact Lhr structure or activity; and (iii) what is the structure of the large C-terminal domain of Lhr?
MATERIALS AND METHODS
Lhr proteins
The open reading frames (ORFs) encoding full-length E. coli Lhr (GenBank accession NP_416170.1) and core EcoLhr-Core (aa 1–877) were PCR-amplified from E. coli K-12 MG1655 genomic DNA with primers that introduced a HindIII site upstream of the start codon and a XhoI site downstream of the native or newly installed stop codons. The PCR products were digested with HindIII and XhoI and ligated into pET28b-His10Smt3 that had been digested with HindIII and XhoI. The resulting pET28b-His10Smt3-EcoLhr plasmids encode polypeptides fused to an N-terminal His10Smt3 tag under the transcriptional control of a T7 RNA polymerase promoter. Proteolytic cleavage with Smt3-specific protease Ulp1 generates a polypeptide with non-native residues SEFELRRQAC at the N-terminus. The ORFs encoding full-length M. smegmatis Lhr (Msmeg_1757), core domain MsmLhr-(1–856) and C-terminal domain MsmLhr-(863–1507) were PCR-amplified from M. smegmatis genomic DNA with primers that introduced a BglII site upstream of the start codon and a NotI site downstream of the native or newly installed stop codons. The PCR products were digested with BglII and NotI and ligated into pET28b-His10Smt3 that had been digested with BamHI and NotI. The resulting pET28b-His10Smt3-MsmLhr plasmids encode polypeptides fused to an N-terminal His10Smt3 tag. Mutant Lhr expression vectors were generated using the Q5 Mutagenesis Kit (New England Biolabs). All plasmid inserts were sequenced to verify that no unintended coding changes were acquired during amplification and cloning. The pET28b-His10Smt3-Lhr plasmids were transformed into E. coli BL21(DE3) cells. Cultures (1-L) amplified from single kanamycin-resistant transformants were grown at 37°C in LB broth containing 60 μg/ml kanamycin until the A600 reached 0.7. The cultures were chilled on ice for 1 h, then adjusted to 2% (v/v) ethanol and 0.5 mM isopropyl-β-d-thiogalactopyranoside and incubated for 16 h at 18°C with constant shaking. All subsequent procedures were performed at 4°C. Cells were harvested by centrifugation and resuspended in 35 ml of buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1 mM DTT [dithiothreitol], 20% glycerol) containing 1 protease inhibitor cocktail tablet (Roche). Lysozyme was added to a concentration of 1 mg/ml and the resulting lysate was sonicated to reduce viscosity. The insoluble material was pelleted by centrifugation at 38 000 g for 45 min. The supernatant was mixed for 1 h with 5 ml of Ni-NTA agarose resin (Qiagen) that had been equilibrated with buffer A. The resin was recovered by centrifugation and resuspended in 40 ml of buffer B (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol). The resin was recovered by centrifugation and resuspended in 40 ml of buffer B containing 3 M KCl. This wash step was repeated twice with buffer B. The recovered resin was poured into a column. After washing the column with 20 ml of buffer B, the bound material was eluted with buffer C (50 mM HEPES-NaOH, pH 7.5, 300 mM KCl, 10% glycerol) containing 500 mM imidazole. The polypeptide compositions of the eluate fractions were monitored by SDS-PAGE. The eluate fractions containing recombinant Lhr were pooled and supplemented with Smt3-specific protease Ulp1 (at a His10Smt3-Lhr:Ulp1, His10Smt3-Lhr-Core:Ulp1 or His10Smt3-Lhr-CTD:Ulp1 ratio of 1000:1) and then dialyzed overnight in 2 L of buffer C containing 1 mM DTT. The tag-free proteins were separated from His10Smt3 by applying the dialysates to a 5-ml Ni-NTA agarose column that had been equilibrated with buffer C; the Lhr proteins were recovered in the flow-through fractions. The flow-through fractions were pooled and diluted to reduce KCl to 90 mM immediately before loading onto a HiTrapQ ion exchange column, which was washed with 100 ml of buffer D (50 mM HEPES-NaOH, pH 7.5, 4 mM DTT, 10% glycerol) containing 50 mM KCl and then developed with a 150-ml linear gradient of 50–1000 mM KCl in buffer D. Peak Lhr-containing fractions eluting at ∼300 mM KCl were pooled and subjected to gel filtration through a Superdex-200 column equilibrated in buffer C containing 1 mM DTT. Peak fractions were pooled, concentrated by centrifugal ultrafiltration, frozen and stored at −80°C. Protein concentrations were determined with the BioRad dye reagent using BSA as the standard.
Nucleoside triphosphatase assay
Reaction mixtures containing (per 10 μl) 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP, and DNA and Lhr as specified were incubated for 30 min at 37°C. The reactions were quenched by adding 2 μl of 5 M formic acid. An aliquot (2 μl) of the mixture was applied to a polyethyleneimine-cellulose TLC plate, which was developed with 0.45 M ammonium sulfate. TLC plates were exposed to a phosphor screen and 32P-ADP formation was quantified by autoradiography using a Typhoon imager (GE Healthcare) and GelAnalyzer 19.1 software (www.gelanalyzer.com).
Streptavidin displacement assay of Lhr translocation on DNA
Synthetic 34-mer oligodeoxynucleotides of otherwise identical nucleobase sequence containing a Biotin-ON inter-nucleotide spacer either at the fourth position from the 5′ terminus or the second position from the 3′ terminus were purchased from Eurofins MWG Operon. These DNAs were 5′ end-labeled with [γ32P]ATP by using T4 polynucleotide kinase and then purified by electrophoresis through a native 20% polyacrylamide gel. Streptavidin–DNA (SA-DNA) complexes were formed by preincubating 100 nM biotinylated 32P-DNA with 4 μM streptavidin (Sigma) in 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, and 1 mM ATP for 15 min at room temperature. The mixtures were supplemented with 40 μM free biotin (Fisher), and the displacement reactions (15 μl, containing 1.5 pmol biotinylated 32P-DNA) were initiated by adding 7.5 or 10 pmol Lhr. After incubation for 20 min at 37°C, the reactions were quenched by adding 4.5 μl of a solution containing 200 mM EDTA, 0.6% SDS, 25% glycerol, and 20 μM of an unlabeled single strand DNA to mask any binding of Lhr to 32P-DNA released from the SA-DNA complex. The reaction products were analyzed by electrophoresis through a 15-cm native 15% polyacrylamide gel containing 89 mM Tris-borate, 2.5 mM EDTA. The free 32P-labeled 34-mer DNA and the SA-DNA complexes were visualized by autoradiography.
Helicase assay
The 5′ 32P-labeled RNA and DNA strands were prepared by reaction of a synthetic 24-mer oligonucleotide with T4 polynucleotide kinase and [γ32P]ATP. The labeled oligonucleotide was separated from ATP by electrophoresis through a 40-cm 18% polyacrylamide gel containing 7 M urea in 89 mM Tris-borate, 2.5 mM EDTA. The oligonucleotide was eluted from an excised gel slice by incubation for 16 h at 4°C in 1 M ammonium acetate, 0.2% SDS, 20 mM EDTA. The oligonucleotide was recovered by ethanol precipitation and resuspended in 10 mM Tris-HCl, pH 6.8, 1 mM EDTA. Labeled RNA or DNA was annealed to a 3-fold excess of a complementary 44-mer RNA or DNA strand to form the 3′-tailed duplex substrates. The annealed duplexes were purified by electrophoresis through a native 12% polyacrylamide gel, eluted from an excised gel slice by incubation for 16 h at 4°C in 150 μl of 10 mM Tris-HCl, pH 6.8, 1 mM EDTA, 50 mM NaCl. Helicase reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 50 mM NaCl, 1 mM CaCl2, 25 nM 32P-labeled 3′-tailed duplex nucleic acid as specified, and Lhr as specified were preincubated for 10 min at room temperature. The reactions were initiated by adding 1 mM ATP and a 40-fold excess of an unlabeled 24-mer oligonucleotide identical to the labeled strand of the helicase substrate. Addition of excess unlabeled strand was necessary to prevent the spontaneous reannealing of the unwound 32P-labeled strand. The reaction mixtures were incubated for 30 min at 37°C and then quenched by adding 1 μl of a 31 mg/ml solution of proteinase K (Sigma-Aldrich). The mixtures were supplemented with 4 μl of 70% glycerol, 0.3% bromophenol blue. The reaction products were analyzed by electrophoresis through a 15-cm 12% polyacrylamide gel in 89 mM Tris-borate, 2.5 mM EDTA. The products were visualized by autoradiography.
Cryo-EM sample preparation and data acquisition
Aliquots (3.5 μl) of MsmLhr-FL (0.3 mg/ml) were applied to UltrAuFoil 300 mesh R 1.2/1.3 grids (Quantifoil), which were glow-discharged for 120 s at 0.37 mBar, 15 mA in a Pelco easiGlow glow discharge cleaning system (TED PELLA). Grids were blotted for 1.5 s with 100% humidity at ∼4°C and flash frozen in liquid ethane using a Vitrobot Mark IV (FEI). Images were collected on a 300 keV Titan Krios electron microscope (FEI) with Gatan K3 Summit detector at Memorial Sloan Kettering Cancer Center. The micrographs were collected at a magnification of 22 500× with a super-resolution pixel size of 0.5032 Å and a defocus range of -1.0 to -2.5 μm. The dose rate was ∼20 e−/pixel/s with a total exposure time of 3 s with 0.075 s subframes (40 total frames) corresponding to a total dose of approximately 53 e−/Å2.
Cryo-EM image processing
Movie frames were motion corrected and 2-fold Fourier cropped to a calibrated pixel size of 1.064 Å using MotionCor2 (9). Contrast transfer function (CTF) parameters were estimated by Ctffind4 (10). Subsequent image processing was performed by RELION 3 (11). Particles were auto-picked without reference and extracted at a 3-fold binned pixel size of 3.192 Å/pixel. After two rounds of reference-free 2D classification, 1 894 929 particles were selected for 3D classification using an initial model as reference generated by RELION. A total of 298 814 particles from the best class were selected for the second round of 3D classification. The best class with 101 323 particles was refined and re-extracted (unbinned, pixel size 1.064 Å/pixel) and polished for 3D refinement. Post-processing in RELION yielded the final EM map at a resolution of 3.6 Å, as estimated based on the gold-standard Fourier shell correlation (FSC) calculations. Local resolution estimates were calculated from two half-data maps using ResMap (12).
Model building and refinement
The model of MsmLhr-CTD was built manually in COOT (13) based on the bulky amino acid side chains in the primary structure. For the other three MsmLhr-CTD protomer structures, the first manually built protomer structure was docked into the density map using UCSF Chimera (14) and then rebuilt manually in COOT. Further refinement of the model was performed in PHENIX (15) using real-space refinement against the same half-map. Structural figures were prepared with Pymol (pymol.org) and ChimeraX (16).
RESULTS
EcoLhr has a homo-oligomeric quaternary structure, whereas EcoLhr-Core is a monomer
To evaluate the enzymatic and physical properties of EcoLhr, we produced full-length (FL) protein and a C-terminal truncation (Core) comprising amino acids 1–877 (equivalent to MsmLhr-Core; Figure 1) under the control of an inducible T7 promoter. The His10Smt3-tagged EcoLhr-FL and Core proteins were purified from soluble bacterial extracts by adsorption to Ni-agarose and elution with imidazole. The tag was removed by treatment with Smt3-protease Ulp1, and the tag-free FL and Core proteins were recovered in the flow-through after a second round of Ni-agarose chromatography. Further purification was achieved by anion exchange chromatography and preparative gel filtration. The native sizes of EcoLhr FL and Core were gauged by analytical gel filtration through a Superdex-200 column that had been calibrated with a mixture of size standards. The 171 kDa EcoLhr-FL polypeptide eluted as a single component with a peak between the 669 and 440 kDa standards (Figure 2A), indicating that it has a homo-oligomeric quaternary structure. By contrast, the 97 kDa EcoLhr-Core polypeptide eluted as a single component peaking between the 158 and 75 kDa standards (Figure 2B), signifying that the Core is a monomer in solution and implying that the large C-terminal domain is responsible for homo-oligomerization of the FL protein.
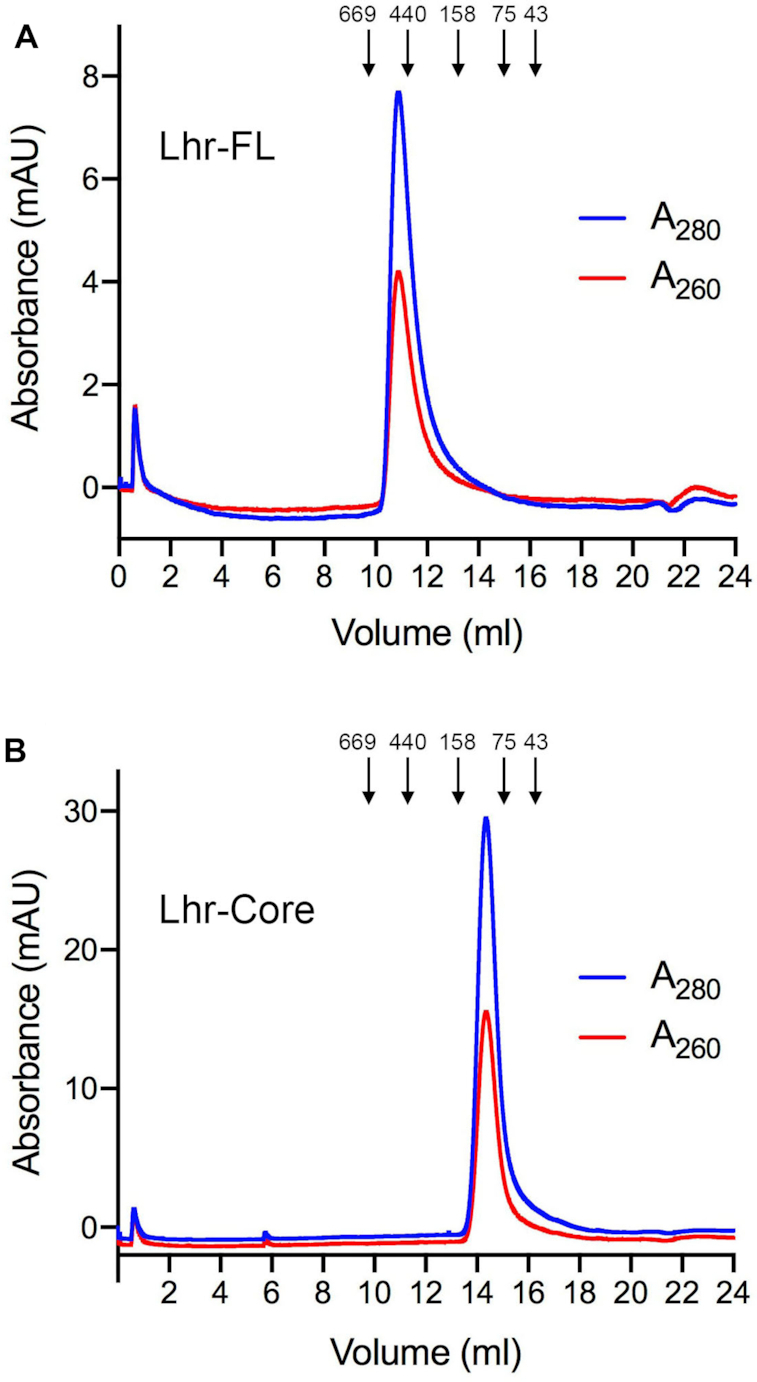
Gel filtration chromatography of EcoLhr-FL and EcoLhr-Core. EcoLhr-FL (panel A) and EcoLhr-Core (panel B) were gel filtered through a 24-ml Superdex-200 column equilibrated with buffer containing 50 mM HEPES-NaOH, pH 7.5, 300 mM KCl, 1 mM DTT, 10% glycerol. The elution profiles shown were monitored continuously by A280 and A260 as a function of elution volume. Arrows denote the elution peaks and native sizes for a mixture of calibration standards: thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ovalbumin (44 kDa).
EcoLhr FL and Core have ATP phosphohydrolase activity
Gel filtration column fractions spanning the protein absorbance peaks were assayed for ATPase activity under reaction conditions optimized previously (5) for MsmLhr (i.e., 1 mM [α32P]ATP as substrate, 1 mM CaCl2 as divalent cation cofactor and 5 μM 24-mer ssDNA as putative DNA cofactor). Activity was gauged by PEI-cellulose TLC of the reaction products and quantified by the conversion of [α32P]ATP to [α32P]ADP. The FL and Core gel filtration fractions displayed ATPase activity that accorded with the abundance of the respective FL and Core polypeptides, as determined by SDS-PAGE analysis (Figure 3A and B).
![ATP phosphohydrolase activity coelutes with EcoLhr. Aliquots (2 μl) of the indicated Superdex-200 column fractions of EcoLhr-FL (panel A) and EcoLhr-Core (panel B) were assayed for ATPase activity in reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP (10 nmol ATP), and 5 μM 24-mer ssDNA. The extents of ADP formation during a 30 min reaction are plotted. The polypeptide compositions of the Superdex-200 column fractions were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown above the ATPase activity profile. The positions and sizes (kDa) of marker polypeptides are indicated on the left.](/dataresources/secured/content-1766036286562-18db5b44-000a-4889-b769-2b04d461587a/assets/gkab145fig3.jpg)
ATP phosphohydrolase activity coelutes with EcoLhr. Aliquots (2 μl) of the indicated Superdex-200 column fractions of EcoLhr-FL (panel A) and EcoLhr-Core (panel B) were assayed for ATPase activity in reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP (10 nmol ATP), and 5 μM 24-mer ssDNA. The extents of ADP formation during a 30 min reaction are plotted. The polypeptide compositions of the Superdex-200 column fractions were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown above the ATPase activity profile. The positions and sizes (kDa) of marker polypeptides are indicated on the left.
Titration of the purified FL and Core proteins showed that: (i) the extents of ATP hydrolysis were proportional to input enzyme and (ii) the two proteins had similar ATPase specific activities (15.5 nmol/pmol for FL and 16.9 nmol/pmol for Core) (Figure 4B). To affirm that the observed ATPase activity inhered to the recombinant EcoLhr proteins, we produced and purified a mutant version of FL and Core in which the putative metal-binding motif II residues Asp179 and Glu180 were replaced by alanine (Figure 4A). The D179A-E180A mutations effaced the ATPase activity of the FL and Core proteins (Figure 4C).
![EcoLhr FL and Core are equally adept at ATP hydrolysis, which is erased by mutating metal-binding motif II. (A) Purification. Aliquots (5 μg) of wild-type (WT) EcoLhr-FL and FL mutants D179A-E180A and W620A (left panel) and aliquots (6 μg) of EcoLhr-Core and Core mutants D179A-E180A and W620A (right panel) were analyzed by SDS-PAGE. The Coomassie blue-stained gels are shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B) ATPase reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP (10 nmol ATP), 5 μM 24-mer ssDNA, and increasing amounts of wild-type EcoLhr-FL or Core as specified were incubated at 37°C for 30 min. The extents of ADP formation are plotted as a function of input Lhr. Each datum is the average of three separate enzyme titration experiments ± SEM. (C) ATPase reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP (10 nmol ATP), 5 μM 24-mer ssDNA, and 100 nM EcoLhr proteins as specified were incubated at 37°C for 30 min. The bars indicate the average extents of ADP formation from three separate experiments ± SEM.](/dataresources/secured/content-1766036286562-18db5b44-000a-4889-b769-2b04d461587a/assets/gkab145fig4.jpg)
EcoLhr FL and Core are equally adept at ATP hydrolysis, which is erased by mutating metal-binding motif II. (A) Purification. Aliquots (5 μg) of wild-type (WT) EcoLhr-FL and FL mutants D179A-E180A and W620A (left panel) and aliquots (6 μg) of EcoLhr-Core and Core mutants D179A-E180A and W620A (right panel) were analyzed by SDS-PAGE. The Coomassie blue-stained gels are shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B) ATPase reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP (10 nmol ATP), 5 μM 24-mer ssDNA, and increasing amounts of wild-type EcoLhr-FL or Core as specified were incubated at 37°C for 30 min. The extents of ADP formation are plotted as a function of input Lhr. Each datum is the average of three separate enzyme titration experiments ± SEM. (C) ATPase reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP (10 nmol ATP), 5 μM 24-mer ssDNA, and 100 nM EcoLhr proteins as specified were incubated at 37°C for 30 min. The bars indicate the average extents of ADP formation from three separate experiments ± SEM.
Requirements for optimal EcoLhr ATPase activity
The ATPase activity was characterized in depth for the full-length EcoLhr enzyme. An added divalent cation was required for ATP hydrolysis (Figure 5A). Comparing various divalent cations at 1 mM concentration, we found that calcium was most effective (Figure 5A), as had been noted previously for MsmLhr (5). Magnesium and manganese were 50–60% as effective as calcium whereas cobalt and nickel were 20–35% as effective. Copper and zinc were ineffective. All subsequent assays were performed with calcium as the divalent cation cofactor. ATPase activity was optimal from pH 6.5 to 9.5 in Tris buffers (Figure 5B). Activity decreased gradually as pH was lowered from pH 6.5 to 5.5, declined to 30% of the peak value at pH 5.0, and was erased at pH 4.5 (Figure 5B). We determined steady-state kinetic parameters by measuring the velocity of ATP hydrolysis as a function of ATP concentration (Figure 5C). From a nonlinear regression curve fit of the data to the Michaelis–Menten equation, we calculated that EcoLhr has a Km of 0.17 ± 0.076 mM ATP and a kcat of 21.2 ± 3.27 s−1. We were surprised to find that omission of the putative ssDNA cofactor had almost no effect on the extent of ATP hydrolysis by EcoLhr (Figure 5D). As a ‘positive control’, we confirmed that the ssDNA was required to activate the ATPase of MsmLhr (Figure 5D), as described previously (5). The A280:A260 ratios of the gel filtered EcoLhr FL and Core proteins were 1.84 and 1.92, respectively (Figure 2), militating against their being contaminated by stoichiometric amounts of co-purifying bacterial nucleic acids.
![Characterization of the EcoLhr ATPase. (A) Divalent cation specificity. Reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM [α32P]ATP, 5 μM 24-mer ssDNA, 100 nM EcoLhr-FL, and 1 mM of the indicated divalent cation (as the chloride salt) were incubated at 37°C for 30 min. The extents of ATP hydrolysis are plotted. Each bar is the average of three separate experiments ± SEM. (B) pH profile. Reaction mixtures (10 μl) containing 20 mM Tris buffer (Tris-acetate pH 4.5, 5.0, 5.5, 6.0 or 6.5 or Tris-HCl pH 7.0, 7.5, 8.0, 8.5, 9.0 or 9.5), 1 mM CaCl2, 1 mM [α32P]ATP, 5 μM 24-mer ssDNA, and 100 nM EcoLhr-FL were incubated at 37°C for 30 min. The extents of ATP hydrolysis are plotted as a function of pH. Each datum is the average of three separate experiments ± SEM. (C) Steady-state kinetics. Reaction mixtures (40 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 5 μM 24-mer ssDNA, 100 nM EcoLhr-FL, and either 0.0625, 0.125, 0.25, 0.5 or 1.0 mM [α32P]ATP were incubated at 37°C. Aliquots (10 μl) were withdrawn at 15 and 30 s and quenched immediately with formic acid. The extents of ATP hydrolysis were plotted as a function of time for each ATP concentration and the initial rates were derived by linear regression analysis in Prism. The initial rates (pmol s−1) were divided by the molar amount of input enzyme to obtain a turnover number V (s−1), which is plotted in the figure as a function of ATP concentration. Each datum is the average of three separate time course experiments ±SEM. A nonlinear regression curve fit of the data to the Michaelis–Menten equation (in Prism) is shown. (D) DNA dependence. Reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP, and 100 nM EcoLhr-FL or MsmLhr-FL as specified, with or without 5 μM 24-mer ssDNA, were incubated at 37°C for 30 min. The bars indicate the average extents of ADP formation from three separate experiments ± SEM.](/dataresources/secured/content-1766036286562-18db5b44-000a-4889-b769-2b04d461587a/assets/gkab145fig5.jpg)
Characterization of the EcoLhr ATPase. (A) Divalent cation specificity. Reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM [α32P]ATP, 5 μM 24-mer ssDNA, 100 nM EcoLhr-FL, and 1 mM of the indicated divalent cation (as the chloride salt) were incubated at 37°C for 30 min. The extents of ATP hydrolysis are plotted. Each bar is the average of three separate experiments ± SEM. (B) pH profile. Reaction mixtures (10 μl) containing 20 mM Tris buffer (Tris-acetate pH 4.5, 5.0, 5.5, 6.0 or 6.5 or Tris-HCl pH 7.0, 7.5, 8.0, 8.5, 9.0 or 9.5), 1 mM CaCl2, 1 mM [α32P]ATP, 5 μM 24-mer ssDNA, and 100 nM EcoLhr-FL were incubated at 37°C for 30 min. The extents of ATP hydrolysis are plotted as a function of pH. Each datum is the average of three separate experiments ± SEM. (C) Steady-state kinetics. Reaction mixtures (40 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 5 μM 24-mer ssDNA, 100 nM EcoLhr-FL, and either 0.0625, 0.125, 0.25, 0.5 or 1.0 mM [α32P]ATP were incubated at 37°C. Aliquots (10 μl) were withdrawn at 15 and 30 s and quenched immediately with formic acid. The extents of ATP hydrolysis were plotted as a function of time for each ATP concentration and the initial rates were derived by linear regression analysis in Prism. The initial rates (pmol s−1) were divided by the molar amount of input enzyme to obtain a turnover number V (s−1), which is plotted in the figure as a function of ATP concentration. Each datum is the average of three separate time course experiments ±SEM. A nonlinear regression curve fit of the data to the Michaelis–Menten equation (in Prism) is shown. (D) DNA dependence. Reaction mixtures (10 μl) containing 20 mM HEPES-NaOH, pH 7.5, 1 mM CaCl2, 1 mM [α32P]ATP, and 100 nM EcoLhr-FL or MsmLhr-FL as specified, with or without 5 μM 24-mer ssDNA, were incubated at 37°C for 30 min. The bars indicate the average extents of ADP formation from three separate experiments ± SEM.
EcoLhr translocates 3′ to 5′ on single-stranded DNA
Notwithstanding that EcoLhr does not require an added nucleic acid cofactor to hydrolyze ATP, the key question is whether, like MsmLhr and other helicases, it has the capacity to couple ATP hydrolysis to mechanical work in the form of DNA translocation and duplex unwinding. To address this issue, we employed a streptavidin (SA) displacement assay of directional DNA translocation (5,6,17,18). 5′ 32P-labeled 34-mer DNA oligonucleotides containing a single biotin moiety, either at the fourth inter-nucleotide from the 5′ end or the second inter-nucleotide from the 3′ end (Figure 6, shown at bottom), were preincubated with excess SA to form a stable SA-DNA complex (Figure 6A, lanes 2 and 8) that was easily resolved from the free biotinylated 34-mer DNA during native PAGE (Figure 6A, lanes 1 and 7). The translocation assay scores the motor-dependent displacement of SA from DNA in the presence of ATP and excess free biotin, which instantly binds to free SA and precludes SA rebinding to the labeled DNA. The rationale of the assay is that directional tracking of the motor along the DNA single strand will displace SA from one DNA end, but not the other. As depicted in Figure 6C, the enzyme acts as a ‘cow catcher’ on a locomotive engine. When moving 3′ to 5′, it can displace SA as it collides with the 5′ biotin-SA. In contrast, a 3′ biotin-SA complex is not expected to be displaced by a 3′ to 5′ translocase because the motor moves away from the SA and simply falls off the free 5′ end. The converse outcomes apply to a 5′ to 3′ translocase, whereby it displaces a 3′ biotin-SA complex but not a 5′ biotin-SA adduct. The instructive finding was that EcoLhr displaced the SA from a 5′ biotin-SA complex on the 34-mer ssDNA to yield the free 32P-labeled 34-mer strand (Figure 6A, lane 11) but was unable to displace SA from 3′ biotin-SA complex tested in parallel (Figure 6A, lane 5). Stripping of the 5′ biotin-SA complex by EcoLhr to liberate free DNA depended on ATP (Figure 6A, lane 9) and calcium (Figure 6A, lane 10). Lhr-Core lacking the large C-domain was also capable of displacing SA from a 5′ biotin-SA complex (Figure 6B, lane 17) in a manner that depended on ATP (Figure 6B, lane 15) and calcium (Figure 6B, lane 16).
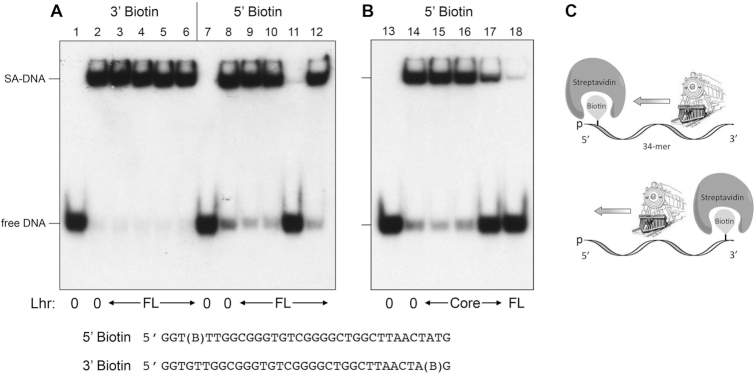
EcoLhr translocates 3′ to 5′ on single-stranded DNA. A schematic representation of the Lhr translocase motor as a cowcatcher that pries apart the otherwise stable streptavidin-biotin complex at the 5′ end of the single strand 34-mer DNA is shown in panel (C). Translocase assays were performed as described under ‘Materials and Methods’ section. Native PAGE analysis of the translocase reaction products is shown in panels (A and B). The species corresponding to SA-DNA complex and free DNA are indicated. The nucleobase sequences of the 5′ 32P-labeled 5′ or 3′ biotinylated 34-mer single strand DNAs are shown at the bottom with (B) signifying the position of the biotin spacer. Translocase reaction mixtures contained 20 mM HEPES-NaOH, pH 7.5, 1 mM ATP, 1 mM CaCl2, 100 nM 32P-labeled 3′ or 5′ biotinylated DNA bound to streptavidin, and 500 nM of Lhr-FL (lanes 3–5 and 9–11), 500 nM Lhr-FL W620A (lanes 6 and 12), 666 nM Lhr-FL (lane 18) or 666 nM Lhr-Core (lanes 15–17). Enzyme was omitted from control reactions in lanes 1, 2, 7, 8, 13, and 14 (indicated by Lhr: 0 below the lanes). Reactions in lanes 1, 7, and 13 were heat denatured at 95°C for 5 min prior to PAGE. ATP was omitted from the reactions in lanes 3, 9, and 15. Calcium was omitted and 1 mM EDTA was added to the reactions in lanes 4, 10, and 16.
Mutation of Trp620 de-couples ATP hydrolysis from SA displacement
The crystal structure of MsmLhr-Core in complex with AMPPNP•Mg2+ and 13-mer ssDNA tracking strand highlighted how the Core C-domain residue Trp597 stacks on a nucleoside near the 5′ end of the ssDNA and likely demarcates the single-strand:double-strand junction during the helicase reaction (6). Whereas mutating Trp597 to alanine had no apparent effect on MsmLhr-Core DNA-dependent ATPase activity, the W597A change effaced its helicase activity and its translocase activity in the SA-displacement assay (6). The corresponding amino acid in EcoLhr is conserved as Trp620 (denoted by the red box and arrowhead in Figure 1). To see if Trp620 plays an analogous role in coupling ATP hydrolysis to mechanical work in EcoLhr, we produced and purified EcoLhr-FL-W620A and EcoLhr-Core-W620A (Figure 4A) and found them to be as active in ATP hydrolysis as the respective wild-type EcoLhr proteins (Figure 4C). However, EcoLhr-FL-W620A was ineffective at ATP-dependent displacement of SA from the ssDNA 5′ biotin-SA complex (Figure 6A, lane 12).
EcoLhr unwinds an RNA:DNA duplex with a DNA loading strand
In light of the translocase activity demonstrated above, we tested wild-type EcoLhr-FL and EcoLhr-Core for helicase activity with a series of 3′-tailed duplex substrates consisting of a 24-bp duplex with a 20-mer 3′ single-strand tail to serve as a loading strand (Figure 7A). The 44-mer loading/tracking strand was a synthetic DNA or RNA oligonucleotide of identical nucleobase sequence (excepting U for T in RNA). The 5′ 32P-labeled 24-mer displaced strand was a DNA or RNA oligonucleotide complementary to that of the 44-mer strand. The helicase assay format entailed preincubation of EcoLhr and labeled nucleic acid substrate, followed by initiation of unwinding by addition of ATP, with simultaneous addition of a ‘trap’ of excess unlabeled 24-mer displaced strand. The trap strand minimizes reannealing of any radiolabeled 24-mer that was unwound by EcoLhr-FL and it competes with the loading strand for binding to any free EcoLhr that dissociated from the labeled 3′-tailed duplex without unwinding it. Consequently, the assay predominantly gauges a single round of strand displacement by EcoLhr bound to the labeled 3′-tailed duplex prior to the onset of ATP hydrolysis. The salient findings from the assay were as follows: (i) EcoLhr-FL unwound 3′-tailed RNA:DNA and DNA:DNA substrates to yield a radiolabeled free single-strand RNA and DNA, respectively, that migrated faster than the input tailed duplex during native PAGE and comigrated with free 24-mer generated by thermal denaturation of the substrates (Figure 7A, lanes Δ); (ii) the extent of unwinding of the RNA:DNA hybrid was greater than that of the DNA:DNA duplex; (iii) EcoLhr-FL failed to unwind an RNA:RNA duplex; and (iv) EcoLhr-FL did not unwind a DNA:RNA hybrid with an RNA loading strand (Figure 7A). Titration experiments showed that the specific activity of the full-length enzyme in unwinding the RNA:DNA hybrid was 4.5-fold greater than its activity in unwinding the DNA:DNA duplex (Figure 7B). These results establish that EcoLhr-FL is a unidirectional motor, powered by ATP hydrolysis, that tracks 3′ to 5′ along a DNA loading strand and unwinds duplexes en route. The EcoLhr-Core protein was able to unwind an RNA:DNA hybrid with a DNA loading strand, albeit less effectively than EcoLhr-FL, and was seemingly unable to unwind a DNA:DNA duplex (Figure 7A).
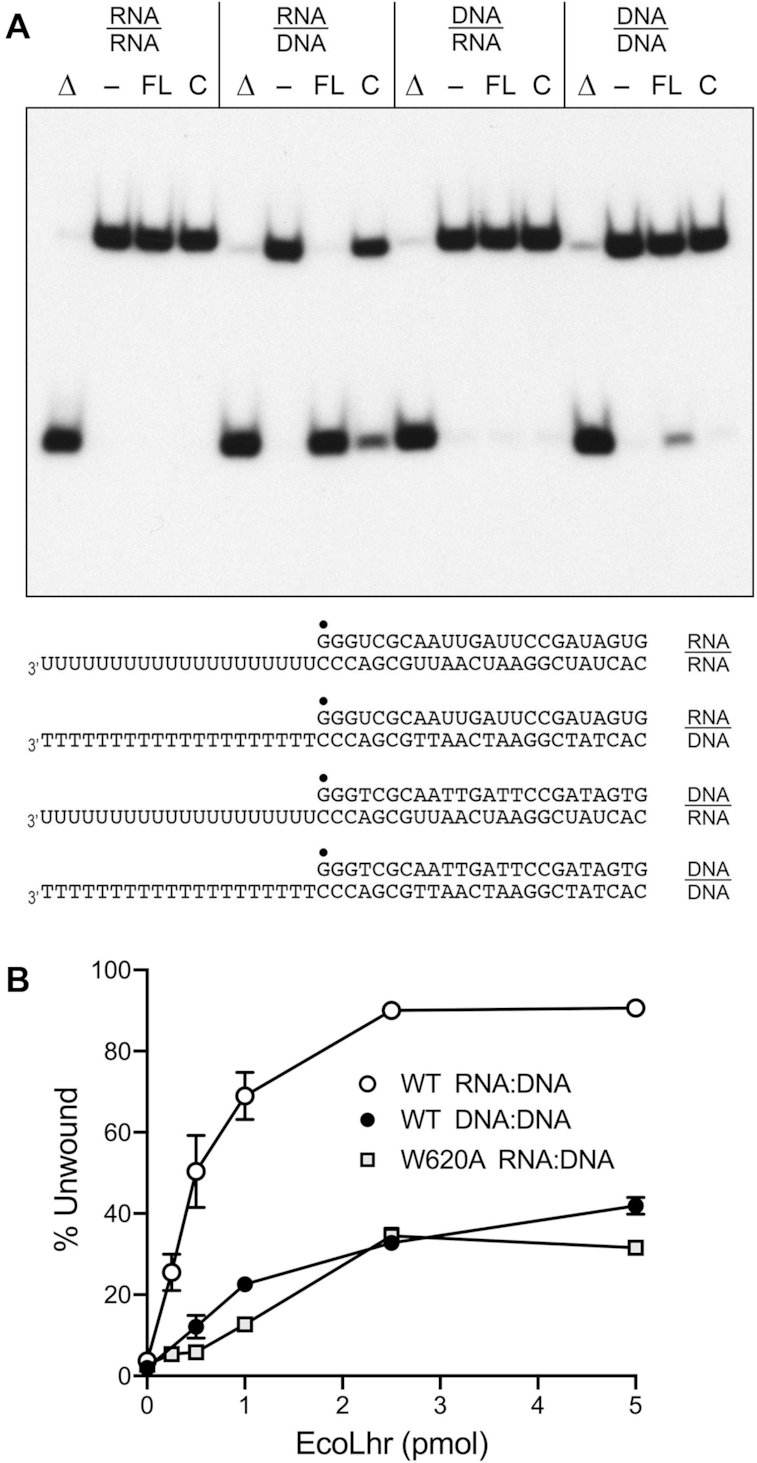
EcoLhr helicase activity with DNA and RNA substrates. (A) Helicase reaction mixtures (10 μl) contained 20 mM HEPES-NaOH, pH 7.5, 50 mM NaCl, 1 mM CaCl2, 25 nM of the indicated DNA:DNA, RNA:RNA, RNA:DNA or DNA:RNA substrates (depicted at the bottom, with the 5′ 32P label denoted by filled circles), 1 mM ATP, and 250 nM EcoLhr-FL (lanes FL) or EcoLhr-Core (lanes C) proteins were incubated at 37°C for 30 min and stopped with the addition of proteinase K. Enzyme was omitted from the reaction mixtures in lanes. Reaction mixtures lacking enzyme that were heat-denatured prior to PAGE are included in lanes Δ. The reaction products were analyzed by native PAGE and visualized by autoradiography. (B) Lhr titration. Helicase reaction mixtures were constituted as in panel (A) with RNA:DNA or DNA:DNA substrates and contained 0, 0.25, 0.5, 1, 2.5, or 5 pmol of wild-type (WT) EcoLhr-FL or EcoLhr-W620A as specified. The extents of duplex unwinding are plotted as a function of input EcoLhr. Each datum is the average of three independent titration experiments ± SEM.
Titration of the EcoLhr-FL W620A mutant for helicase activity on the RNA:DNA hybrid substrate showed that its specific activity was 7-fold less than that of the wild-type enzyme (Figure 7B). The residual helicase activity of the W620A protein stands in contrast to the apparent absence of SA displacement activity noted above. This disparity can be rationalized by positing that: (i) the amount of mechanical force needed to strip SA from biotin is greater than that required to disrupt a nucleic acid base pair; (ii) the W620A mutation does not preclude DNA translocation, which obviously is necessary for duplex unwinding, but rather affects the efficiency of chemo-mechanical coupling and, potentially, the force generated during the power stroke.
MsmLhr and its C-terminal domain have homo-oligomeric quaternary structures
To re-evaluate the quaternary structure of MsmLhr-FL in light of the present findings for EcoLhr, we purified MsmLhr-FL, MsmLhr-Core and the MsmLhr C-terminal domain (CTD; aa 863–1507) and gauged their native sizes by analytical gel filtration through Superdex-200. The 162 kDa MsmLhr-FL polypeptide eluted as a single component with a peak between the 669 and 440 kDa standards (Figure 8), indicating that it has a homo-oligomeric quaternary structure. This conclusion supersedes inferences from glycerol gradient sedimentation analysis of MsmLhr-FL in which a monomeric peak of MsmLhr-FL protein and associated ATPase/helicase activities was observed that sedimented between catalase and BSA internal standards (5). In retrospect, we can see that the monomer form was recovered in low yield relative to the protein applied to the glycerol gradient and that any homo-oligomeric forms of MsmLhr-FL would have pelleted under the conditions used for glycerol gradient sedimentation (5). Moreover, the glycerol gradient experiment was performed in the presence of 0.05% Triton X-100 (not present during our gel filtration experiments), which could have promoted an oligomer-to-monomer transition. The 93 kDa MsmLhr-Core polypeptide eluted as a single component peaking between the 158 and 75 kDa standards (Figure 8), confirming the evidence from velocity sedimentation and crystallography that the MsmLhr-Core is a monomer. The key finding here is that the 69 kDa MsmLhr-CTD eluted as a single component peaking between the 440 and 158 kDa standards (Figure 8), signifying that the CTD per se has a homo-oligomeric quaternary structure.
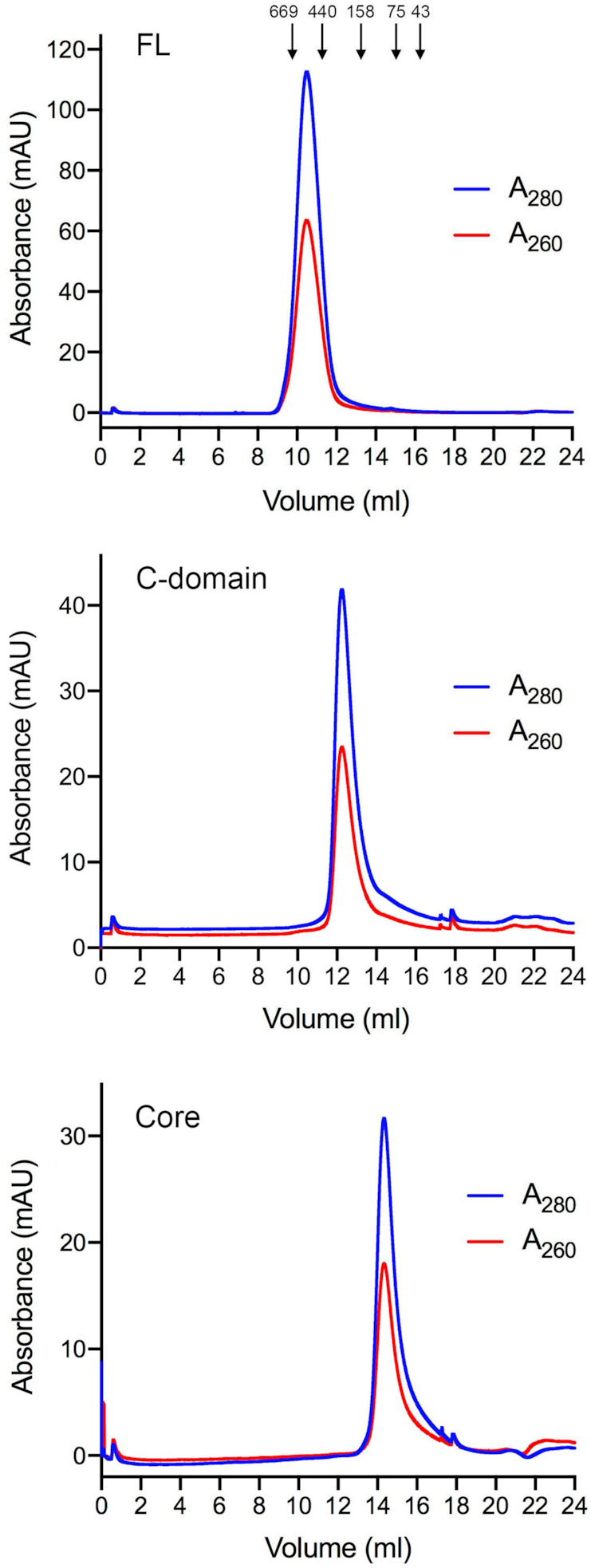
Gel filtration chromatography of MsmLhr FL, Core and CTD. MsmLhr-FL (top panel), MsmLhr-CTD (middle panel) and MsmLhr-Core (bottom panel) were gel filtered through a 24-ml Superdex-200 column equilibrated with 50 mM HEPES-NaOH, pH 7.5, 300 mM KCl, 1 mM DTT, 10% glycerol. The elution profiles shown were monitored continuously by A280 and A260 as a function of elution volume. Arrows denote the elution peaks and native sizes for a mixture of calibration standards: thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ovalbumin (44 kDa).
Cryo-EM structure of MsmLhr-CTD
We purified full-length MsmLhr and endeavored to determine its structure via cryo-electron microscopy (cryo-EM). The cryo-EM images sufficed to solve the structure of the CTD assembly at an overall resolution of 3.6 Å (Supplementary Figure S1 and Table S1). The Lhr-Core was poorly resolved and could not be modeled. The 3D map of the CTD was generated and refined without imposing symmetry in order to avoid bias to the oligomeric state. The 3D reconstruction demonstrates that the CTD forms a tetramer in a tail-to-tail arrangement (Figure 9A) with 2-fold symmetry about the y-axis. An atomic model for the CTD polypeptide chain from Leu893 to Leu1506, with side chains and good overall stereochemistry, was built into the electron density (Supplementary Figure S1E and Figure 9B). The continuous CTD polypeptide is punctuated by two gaps (aa 1046–1052 and aa 1251–1276) corresponding to disordered surface loops. Whereas we detected low-resolution electron density emanating from the N-termini of the CTD polypeptides (Figure 9A), it was of insufficient quality to permit modeling of the Lhr-Core domain.
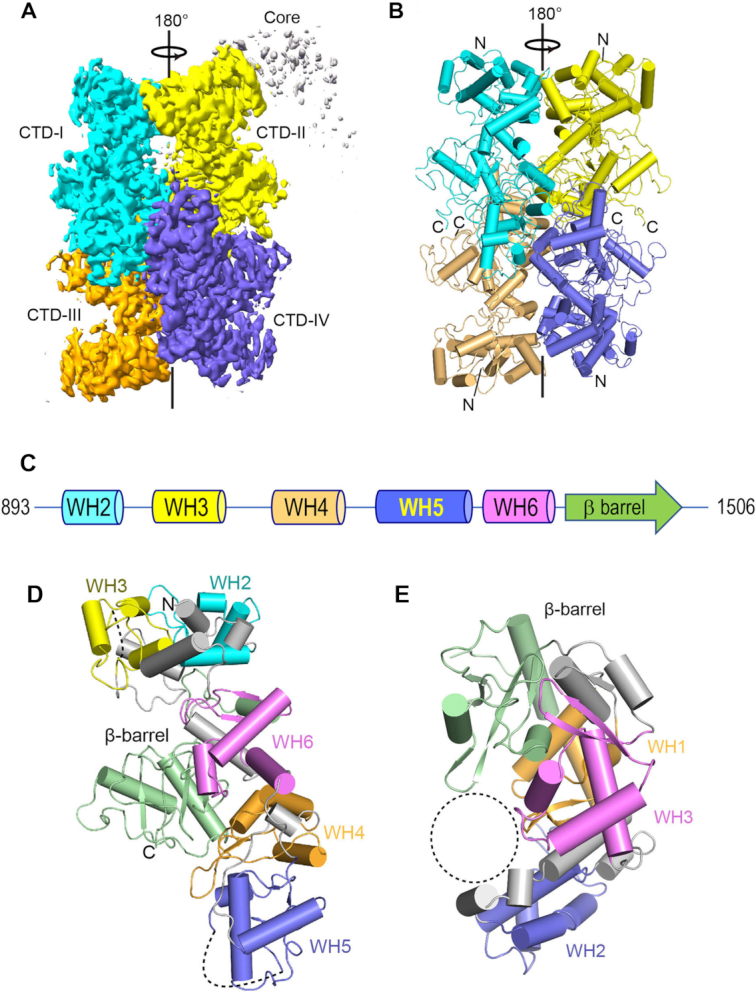
Cryo-EM structure of the MsmLhr CTD. (A) Overview of the cryo-EM map of MsmLhr CTD tetramer, colored by protomer, at 0.017 contour level. (B) Cartoon representation of MsmLhr CTD tetramer structure, colored by protomer as in panel (A), with helices depicted as cylinders. The amino (N) and carboxy (C) termini of the subunits are indicated. (C) Color-coded modular architecture of the MsmLhr CTD protomer. WH denotes winged-helix modules. (D) Cartoon representation of the MsmLhr CTD protomer structure, colored by modules as in panel (C). The disordered gaps in the structure are shown as dashed lines. (E) Tertiary structure of the DNA glycosylase AlkZ (pdb: 5UUJ), colored by modules. The dashed circle indicates the presumptive DNA binding site in AlkZ.
The tertiary structure of the CTD monomer consists of five tandem winged-helix (WH) modules (named WH2 to WH6 in Figure 9C, in series to the WH1 winged-helix module of Lhr-Core) and a C-terminal β-barrel module that are organized as two structural sub-domains (Figure 9D). The N-terminal subdomain consists of the tandem WH2 and WH3 modules that pack closely against each other. The WH4, WH5 and WH6 modules, and the β-barrel domain comprise a separate C-shaped subdomain. WH folds consist of three α-helices followed by two loops or ‘wings’ and two β strands forming an antiparallel β sheet (19). The WH fold is a subtype of the helix-turn-helix fold, which is found commonly in DNA-binding proteins (20).
The four Lhr-CTD protomers (which we refer to as CTD-I, -II, -III and IV) interact to form a cylindrical tetrameric quaternary structure 87 Å in diameter and 110 Å long (Figure 9B). The contact surfaces formed by CTD-I and CTD-IV (front view in Figure 9A) and (pseudo symmetry-related) CTD-II and CTD-III (back view in Figure 9A) are the main subunit interfaces, with contact surface areas of 768 and 669 Å2, respectively (21). The inter-subunit contacts are mainly via hydrogen bonds or salt bridges between Arg1206 and Gly1149-O, Arg1286 and Asp1237, Ser1288 and Arg1286, Tyr1203 and Gly1201-N, and His1199-Ne and His1199-O (Supplementary Figure S2A). Additional interfaces formed by CTD-I and CTD-II (top view in Figure 9A) and (pseudo symmetry-related) CTD-III and CTD-IV, comprise contact surface areas of 612 and 567 Å2, respectively, wherein Asp1366 makes a salt bridge to Arg915; a hydrogen bond is made between the Ala917 amide and the Gly1314 carbonyl; and Thr1100 makes a hydrogen bond to Thr1100 in the neighbor protomer (Supplementary Figure S2B). The interfaces between CTD-I and CTD-III (left side of Figure 9A) and CTD-II and CTD-IV (right side of Figure 9A) are relatively small, with contact surface areas of 369 and 286 Å2, respectively. The amino acids that contribute to the subunit interfaces in the MsmLhr CTD tetramer are denoted by black dots above the primary structure in Figure 1. Most of the interfacial contact points are conserved in EcoLhr.
Structural homology of Lhr-CTD with DNA glycosylase AlkZ
A DALI (22) search of the protein data bank with the Lhr-CTD model recovered as the ‘top hit’ a structure of AlkZ (Z score 16.6; rmsd 4.7 Å at 293 Cα positions; 15% amino acid identity), a novel bacterial DNA glycosylase that recognizes and excises inter-strand DNA crosslinks between major groove purine bases generated by the natural product toxin Azinomycin B (23,24). The crystal structure of the monomeric 371-aa AlkZ protein (pdb 5UUJ) consists of three WH modules and a terminal β-barrel that adopt a C-shaped fold (Figure 9E). The WH1, WH2, WH3 and β-barrel modules of AlkZ are organized spatially akin to the WH4, WH5, WH6 and β-barrel modules of Lhr-CTD (Figure 9D and E). The second-best ‘hit’ (Z-score 7.5; rmsd 3.2 Å at 63 Cα positions) was to a 90-aa Acidithiobacillus ferrooxidans transcriptional repressor (pdb: 6J05) composed of a single WH module, followed by a long list of WH proteins with lower Z scores.
DISCUSSION
The present study extends our understanding of the Lhr clade of SF2 helicases in three key respects. First, we demonstrate that the 1538-aa EcoLhr protein is a bona fide ATPase, translocase and ATP-dependent helicase. Like MsmLhr, EcoLhr translocates 3′ to 5′ on ssDNA and unwinds 3′ DNA-tailed duplex nucleic acid, either a duplex DNA or an RNA:DNA hybrid, with RNA:DNA being the preferred substrate. The ATPase and translocase activities of EcoLhr inhere to its N-terminal 877-aa Core domain. Whereas MsmLhr requires ssDNA to activate its ATPase, the EcoLhr ATPase activity is unaffected by exogenous ssDNA; this property might account for initial findings that extracts of E. coli induced to overexpress EcoLhr did not display increased nucleic acid-dependent ATPase activity vis-à-vis uninduced control extracts (7).
Second, we present evidence via gel filtration that full-length EcoLhr and MsmLhr have homo-oligomeric quaternary structures in solution, which are clearly distinct from the monomeric states of their respective Lhr-Core domains. The inference that the Lhr CTD is responsible for homo-oligomerization is supported by the finding that the MsmLhr CTD per se has a homo-oligomeric quaternary structure.
Third, we applied cryo-EM methods to solve the structure of the CTD component of full-length MsmLhr. The 614-aa CTD protomer is composed of a series of five WH modules and a C-terminal β-barrel module that adopt a distinctive tertiary structure. The cryo-EM experiments revealed that the CTD assembles into a unique homo-tetrameric quaternary structure stabilized by multiple inter-subunit interaction surfaces. The helicase domains are present and seen to emanate from the N-termini of the CTD subunits of the CTD tetramer, but the helicase core (for which a crystal structure is available) could not be modeled because of insufficient cryo-EM density. We surmise that the Lhr Core helicase domains are positionally dynamic with respect to the central CTD tetramer.
Comparison, here and previously (5), of the biochemical activities of the full-length versus Core MsmLhr and EcoLhr enzymes indicates that the CTD abets Lhr’s duplex unwinding function. It is not clear whether this salutary effect of the CTD occurs simply by virtue of its ability to oligomerize the helicase domain, i.e., in a scenario whereby a Lhr-Core oligomer is better at duplex unwinding than a Lhr-Core monomer. Alternatively, the CTD itself might interact with nucleic acid to facilitate action of the Lhr Core helicase. Our efforts to date to detect binding of the purified MsmLhr-CTD protein to ssDNA or duplex DNA in a gel shift assay format have not been fruitful.
A novel insight from the CTD cryo-EM structure is the partial homology of the Lhr CTD protomer to the monomeric AlkZ protein (24). The DNA glycosylase activity of AlkZ protects Streptomyces sahachiroi, the bacterium that produces the toxin azinomycin B (AZB), from lethal major groove guanine-N7 DNA inter-strand crosslinks formed by AZB (23,25). Treatment of budding yeast Saccharomyces cerevisiae with AZB triggered transcriptional upregulation of several genes involved in DNA repair and recombination (26). To our knowledge, there are no reports on the effects of AZB on mycobacteria. The antitumor activity of AZB is cited as comparable to that of mitomycin C (MMC) (27). MMC is an antibiotic/antitumor toxin that induces minor groove DNA inter-strand crosslinks between guanine nucleobases (28). Lhr is strongly upregulated at the transcriptional level in response to treatment of mycobacteria with MMC (2–4) and deletion of the lhr gene sensitizes M. smegmatis to killing by transient exposure to MMC (29). It is tantalizing to think that Lhr might be a novel multi-functional ICL repair factor composed of a helicase enzyme in the Lhr Core and a possible AlkZ-like ICL-recognition component within the Lhr CTD. At present, the basis for DNA recognition by AlkZ is uncharted because a structure is lacking for AlkZ in complex with DNA. However, Mullins et al. (24) have speculated as to potential catalytic residues and suggested a candidate DNA interaction surface in the concavity of the AlkZ fold (Figure 9E). To our inspection, the putative AlkZ glycosidase active site residues are not conserved in MsmLhr-CTD and the counterpart of the suggested AlkZ DNA binding surface would be occluded in the context of the CTD homo-tetramer. Thus, the functional significance of the Lhr-CTD/AlkZ homology remains speculative for now. Also in the realm of conjecture is the prospect that the preferred RNA:DNA helicase activity of Lhr might be pertinent to its function in evading the toxicity of MMC, by acting on inter-strand crosslinks between guanine nucleobases in the context of RNA:DNA hybrids, e.g., in RNA-loops or RNA-primed Okazaki fragments.
DATA AVAILABILITY
Atomic structure models of MsmLhr-CTD have been deposited in the PBD and EMDB databases under ID codes 7LHL and EMD-23345, respectively.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [AI64693to S.S.]; NCI [P30CA008748]. Funding for open charge: National Institutes of Health [AI64693].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
21.
23.
24.
25.
26.
27.
28.
29.