 Single-molecule insight into stalled replication fork rescue in Escherichia coli
Single-molecule insight into stalled replication fork rescue in Escherichia coli
Nucleic Acids Research



- Altmetric
DNA replication forks stall at least once per cell cycle in Escherichia coli. DNA replication must be restarted if the cell is to survive. Restart is a multi-step process requiring the sequential action of several proteins whose actions are dictated by the nature of the impediment to fork progression. When fork progress is impeded, the sequential actions of SSB, RecG and the RuvABC complex are required for rescue. In contrast, when a template discontinuity results in the forked DNA breaking apart, the actions of the RecBCD pathway enzymes are required to resurrect the fork so that replication can resume. In this review, we focus primarily on the significant insight gained from single-molecule studies of individual proteins, protein complexes, and also, partially reconstituted regression and RecBCD pathways. This insight is related to the bulk-phase biochemical data to provide a comprehensive review of each protein or protein complex as it relates to stalled DNA replication fork rescue.
INTRODUCTION
Genome duplication is inherently accurate and highly processive (1). In Escherichia coli, DNA replication initiates at oriC and generates two replication forks that move bidirectionally away from one another until their progress is permanently impeded in the terminus region, with the two daughter molecules subsequently resolved by site-specific-recombination or decatenation (2–6). However, each of the two forks generated seldom makes it to the terminus without encountering problems that could potentially be life-threatening to the cell. Each fork is thought to stall or collapse entirely at least once per cell cycle and possibly even more frequently (7–16). Fork stalling is the result of the advancing replisomes encountering physical impediments to progression, or the replisomes experiencing a shortage of DNA synthesis precursors (17–19). Impediments to progression include proteins bound to the DNA ahead of the replication fork such as repair enzymes, repressors or RNA polymerase (either alone or coupled to ribosomes), noncoding lesions in the template DNA, unusual secondary structures that arise in the DNA, R-loops, and either single- or double-strand breaks (14–18,20–22). Impeded forks must be rescued and a failure to do so is a lethal event. Consequently, the accurate and faithful duplication of the genome relies on the DNA replication, repair and genetic recombination machinery working closely together (7,14,23–25). This follows because many of the proteins involved in repair and recombination play critical roles in rescuing stalled or collapsed forks. In some instances, stalled forks can be directly restarted by components of the replisome itself and, because these processes do not require the recombinational repair machinery and have yet to be dissected at the single-molecule level, they will not be discussed herein (26–29).
An impeded fork is one in which net forward progression has been prevented. In this review, we define a stalled fork as one in which the DNA remains intact but the progression of the replisome is blocked. The second kind of impeded fork is one which is termed collapsed or broken. Here, one or more fork arms are no longer connected to the parental duplex. In either of these impeded states, polymerase uncoupling and or dissociation of one or more replisome components can occur (30). The impeded fork can be rescued by the enzymes of recombination of the gap (RecF) repair pathway (8,24). An additional pathway for the rescue of stalled forks involves regression of the newly replicated strands of DNA at the stalled fork to form a Holliday junction-like structure or ‘chicken foot’ intermediate in a reaction catalyzed by the DNA helicase RecG (Figure 1A and (31–34)). This structure can be further processed in a reaction known as branch migration by the multi-subunit enzyme RuvAB, or the junction may be cleaved by the dimeric Holliday junction resolvase RuvC (35). In contrast, when the replisome encounters a double-strand DNA break or a nick in the leading strand, forward progress is also impeded as the replisome polymerizes off the ends of the duplex, the fork structure no longer exists and resurrection of the structure requires rescue by the RecBCD pathway (Figure 1B and (8,24)). Here, processing of the nascent double-strand break by the helicase-nuclease RecBCD occurs and this is followed by strand invasion catalyzed by DNA strand exchange protein RecA (36,37).

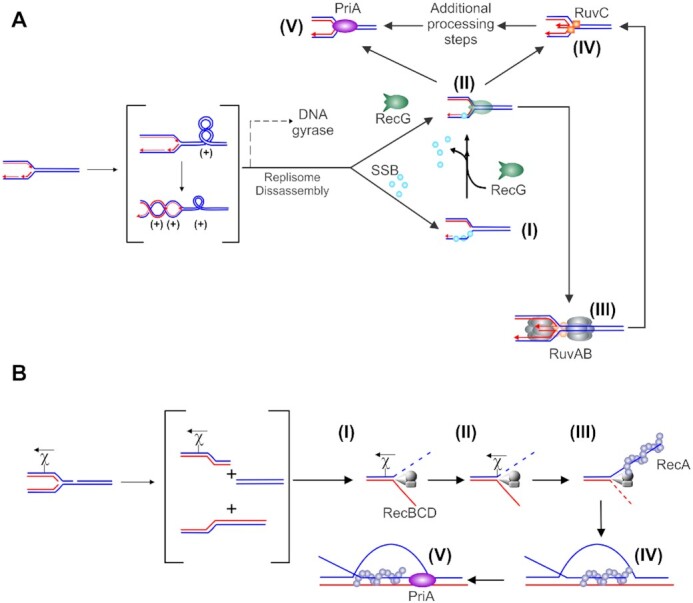
Impeded DNA replication forks can be rescued by different enzymatic pathways. (A) Rescue of unbroken, stalled forks is initiated by RecG. A schematic showing replicating DNA with the fork encountering a block, leading to replisome disassembly (adapted from references (105,264)). If a gap, for example, occurs in the lagging strand, it will be rapidly bound by SSB (step (I), blue spheres). RecG then binds and couples fork regression to the displacement of SSB (II). The resulting Holliday Junction is bound by RuvC that cleaves the DNA into a substrate for the replication restart DNA helicase, PriA (steps IV and V). Alternatively, once the replisome disassembles, DNA is released and is free to extrude the chicken foot to which RecG (colored green) binds with high affinity (II). The dashed line in the upper scenario indicates a potential alternate path if DNA gyrase were to act first. RecG could then bind to the (−)scDNA that may have regressed or, assist regression similar to that observed for (+)scDNA (115). This results in the formation of a Holliday Junction to which RuvAB binds and processes (III) into a substrate for RuvC (IV). Cleavage by RuvC followed by additional processing could form a PriA substrate. Alternatively, RecG could regress forks into the PriA substrate directly. (B) Rescue of collapsed broken forks is performed by the RecBCD pathway. The nascent dsDNA end is bound by RecBCD (I) which rapidly unwinds and degrades up to chi (II). The enzyme pauses at this sequence, is reversibly altered and once translocation resumes, it loads RecA onto the 3′-terminated strand (III). Once a filament forms, RecA invades a target duplex, searches for and locates the homologous target and the exchange of strands results in the formation of a displacement loop (D-loop; IV). The 3′-end of the invading ssDNA in the context of the D-loop is a substrate for PriA (V). Other fork rescue pathways have been proposed in addition to what is shown in these schematics. For these, which are yet to be interrogated at the single molecule level, the reader is referred to (265–267).
The endpoint of each fork rescue mechanism is the formation of a substrate for the DNA helicase PriA which loads the replicative, homo-hexameric DNA helicase DnaB onto the template lagging strand arm of the fork, leading to the resumption of replication (38,39). When forks are processed by RecG and RuvABC, the endpoint is a fork with a 3′-OH on the nascent leading strand the preferred fork substrate for PriA (40,41). When forks are processed by RecABCD, the endpoint is the formation of a displacement loop (D-loop) to which PriA also binds with high affinity (41). As for forks, DnaB is also loaded onto the template lagging strand arm, leading to the resumption of replication (39).
In addition to the enzymes of the DNA repair and recombination machinery being important, the single-strand binding protein (SSB) is also a key player in fork rescue (42). This protein is essential to all aspects of DNA metabolism in Escherichia coli (43–47). It is present at >25 copies per fork, binds to and protects exposed single-stranded DNA (ssDNA), and also interacts with multiple proteins at forks, including replisome components and repair enzymes including those involved in fork rescue.
In this review, we focus on single-molecule studies of the enzyme systems involved in the rescue of impeded replication forks. These proteins are discussed in order of their likely involvement in fork rescue. For rescue of stalled forks via regression, these are SSB and the DNA helicases RecG and RuvAB, followed by the resolvase, RuvC. For the rescue of collapsed or broken forks by DNA processing and strand invasion to create D-loops, these are the DNA helicase RecBCD and the recombinase RecA. As RecA filament formation can also be facilitated by recombination mediators, these are discussed in the RecA section.
Single-molecule methods
To enable the reader to understand how data are derived from the various single-molecule studies cited we first present a brief introduction to the five approaches employed. These approaches are presented in their simplest formats and the reader should be aware that they have been modified or combined with other approaches to increase understanding. The tethered particle motion assay (TPM) is simple and involves the attachment of a single DNA molecule to a polystyrene bead at one end and the coverslip surface to the other (48). The Brownian motion of the bead is restricted to a hemispherical region by its DNA tether (Figure 2A, left). The size of the region decreases once a DNA-binding protein induces changes in the DNA (Figure 2A, right). Beads are imaged using one of several different modalities with changes in their position being monitored using centroid tracking or frame averaging (49,50). TPM was used to image RecA filament assembly on SSB-coated ssDNA and rotation of duplex DNA by RuvAB (51,52). Fluorescent energy transfer (FRET) is an established single-molecule technique that frequently uses oligonucleotide length DNA molecules (53). Here donor and acceptor dye molecules are placed at test positions so that the binding and/or activity of the protein being studied induces a change in the distance between the two dye molecules (Figure 2B). In one conformation, dye molecules are spaced far apart and fluoresce independently of one another in the low or no FRET position. When protein is introduced, dye molecules can be brought into proximity and nonradiative energy transfer between two fluorescent molecules takes place, typically referred to as high FRET. The extent of the FRET signal reports the intervening dye distance, estimated from the ratio of acceptor to total emission intensity. To permit clear visualization of single dye molecules, total internal reflectance microscopy (TIRFM) is used for imaging. The FRET/TIRFM approach has been used for numerous proteins including RecA, RuvC and SSB (54–56).
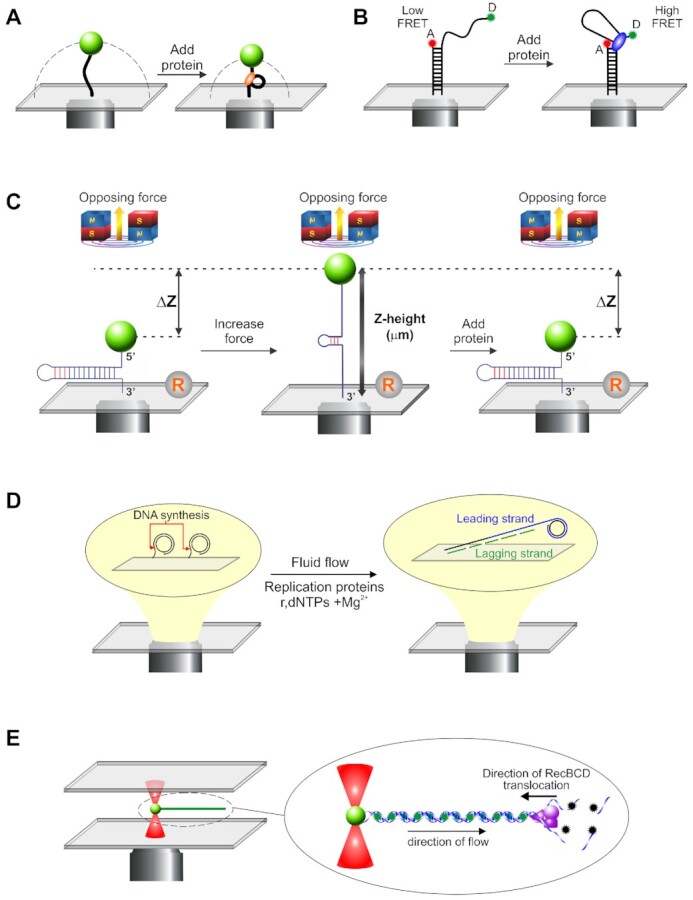
A range of single molecule techniques are used to understand fork rescue enzymes. (A) Tethered particle motion (TPM): Here, small polystyrene beads are attached to single DNA molecules which are in turn bound to a coverslip surface. The Brownian motion of the bead is constrained to a hemi-spherical region (the dashed line) by the DNA (left panel). When protein is introduced, changes in the DNA length decrease the size of constrained region of the bead (right panel). Studies can be done in simple, single channel flow cells or more complex designs using buffer wells (268,269). (B) Fluorescence resonance energy transfer (FRET) combined with TIRFM: Here, DNA substrates are typically <100 bases in length. They are attached to coverslip surfaces with donor and acceptor fluorescent dyes attached to the DNA at test positions and exposed in solution. When dye molecules are spaced far apart, their interaction is negligible and no resonance energy transfer can be observed (low FRET, left panel). When protein is bound, changes in the DNA can decrease the spacing between the two dye molecules such that fluorescence energy transfer occurs (high FRET, right panel). To facilitate imaging with high signal to noise, total internal fluorescence microscopy (TIRFM) is used to illuminate and visualize dye molecules, with reactions typically performed in single channel flow cells (269). (C) Magnetic tweezers: In this approach, single DNA molecules are sandwiched between super-paramagnetic beads and a coverslip the surface. The position of the beads can be carefully controlled by the application of a magnetic field, with the amount of force applied being proportional to the strength of the field. In the example shown, the increase in force is associated with unwinding of the hairpin (middle panel). The position of the super-paramagnetic bead changes as proteins exert their effects on the DNA, here duplex rewinding (right panel). Changes in Z-height are determined relative to a reference bead fixed to the surface (R). MT studies have employed single channel or disturbance-free flow cells (59,269). (D) TIFRM combined with fluid flow: In this technique, single DNA molecules are attached to the surface of a coverslip. In the example shown, this a circular template with one strand attached (left panel). When replication components are introduced using fluid flow, DNA synthesis can be visualized as the DNA is extended by the flow (right panel). TIRFM is used to image the attached molecules using either DNA dyes or fluorescent tagged proteins. Reactions are visualized using single or triple channel flow cells, with each channel having single inlet and outlet ports, or using laminar boundary-steering flow cells (61,269–271). (E) Optical tweezers combined with laminar flow and fluorescence microcopy. Here, optical tweezers are used to trap and manipulate 1 μm diameter polystyrene beads attached to single DNA molecules (left panel). Fluid flow past the bead stretches the DNA out, like a rope behind a boat. In the example shown, the DNA is visualized using the fluorescent dye YOYO-1 (inset). Changes in the length of the DNA corresponding to DNA unwinding and degradation by RecBCD occur as a result of dye displacement. Green stars, fluorescing dye molecules; black stars, nonfluorescent dye molecules. Alternatively, proteins can be fluorescent tagged or, both proteins and DNA tagged with different wavelength fluorophores to enable visualization by wide-field epifluorescence microscopy. Reactions are done in multi-stream microfluidic chambers (269).
Magnetic tweezers are a more sophisticated version of the TPM assay (57,58). Here single- or double-stranded DNA, 500–3000 bases in length is sandwiched between a superparamagnetic bead and a coverslip surface within a flow cell (Figure 2C, left panel). A pair of magnets is placed above the sample chamber but as close as possible to apply the magnetic field gradient. As the gradient increases, the bead experiences an upward force, which in this case causes the hairpin to unzipper (Figure 2C, middle panel). The addition of a DNA helicase like RecG results in duplex rewinding which is observed as a decrease in bead Z-height (right panel). This experimental approach was used to study fork regression by RecG, SSB binding to ssDNA and RecA filament formation (34,59,60). A rolling-circle DNA synthesis assay that uses flow to extend DNA molecules and TIRFM to image single DNA molecules being replicated was developed (61). Here, partial duplex, circular DNA is attached to the surface of a flow cell (Figure 2D, left panel). When replication components are introduced under conditions of flow, replication initiates at the ss/dsDNA fork junction, increasing the nascent duplex DNA length (Figure 2D, right panel). Reactions can be visualized using fluorescent dyes with a high affinity for dsDNA or proteins can be tagged in various ways, or a combination of both can be used. This approach has been used to dissect the DNA replication process, including revealing SSB dynamics (62). Finally, optical tweezers combined with laminar flow and wide-field epifluorescence microscopy has been used to interrogate RecA filament assembly, the unwinding of DNA by RecBCD, and SSB behavior (60,63,64). In this approach, an infrared laser beam is focused to a position in the focal plane forming a Gaussian trap (Figure 2E, left panel). Here polystyrene beads are optically trapped, and if a single DNA molecule, bound to fluorescent dyes is attached, it will be extended by fluid flow past the bead. The labeled DNA appears as a white string against a black background. By performing assays in multi-stream laminar flow cells, the trapped complex can be translated into adjacent streams containing protein to initiate reactions, in this case, RecBCD (Figure 2E, right panel). Both DNA and proteins can be labeled with different wavelength fluorophores, and the system can be modified to have dual optical traps (65–67).
SSB
SSB is an essential protein functioning in all aspects of DNA metabolism in Escherichia coli (43–47). The protein exists as a homo-tetramer with a monomer MW of 18 844 Da (68). Each monomer is divided into two domains defined by proteolytic cleavage: an N-terminal domain comprising the first 115 residues and a C-terminal tail spanning residues 116–177 (69). The N-terminal domains are responsible for tetramer formation and binding to ssDNA that is mediated by the four oligonucleotide-oligosaccharide binding folds (OB-folds) in the tetramer. Here, ssDNA binding by the OB-folds results in the wrapping of the polynucleotide around the SSB tetramer and depending on the solution conditions, results in different binding modes (Figure 3A and (67,70,71)). OB-folds are also responsible for binding to the linker domain of nearby SSB tetramers, resulting in cooperative ssDNA binding (72,73). The disordered C-terminal tail can be further subdivided into two regions: a sequence of approximately 50 amino acids that has been called the intrinsically disordered linker or linker (43,47,73,74). This is immediately followed by the acidic tip or tip, which is the last 8–10 residues of SSB. The linker is responsible for mediating protein–protein interactions using a mechanism similar to that employed by Src homology three domains binding to PXXP ligands, while the acidic tip uses long-range electrostatic interactions to maintain the structure of the C-terminal domain in an active conformation (72,75–78).
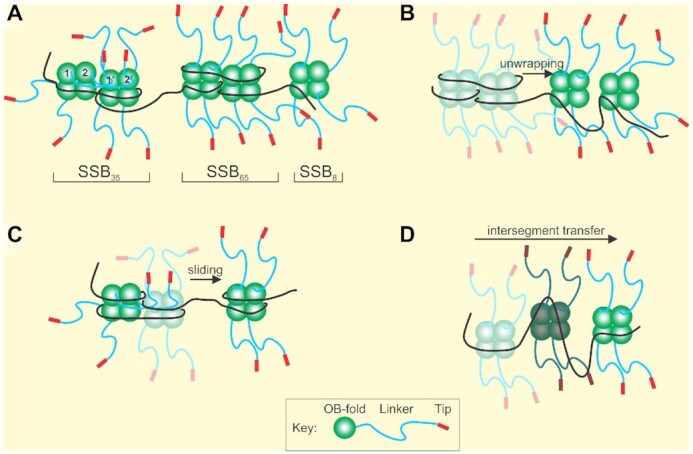
SSB forms dynamic structures on single-stranded DNA. (A) SSB can interconvert between ssDNA binding modes defined by the number of nucleotides of ssDNA occluded by a tetramer. (B) SSB complexes undergo unwrapping and wrapping transitions during complex assembly. Tetramers are colored as faded complexes (wrapped) to show transition to unwrapped complexes (bright colors). (C) SSB protomers can slide on ssDNA. (D) SSB tetramers relocate on ssDNA via intersegment transfer. A single tetramer is shown in three positions indicated by different shades of color as it moves between ssDNA segments. Details are described in the SSB section.
The protein has dual roles that are intimately connected. First, SSB protein binds to and protects single-stranded DNA (ssDNA) intermediates generated during DNA processing such as those found during DNA replication, repair and DNA strand exchange. Second, it binds to as many as nineteen proteins temporally and spatially, to both store and target enzymes to the DNA when needed (79,80). These roles are connected via the linker domain of the protein, binding to OB-folds present in both SSB and its binding partners (Bianco, submitted and (32,42,72,82). When linker/OB-fold interactions take place between SSB tetramers, cooperative ssDNA-binding results (Figure 3A, SSB35 mode and (72,73)). When these interactions occur between an SSB tetramer and an interactome partner, loading of that protein onto DNA can take place (42,83,84).
SSB binds tightly and cooperatively to ssDNA with a Kd = 1.45 × 10−7 to 4.04 × 10−9 M, requiring concentrations of salt as high as 2 M for dissociation (51,60,85). This tight binding is used to advantage by the DNA helicases RecG and PriA to gain access to otherwise busy stalled replication forks to outcompete other proteins. Here, atomic force microscopy was used to show that SSB bound to forks increases the binding of each enzyme to DNA with each helicase separately loaded onto fork arms and in the process, being remodeled by SSB (83,84).
Depending on solution conditions such as varying [NaCl], temperature, pH and applied force, each tetramer can typically occlude 17–65 nts of ssDNA, or as recently revealed, 8 nt only, and the protein can also form octamers (Figure 3A and (47,60,66,67,86,87)). DNA molecules can be partially or completely covered by tightly bound SSB depending on protein concentration, and somehow, other proteins must gain access to the ssDNA for subsequent processing (88,89). In vivo, there are ∼2000 SSB tetramers per cell so the protein is present in excess over nascent single-stranded DNA regions which it completely covers in a cooperative fashion (90). To provide access to other proteins, either SSB must completely disengage from the DNA or SSB–DNA complexes must be dynamic with tetramers rearranging on the nucleic acid lattice first before finally disengaging.
Multiple single-molecule studies have revealed the dynamic behavior of SSB in different ways (Figure 3B–D). FRET studies using oligonucleotide length DNA molecules revealed that SSB–ssDNA binding modes can interconvert in a salt-dependent manner; that the protein can slide on DNA and also undergo intersegment transfer (54,55,91,92). A combinatorial study using both optical and magnetic tweezers and long ssDNA substrates revealed reversible intramolecular condensation that also involved switching between binding states and possibly tetramer–tetramer interactions as well (60). Recently, dual optical tweezers approaches were used to demonstrate the individual binding modes of SSB, that ssDNA-binding by SSB is biphasic, with the initial ssDNA wrapping events being followed by unwrapping events as protein density on the DNA increases and, that the unwrapping energy cost increases as more ssDNA is successively unraveled (66,67). These studies also uncovered two new binding modes of SSB where the protein binds only 8 nt or separately, 17 nt of ssDNA, called SSB8 and SSB17, respectively, distinguishing them from the well-established SSB35 and SSB65 binding modes.
In addition to the changes in binding modes induced by solution conditions, the binding state of SSB can also be altered by the binding of the recombination mediator protein RecO and the mediator complex RecOR (both part of the RecF pathway). An important component of the endpoint of the continually changing SSB–ssDNA landscape is to provide access for subsequent enzymes as shown for RecA (54,67). Here, TIRF microscopy employing fluid-stretched ssDNA was used to demonstrate that dimers of RecA take advantage of small gaps in the dynamic SSB–ssDNA complexes to nucleate nucleoprotein filaments which then grow more rapidly in the 5′-3′ direction by monomer addition (93). Both nucleation and filament growth are enhanced by RecO and RecR, consistent with the ability of RecO to bind to the linker domain of SSB and influence the binding state of the protein (reorganize and/or displace), as well as the ability of RecO to bind exposed ssDNA previously occupied by an SSB tetramer (82,94–96).
At replication forks, there is on average 0.5–1 kb of ssDNA available (3). Using a site size of 40 nucleotides occluded per tetramer, there would be on average 25 tetramers bound per fork, predominantly on the lagging strand. As replisomes advance, SSB would likely be recruited from the bulk solution as the duplex is unwound but as SSB–ssDNA complexes are dynamic, it is conceivable that transfer between ssDNA regions within the immediate proximity of the fork may also occur. To address this, TIRF studies employing fluid-stretched DNA and fluorescent proteins were done (97). As expected, SSB molecules are recruited from the solution to bind to nascent ssDNA produced by DnaB. Also, ssDNA-bound SSB is recycled from one Okazaki fragment to the next on the lagging strand arm of the fork (62). Recycling may involve intersegment transfer or, tetramer–tetramer binding or, a combination of both, as tetramers relocate from one location to the next.
In contrast to the above-mentioned situations where SSB must, for the most part, remain associated with the DNA but still provide protein access to a dynamic SSB–DNA complex, some reactions such as DNA replication fork rescue require that SSB be completely and rapidly disengaged from the DNA for the reaction to proceed. To achieve displacement, an actively translocating motor protein must generate sufficient force to dislodge SSB tetramers. This follows because at least 10 pN of force are required to fully disengage a single SSB tetramer from the DNA (55). During fork regression, RecG couples fork arm DNA unwinding and duplex rewinding to the efficient displacement of multiple SSB tetramers (described below in more detail; (34,98)). Displacement requires functional linker domains of SSB, as mutants lacking the linker require 14-times longer to be dislodged by the advancing RecG helicase. Similarly, in DNA strand exchange where contiguous RecA nucleoprotein filaments are required, both SSB rearrangement and displacement are required. While rearrangement facilitates nucleation, filament extension results in SSB tetramers being dislodged. Both rearrangement and dissociation may require functional SSB linker domains (93,98). The requirement for the linker domain at this stage of protein function is consistent with a requirement for cooperative tetramer-tetramer interactions mediated by SSB linker/SSB OB-fold interactions being involved in complex rearrangement and/or dissociation, in addition to their involvement in ssDNA binding (72,82,98).
RecG
The RecG DNA helicase is a powerful, monomeric enzyme with potent biological activity that acts on nascent stalled forks. It is tightly regulated in vivo by restricting copy number to 10–15 molecules per cell and by binding to SSB where it is sequestered at the inner membrane until needed similar to DnaA, RecA, LacI and the RNA degradosome (80,90,99–104). When forks stall, it is rapidly delivered to forks presumably by SSB where the data show it can bind DNA directly or, to SSB bound to the fork (34,83). In the former, this could involve RecG being transferred to the DNA by SSB while in the latter, SSB to SSB transfer of the enzyme may be involved, but this is an open question (83).
Once RecG is bound to the fork substrate, it catalyzes an efficient, unidirectional fork regression reaction (Figures 1A, 4A and (34)). To achieve this, the specialized attributes of RecG come into play. The enzyme couples dsDNA unwinding to duplex rewinding and in the process generates sufficient force to clear the fork of bound obstacles. These specialized attributes were demonstrated using a combination of optical and magnetic tweezers and a 1200 bp hairpin DNA substrate (34,98).
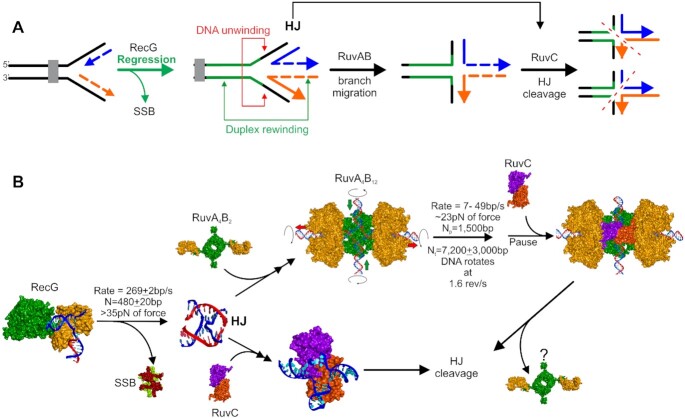
Stalled replication forks are rescued in a RecG-dependent manner. (A) Schematic of the stages of rescue. The progress of the fork is stalled by an impediment. RecG binds to the fork and catalyzes unidirectional fork regression, displacing SSB (if bound to ssDNA regions). Here RecG couples dsDNA unwinding to duplex rewinding to generate a chicken foot intermediate or Holliday Junction. RuvC can bind to the HJ in one of two configurations producing different cleavage products. Alternatively, RuvAB can bind the HJ and drive branch migration. Once RuvAB pauses or dissociates, RuvC will bind and cleave the junction. (B) Mechanistic insight into stalled DNA replication fork rescue gained from single-molecule studies of individual proteins. Crystal structures or structural models are shown for each partner represented as Connolly surfaces. RecG (helicase domains in green and wedge domain in orange (107)) binds to the fork, drives regression and displaces SSB tetramers (subunits colored light green and maroon (272)). The released HJ can adopt one of four configurations (only one X-junction is shown). In one pathway, RuvA tetramers (green) bound to 2 RuvB monomers(orange) bind to the HJ and assemble the complete branch migration complex with the HJ in a planar configuration (273). Branch migration, coupled to DNA rotation ensues, and when the enzyme complex pauses (or dissociates) the RuvC dimer binds. Alternatively, RuvC (each subunit in a different color; (274)) can bind directly to the HJ produced by RecG. In each pathway, RuvC binding leads to HJ cleavage at the RuvC consensus sequence which occurs every 25 bp in the genome. Details of these pathways are discussed in the RecG, RuvAB and RuvC sections.
Here RecG demonstrated an 8-fold preference for forks with a gap in the nascent leading strand consistent with multiple bulk-phase studies and the crystal structure (33,105–107). Once bound, RecG catalyzed unidirectional regression at a 269 ± 2bp/s for an average distance of 480 ± 20 nt in the absence of SSB (Figure 4B). Subsequent work showed that when fork arms are bound by SSB, it takes RecG 2.3 ± 0.6 s to initiate regression which then ensues at a 2.5-fold slower rate of 110 ± 6 bp/s (98). When a more tightly-bound version of SSB lacking the C-terminal domain was used, the length of the loading step increased 14-fold to 32 ± 7 s, but the regression rate was unaffected.
During regression, RecG can generate >30 pN of force. By extrapolation of the data in Manosas et al., RecG is predicted to stall at ∼50 pN (34,108). The ability of this monomeric enzyme to work against such a large opposing force is significant, given that the multi-subunit RNA polymerase stalls completely at 30–35 pN (109). The large force generated is essential to completely clear the fork arms of multiple SSB protomers and should be sufficient to displace dsDNA-binding proteins such as nucleoid-associated proteins, repressors and possibly even RNA polymerase, if required. Finally, using a modified 600 bp, 3-duplex arm fork, Manosas et al. demonstrated that RecG efficiently regresses forks into Holliday junctions, which can be subsequently further branch migrated by RuvAB or cleaved by RuvC, consistent with bulk-phase studies (Figure 4A and B (110), and see the RuvAB and RuvC sections below). This result is significant as it demonstrated the formation of an HJ or chicken foot intermediate by RecG, a key intermediate in most fork rescue models (111,112).
While RecG can bind to and process forks in the absence of SSB, the presence of SSB already bound to the fork affects fork regression in several ways, in addition to slowing down the overall regression rate (83,98,113). First, SSB enhances the binding of RecG to forks but as the linker domain of SSB binds the OB-fold of RecG and this domain of the helicase is required to bind to forks with specificity, RecG is initially loaded onto the parental duplex (107,114). This effect has been termed ‘SSB-remodeling of RecG’ and requires that the helicase domains facilitate the initial binding of the enzyme to DNA. Next, the helicase scans 36 ± 13 bp of the parental duplex ahead of the fork using thermal sliding, thereby testing the integrity of the fork. Finally, once RecG returns to the fork, it displaces SSB and facilitates unidirectional, ATP-hydrolysis-dependent fork regression (34,83,113).
RuvAB
The RuvAB complex is most well-known for branch migration of Holliday junctions (HJ), which are central, four-stranded homologous recombination intermediates (35). RuvAB is also implicated in the later stages of stalled DNA replication fork rescue as HJs form as the result of either positive torsional strain- or, RecG-dependent, regression of forks (Figures 1A and 4B, top pathway and (34,112,115)). Thus, it is likely one of the next enzyme complexes to participate in stalled replication fork rescue.
The active branch migration complex consists of at least one symmetric tetramer of RuvA protein that binds one face of the Holliday junction and two homo-hexameric rings of RuvB which function as chemo-mechanical motors to drive branch migration (116–120). It is important to note that while RuvB is classified as a DNA helicase, it does not catalyze strand separation on its own. Instead, RuvAB is the ATP-dependent Holliday junction helicase, with RuvB functioning as a DNA pump that pushes DNA through the central hole in the RuvB hexamer and across the surface of RuvA where strand separation occurs (121,122).
In the absence of divalent metal ions, Holliday junctions adopt a fully extended conformation that resembles a plus sign and there is no coaxial stacking between the component helices. This is known as the planar, open X-form (123). However, HJs are dynamic structures that fluctuate between at least four different conformations in the presence of divalent metal cations (124–126). Here, the helices undergo coaxial stacking so that the resulting structures have 2-fold symmetry. These stacked X-junctions can exist in either parallel or anti-parallel conformations (2 for each, dictated by how the helices stack). Single-molecule fluorescence resonance energy transfer was used to show that RuvA binding halts these conformational dynamics converting the HJ into an open planar configuration which is a requirement for efficient branch migration (127–129). DNA binding is mediated by domains I and II of RuvA with the third domain binding to RuvB and regulating branch migration (130).
For assembly of RuvAB onto a HJ, two domains III of the stable RuvA tetramer bind to a RuvB monomer each and the complex binds to the HJ in a reaction that requires only Mg2+-ions (Figure 4B top pathway and (118,131–133)). Thereafter, the remaining 10 monomers of RuvB bind to complete the formation of the opposed hexameric rings sandwiching the RuvA-HJ complex. Stable RuvAB-HJ complex formation requires both ATP binding and hydrolysis (134). Thereafter, ATP-hydrolysis dependent branch migration ensues (Figure 4A).
To quantitate branch migration, three separate studies were done, two using magnetic tweezers and the third using tethered particle motion (135–137). The results show that unidirectional branch migration proceeded at 7–49 bp/s with a processivity of 7200 ± 3000 bp (Figure 4B). The reaction continues unabated for on average 1500 bp before changing rates or pausing for as long as 10–20 s. It is conceivable that during these sequence-independent pauses, the complex is waiting for RuvC dimers to bind but this remains an open question. In contrast to pauses, branch migration is impeded at sequence heterologies as short as 20 bp, resulting in dissociation of RuvAB or in some cases, bypass following the extended pause. Dissociation of RuvAB at these heterologies may be followed by binding of RuvC or this may be a signal to other processing enzymes that DNA in the vicinity of forks contains imperfections requiring repair before the resumption of DNA replication. These studies also showed that RuvAB can work against opposing forces as large as 23 pN, and this is sufficient to displace nucleoid-associated proteins, repressors and possibly even RNA polymerase (138,139). This is an important facet of RuvAB function as these proteins may rapidly rebind to nascent heteroduplex fork arms once replisomes pass by and they could impede repair processes and thus require removal.
Previous work suggested that RuvB functions as a molecular pump driving branch migration while rotating the DNA (140). To test this, a novel, tethered particle method was developed (52). Here a HJ was sandwiched between 2.5 kB arms of homologous DNA. One end was attached to a coverslip surface while the other was attached to 850 nm, streptavidin-coated magnetic beads tagged with 20 nm fluorescent beads at a low density. In this assay system, the DNA attached to the bead is free to rotate the bead as branch migration proceeds. Results show that in the presence of RuvAB and ATP, the fluorescent beads rotated. This means that the DNA rotated at 1.6 revolutions/s as branch migration proceeded linearly at 10 bp/s. When an HJ is placed between two constrained segments of the chromosome, DNA rotation may limit the processivity of branch migration as torsional strain accumulates.
RuvC
The RuvC dimer cleaves Holliday junctions resulting in their resolution. Consequently, the enzyme is known as a resolvase (123,141). Junction binding is sequence-independent, but structure-specific and occurs with a 103- to 104-fold higher affinity than duplex DNA (142,143). Cleavage, however, demonstrates sequence specificity and occurs via the introduction of two symmetric, 5′-phosphorylated endonucleolytic cleavage events near the center of the HJ, at the consensus tetrameric sequence 5′-(A/T)TT↓(G/C), where ↓ represents a cleavage site close to the crossover point (Figure 4B and (141,142)). For symmetric cleavage to occur the individual events may be simultaneous or require the coordination of sequential cleavage events. The data support the latter model as the two active sites can be uncoupled (144–146). Importantly, the two incisions occur within the lifetime of a single protein–DNA complex (144). To ensure that both strands of the junction are cleaved before the enzyme dissociates, the rate of second-strand cleavage is accelerated by several orders of magnitude compared to the first (144). The increased flexibility of a nicked HJ was proposed to accelerate the second cleavage reaction by promoting the placement of the DNA in the second active site leading to cleavage (123).
Recent biochemical data show that RuvC preferentially cleaves HJs formed by RecG following the regression of stalled forks (Figures 1A, 4A and B, bottom pathway; (147)). It is conceivable that this may be the preferred pathway for fork rescue with additional branch migration by RuvAB and possible subsequent cleavage by RuvC, representing an alternative pathway. In both pathways, RuvC cleaves naked DNA and in the RuvAB pathway, this may involve sequence scanning of the junction held in a planar configuration by RuvA, with target recognition leading to RuvA dissociation and cleavage by RuvC (148).
Once RecG (or RuvAB) dissociates from the DNA, the nascent HJ is free to change conformation or spontaneously branch migrate (34,124–126). Thus, RuvC binding must trap the junction, control branch migration and facilitate cleavage by inducing structural tension in the DNA. It was shown that RuvC binding alters the HJ structure to an unfolded, tetrahedral conformation with twofold symmetry that resembles an ‘X’ and is known as the open-X conformation (Figure 4B, bottom pathway and (149)). In the absence of these conformational changes, resolution does not take place because they position the DNA strands in the correct position for cleavage to occur. The combination of conformational changes and relief of protein-induced structural tension in the DNA was shown to facilitate the coordination of the two cleavage events.
Studies with RuvAB showed that the enzyme paused frequently during branch migration, that these pauses did not correlate with the presence of RuvC consensus sequences but could be associated with RuvC binding (135–137). Similarly, RecG regresses forks on average 480 ± 20 nt and then dissociates in a sequence-independent manner (34). How then does RuvC manage to cleave at its consensus sequence? To address this, a FRET study using model junctions and RuvC in the presence of calcium as the divalent metal ion was done (calcium does not support cleavage) (56). The authors found that once bound, RuvC can convert to a short-lived partially dissociated intermediate where the HJ can undergo dynamic switching and possibly limited branch migration. During this partially dissociated state, it is conceivable that RuvC is sampling the sequence and once the consensus is located, tight binding occurs leading to HJ resolution. However, analysis of the E. coli genome shows that the total number of RuvC consensus sequences (all four possible combinations) is 180 083 or one every 25 bp (analysis by PB using Patmatch at EcoCyc (150)). This means that once RuvC binds to a HJ where it protects 20 bp; it will encounter the consensus sequence almost immediately each time it moves the distance of a footprint (151).
RecBCD
RecBCD enzyme is a multi-functional, enzyme involved in genetic recombination, DNA repair, maintenance of cell viability, and degradation of both foreign and damaged DNA (Figure 1B, 5 and (37,152,153)). The enzyme is both a destructive and sequence-independent exo- and endonuclease, and a highly processive, ATP-dependent, bipolar DNA helicase (154–157). It comprises three subunits: RecB (3′-5′ DNA helicase and nuclease), RecD (5′-3′ DNA helicase) and RecC (structural subunit that mediates strand separation; reads the sequence of the unwound 3′-terminated strand relative to the entry point of the enzyme; responsible for recognition of chi; see below) (157–161). The degradation of DNA is coincident with the unwinding of dsDNA, occurs as a result of endonucleolytic cleavage of the nascent, unwound ssDNA, and is more frequent on the 3′-terminated than on the 5′-terminated strand relative to the entry point of the enzyme (162). Studies using optical tweezers combined with laminar flow cells and video-fluorescence microscopy showed that the enzyme unwinds dsDNA at rates >1000 bp/s with a maximum processivity of 43 000 bp and no detectable pausing (Figure 5 and (63)). If an advancing replisome encountered a nick and dissociated from the DNA exposing a dsDNA end, RecBCD would bind with high-affinity and could potentially cause significant damage to the chromosome if it were to unwind and degrade 43 kB/binding event each time. Therefore, there must be a mechanism in place to regulate RecBCD and to facilitate the activity of the next protein in the repair pathway, RecA.
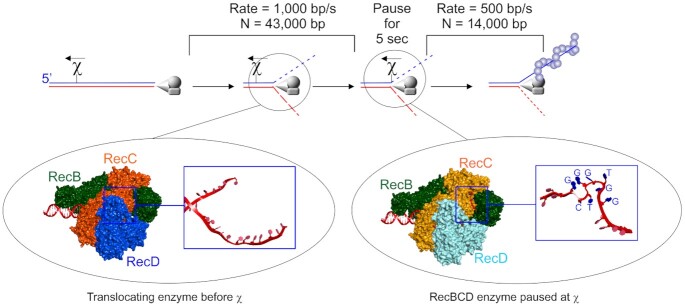
The rapidly translocating RecBCD enzyme is regulated by chi-sites. Top panel, a schematic of a broken dsDNA molecule with RecBCD bound to one end. In the presence of ATP, the enzyme unwinds and degrades the duplex asymmetrically up to a correctly positioned chi-sequence where it pauses for up to 5 s and is modified in multiple ways. Once translocation resumes, RecA is loaded onto the 3′-terminated strand. Left inset, the translocating enzyme before χ-recognition with the unwound strands of DNA (red strands in the blue box) passing through channels in RecC and being translocated upon by RecB and RecD, upper and lower strands respectively (157). Right inset, the enzyme paused at chi, with the unwound ssDNA containing the χ-sequence in its unusual folded structure (red strand in the blue box with the χ-sequence indicated (169)). Details can be found in the RecBCD section.
This mechanism involves an 8 base DNA sequence element known as chi (chi = χ = crossover hotspot instigator) (163). Chi sites are hot spots for genetic recombination that are recognized by RecBCD (164–166). Recognition of this 8-base element is orientation-dependent as the holoenzyme must approach χ from the 3′ side for recognition to occur and elicit changes in RecBCD (165,167). In vitro assays established that Chi is recognized by the translocating RecBCD enzyme as the single strand of DNA containing the sequence 5′-GCTGGTGG-3′ and this was confirmed in a recent crystal structure (Figure 5 and (168,169)). The structure shows that RecC is responsible for recognition but instead of reading a linear ssDNA sequence, chi is recognized as a uniquely folded DNA structure that is stabilized by intra-sequence contacts between the first and fourth guanine residues of the hotspot (Figure 5, right inset).
The effects of chi-recognition by RecC on the translocating RecBCD enzyme are complex and manifest multiple changes in the three-subunit complex. Many of these changes were suggested in bulk-phase studies and were demonstrated using single-molecule approaches (Figures 2E and 5). They are presented sequentially but their temporal occurrence remains to be established. First, bulk-phase and single-molecule studies show chi-recognition causes the translocating enzyme to pause for up to 5 s (170,171). Second, the nuclease activity of RecB is attenuated on the 3′-strand relative to the entry point of the enzyme (the χ-strand; Figure 5 and (170)). This results in the generation of the last cleavage event 4–6 nucleotides to the 3′-side of the χ sequence on the DNA strand containing the χ sequence (164,165). Third, the enzyme switches lead subunits so that once translocation and unwinding resume, RecB is the lead motor, whereas, before chi, RecD was the lead motor (although both subunits were active (154,161,172). Fourth, post-χ translocation occurs at two-fold lower rates, presumably because RecD is inactivated in response to chi-recognition although it remains associated with the advancing RecBC complex (171,173–175). Fifth, the nuclease activity on the RecD or 5′-terminated strand relative to the entry point of the enzyme is upregulated (167,174). This is essential to the formation of 3′-ssDNA overhangs that RecA uses to invade target DNA. Sixth, chi-modified RecBCD loads RecA onto the now, intact 3′-ssDNA mediated by the RecB nuclease domain that is positioned at the end of the enzyme opposite to the dsDNA entry point (Figures 1B, 5 and (169,176–179)). Not surprisingly, RecA-mediated recombination events are stimulated primarily to the 5′ side of the χ-site, the region of DNA where the 3′-terminated or χ-containing strand is now intact (163,166,180,181). Although RecA binds preferentially to sequences rich in GT, it is loaded onto the ssDNA loops produced by the translocating, chi-modified RecBCD and not onto the χ-site, which is still bound to the RecC subunit (Bianco and Kowalczykowski; unpublished; (169,182,183)). Once RecBCD dissociates from the DNA, the changes elicited by chi-recognition are lost and the enzyme reverts to the unmodified, pre-χ state (170,184).
In the absence of chi sequences, dsDNA is effectively destroyed by the nuclease activity of RecBCD (152,185). Thus, the frequency of chi sites in the genome (1009), their clustering and orientation bias, play important roles in regulating the nuclease activity of the enzyme to ensure the survival of the cell (37,186–189). This ensures that on each arm of the chromosome, RecBCD encounters an appropriately oriented chi site essentially pointing back toward the origin, approximately every 5000 bp. The high density of chi-sites is required because the efficiency of chi-recognition is only 30% and this ensures that RecBCD is switched from a destructive force into a recombinogenic enzyme within 5–15 s (63,170,190). Therefore, once forks break apart and dsDNA ends are exposed, RecBCD will bind and proceed to unwind and degrade the DNA up to an appropriately oriented χ-site in 5 s or less. The processivity of RecBCD that has recognized and responded to chi is reduced 3-fold relative to the enzyme on DNA without chi, consistent with the decay in recombinogenic frequency as a function of genetic distance (171,191). This final change elicited in the enzyme by χ-recognition ensures that recombination catalyzed by RecA to invade a target and create a D-loop to which PriA can bind, happens quickly and efficiently and large tracts of the genome are not unwound and degraded unnecessarily once χ-recognition has taken place.
RecA
RecA is the prototypical recombinase and is required for genetic recombination and DNA repair (192–194). In vitro, the protein catalyzes D-loop formation where a single-stranded DNA molecule is paired with a homologous target in duplex DNA (Figure 1B and (195,196)). This reaction is relevant to the rescue of stalled replication forks as it essentially resurrects the structure of the fork permitting PriA binding and ultimately resulting in replication restart (39,197). RecA also catalyzes a reaction known as DNA strand exchange, but this reaction is relevant for homologous recombination in vivo involving long DNA molecules such as F-factors, bacteriophage DNA and long stretches of the genome are involved (198,199). RecA has also been proposed to play more direct roles in the rescue of stalled forks. One model suggests its role is to bind to exposed ssDNA to protect it resulting in what is called stabilization, and a second role is in the actual process of fork regression (7,200). While initial studies demonstrated RecA could catalyze fork regression when SSB is added after RecA, later work demonstrated that this reaction is blocked by SSB when it is added to reactions before the recombinase (201,202). It has also been proposed that binding of RecA to torsionally constrained dsDNA that induces torsion in the DNA could be used to drive fork regression but this remains to be unambiguously demonstrated (203).
The active form of RecA is a right-handed, helical, nucleoprotein filament formed on ssDNA, with the protein in the high-affinity ssDNA-binding state induced by the binding of ATP, ATP-γ-S or ADP-AlF4− to the RecA-DNA complex (193,194,204–209). RecA protein has a natural propensity to form filaments that it does so in the presence of the nonhydrolyzable ATP analog ATP-γ-S in the absence of DNA and, in the presence of DNA and ADP (210–212). However, while these filaments are inactive, this information is included to remind the reader of this natural propensity of the protein to self-associate with a cooperativity parameter (ϖ) at least as high as 104, and this is critical for its function (213).
In vivo, filament formation can occur on ssDNA formed when (i) DNA damage produces single-strand gaps embedded within the genome such as those thought to occur in the vicinity of stalled forks; (ii) RecBCD processes dsDNA resulting from fork collapse into 3′-tailed duplexes in a chi-dependent manner; (iii) dsDNA from collapsed forks is processed by the combined actions of the RecFOR pathway enzymes; or (iv) when DNA polymerases stochastically uncouple during unperturbed DNA replication (214–217). In each scenario, the transient ssDNA regions formed are most likely to be rapidly bound by SSB (218,219). The initial binding of ssDNA by SSB coats the DNA and this removes secondary structure that impedes complete RecA nucleoprotein filament formation (220–223). In this context, SSB functions both as a mediator of filament assembly by the formation of an optimal ssDNA scaffold to which RecA can efficiently bind, as well as a natural competitor of RecA that necessitates the requirement for SSB dynamics on ssDNA in addition to facilitated loading by either RecBCD or the RecFOR proteins (214,221,224–227).
While dsDNA is abundant in the cell, it is not typically bound by free RecA with the nucleation step likely rate-limiting (211). However, under the right experimental conditions, RecA can be forced to bind dsDNA forming nucleoprotein filaments, with multiple studies providing insight into the behavior of RecA and filament function, as explained below.
To form a filament, RecA protein, in the presence of ATP, polymerizes onto either ss- or dsDNA in a highly cooperative fashion, and on ssDNA in the 5′-3′ direction (211,228–231). Nucleoprotein filaments whose length is defined by the length of DNA available, consists of RecA, ATP and DNA assembled with a 6:6:18 stoichiometry (RecA monomer:ATP:nt of ssDNA (or bp of dsDNA)) per filament turn and this is easily observed in recent crystal structures (228,232,233). The binding of RecA to DNA stretches the DNA molecule to ∼150% that of B-form DNA and, in the case of dsDNA, the helix is also unwound (64,230). This stretched form of DNA is comparable in length to overstretched and S-DNA, to which RecA binds 5-fold faster than B-DNA (203,234–238). Within filaments, the longitudinal stiffness of the DNA is also increased (205). Collectively these results suggest that filament formation on dsDNA relies on the coupling of DNA conformational fluctuations to both protein–DNA and protein–protein interactions. Further, the binding of RecA to DNA not only extends the nucleic acid but also stiffens it as well and this has important ramifications for DNA strand exchange where it facilitates base position in preparation for the homology search (232). The rigidity and stability of RecA nucleoprotein filaments may contribute significantly to the role of the protein in protecting exposed regions of ssDNA at forks. It is also critical for the binding of the LexA repressor, whose induced, auto-catalytic cleavage is required when the genome encounters significant damage and the SOS response must be induced (239,240).
Importantly, RecA nucleoprotein filaments are not static, terminal structures. Instead, they are dynamic structures controlled by (1) ATP binding, hydrolysis and product release which regulate filament formation, function and dissociation, and (2) protein mediators that facilitate both assembly and disassembly of filaments (see below (59,241,242)). As stated above, ATP binding to RecA-DNA complexes converts RecA into the high-affinity ssDNA-binding state. Here, the DNA in the filament center is stretched, RecA filaments are fully active for all biological function, are stable provided ADP does not accumulate, and are competent for ATP hydrolysis, with the minimum cluster size required for ATP hydrolysis being 10 RecA monomers bound contiguously on ssDNA (222,243). This minimum complex size required for the most basic activity of RecA must be taken into consideration when designing single-molecule substrates.
The hydrolysis of ATP and product release play important roles in various aspects of DNA strand exchange (194). They also play an important role in facilitating the formation of complete and contiguous nucleoprotein filaments (Figure 6A). This follows because RecA can nucleate filament formation at multiple locations on DNA (64). Since it is highly unlikely that these will be in register, ATP hydrolysis is used to redistribute RecA without complete disassembly of filaments (244–247). This re-establishes the filament register so that complete filaments can be formed. This makes sense because it is well known that ATP hydrolysis occurs throughout the length of a filament without resulting in net RecA dissociation, but once the ADP:ATP ratio reaches a critical level, filaments dissociate (243). The dynamic behavior of assembled filaments associated with ATP hydrolysis has been observed using different approaches (241,245,248,249).
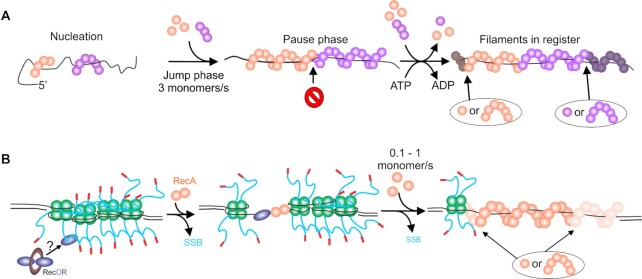
RecA nucleoprotein filament assembly is a dynamic, multi-step process. (A) Filament formation on ssDNA. Nucleation occurs at multiple random sites with filament growth occurring bidirectionally, but more rapidly in the 5′ to 3′ direction. The initial growth results in a jump phase where the ssDNA within the growing filaments is stretched. Once the orange filament growth is impeded by the purple filament which is out of register, a pause phase occurs. ATP hydrolysis coupled to RecA protomer dissociation re-establishes register. Additional RecA binding (monomers to hexamers) results in filament growth in the next jump phase. The darkened RecA subunits indicate the positions to which free RecA will bind. (B) Filament formation on SSB-coated ssDNA. Four SSB tetramers are bound to an ssDNA gap. Here, RecO initiates the process by binding to the linker domain of one SSB monomer. This leads to tetramer dissociation exposing ssDNA to which a RecA dimer can bind to nucleate filament formation. Filament growth occurs bidirectionally but faster in the 5′ to 3′ direction indicated by the lighter RecA subunits where free RecA will bind. The overall rate of filament growth on SSB-coated ssDNA is 10-fold slower as RecA takes advantage of SSB dynamics to overcome the SSB barrier, displacing tetramers. For additional details see the section on RecA, with details of SSB dynamics in the SSB section.
How does RecA form a nucleoprotein filament? Using dsDNA, it was shown that the protein nucleates filament formation in an ATP-dependent manner at multiple locations on the nucleic acid polymer, forming discrete clusters of 4–5 RecA monomers likely bound in juxtaposition within the cluster (64,248). The formation of nucleation sites can be enhanced by the application of force creating non-B-form conformations to which RecA binds with a greater affinity (203,235–238,250). Once nucleated, the extension phase where filaments grow in length, ensues at a rate of 2–20 monomers being added per second, preferentially to the 3′-end of the existing filament clusters, although growth does occur bidirectionally (64,203,248). ATP binding but not hydrolysis is required for both the nucleation and extension (growth) phases.
On naked ssDNA, the nucleation unit is a multimer involving 4–6 monomers of RecA which forms filaments rapidly at a rate of 3 ±1 monomer s−1 following a lag phase that is likely due to slow nucleation (Figure 6A and (59,241)). During the growth phase, jump-pause dynamics were observed and this was interpreted as filaments polymerizing into each other following nucleation at multiple sites. When ssDNA is coated by SSB, the lag phase is >10-fold longer, filament growth is up to 10 times slower (rate is 0.1–1 monomer/s), and occurs without jump-pause dynamics (Figure 6B and (59,66,93)). Filaments are nucleated as a dimer of RecA and grow bi-directionally but 1.6-fold faster in the 5′-3′ direction (93). Two studies found that filament growth occurs by adding one monomer at a time (93,251). In contrast, a separate study found that filament growth occurs with either 5 or 6 RecA molecules being added at a time (241).
The SSB inhibition of filament formation is consistent with the protein imposing a large energy barrier at both the nucleation and filament growth phases. Notably, when SSB is present, the jump-pause kinetics observed on naked ssDNA are absent, consistent with SSB removal being the rate-limiting step in the filament growth phase (59). Therefore, SSB tetramers must either dissociate, slide on the DNA or, RecA must take advantage of the stimulated unwrapping and dissociation behavior of SSB to first provide nucleation sites and then to permit filament polymerization (54,60,66,67,71). However, as SSB likely coats ssDNA-gaps completely in vivo, and there are limitations imposed on the intrinsic SSB mobility in ssDNA gaps by flanking duplex regions, the dynamic behavior of SSB alone is insufficient to facilitate RecA filament nucleation and polymerization. This requires the activity of the proteins of the RecF pathway, in particular RecF, O and R to enhance the ability of RecA to form filaments in gapped duplexes (Figure 6B and (225,227,252)). Single-molecule studies using ssDNA molecules with either free ends or large ssDNA gaps (8000 nt) revealed that RecO, and RecOR are the key players of the RecF pathway in RecA filament formation (60,96). The results showed that RecOR increased the rate of nucleation and this was further enhanced by the addition of RecF. RecOR also accelerated the extension phase RecA filament formation which was RecF-independent. It was also found that ssDNA binding by RecO displaces SSB from the DNA. Collectively, the mechanism of RecA filament formation on SSB-coated ssDNA, in the presence of mediators, requires two components. First, the mechanism of SSB displacement involves the binding of SSB to RecO via SSB linker domain/RecO OB-fold interactions and RecO binding directly to ssDNA via its OB-fold (82,94,95). Second, the dynamic behavior of SSB tetramers to expose short ssDNA regions is taken advantage of permitting RecO binding which results in SSB displacement (54,66,71). RecA will nucleate filaments in these ssDNA gaps vacated by SSB (54,60,93).
Once RecA nucleoprotein filaments have performed their function, they require disassembly. This could be accomplished by ATP hydrolysis alone (253,254). However, this may not occur rapidly enough as several DNA helicases have been shown to disrupt filaments far more rapidly. These include the PcrA and UvrD enzymes (255–258). Thus within a few minutes, complete filament disassembly is achieved and additional DNA processing enzymes can take over, fork rescue can be completed and DNA replication can resume.
The final reaction catalyzed by RecA that is relevant to stalled replication fork rescue is D-loop formation. This reaction consists of three experimentally distinguishable steps: (i) presynapsis, (ii) synapsis and (iii) DNA heteroduplex extension or branch migration, which if it does occur at all, is limited in D-loop formation, in contrast to typical DNA strand exchange reactions (193,194). Presynapsis is where RecA protein assembles onto the ssDNA to form the nucleoprotein species that is active in the homology search. Synapsis is characterized by initially random nonhomologous contacts occurring between the presynaptic complex and naked dsDNA, the search for homology, homologous pairing, and finally the conversion from paranemic to plectonemic joint molecules.
Typically, the ability of RecA to catalyze D-loop formation has been assayed with a short ssDNA and a negatively supercoiled DNA plasmid in bulk-phase assays (195). To visualize this reaction at the single-molecule level, linear duplex targets are constrained between magnetic beads and glass surfaces in magnetic tweezer instruments (259,260). Results show that multiple RecA filaments invaded the target at multiple sites, generated torsion in the target, forming a synaptic complex 79 ± 6 bp in length, located the site of homology, and exchanged 79 ± 6 bp of DNA with the displaced strand wrapped around the nascent heteroduplex DNA. The resulting exchange complex was characteristic of single-molecule D-loops (261). ATP hydrolysis occurred throughout the reaction, providing the ability of RecA filaments to dissociate and redistribute to bypass the accumulation of topological stress as proposed (194). Recent single-molecule studies of synapsis and the homology search revealed that RecA uses ‘intersegmental contact sampling’ to locate the site of homology in 30–120 s (262).
SUMMARY AND OUTLOOK
The rescue of stalled DNA replication forks leading to the resumption of DNA replication is essential to cell survival (8). Here, we have discussed the existing single-molecule insight into the proteins and macromolecular machines responsible for fork rescue using two distinct pathways. While the majority of studies have utilized reductionist biochemistry by focusing on each protein or enzyme working alone, recent studies have begun to appear where multiple components of repair pathways are reconstituted on single molecules of DNA. It is anticipated that as more knowledge is gained of the individual players, more reconstituted pathway studies will appear producing more complete pictures of the range of events that transpire to rescue stalled forks in vivo. In the absence of these studies, the current knowledge gained using single-molecule techniques can be assembled to provide insight into how rapidly forks can be rescued.
The current knowledge gained indicates that once forks stall, the repair pathways act rapidly to resurrect forks. For fork regression, it is unclear how rapidly RecG associates with SSB at forks, but it is <5 min (263). Once bound to the fork, RecG-catalyzed regression takes 2 s in the absence of SSB. However, when SSB is present it takes 7 s: 2.3 s to load and 4.4 s to regress the fork 480 bp. When more tightly bound obstacles are present, the loading step is delayed to 32 s but regression is unaffected, so complete regression could require as long as 37 s. If RuvAB binds to the extruded HJ, branch migration would proceed at 49 bp/s to the first pause some 1500 bp away (to permit RuvC binding) would take an additional 30 s (135–137). Therefore, regression, coupled to branch migration to move the fork away from the site of DNA damage can occur as rapidly as 67 s. Once RuvC binds to either regressed or branch migrated forks where it protects ∼20 bp, it will locate and cleave at its consensus sequence very rapidly (<1–2 s) as these occur every 25 bp in the genome (see RuvC section). In contrast, if the regression repair pathway consists of RecG followed by RuvC, the formation of a PriA substrate takes <1 min. If the sequential actions of RecG, RuvAB and RuvC are required, PriA substrate formation will require a little over one minute. These are likely best-case scenarios as the binding of other proteins to the DNA likely will limit the overall reaction rate.
In contrast, if the structure of the fork collapses and a dsDNA end is exposed, RecBCD will bind to the end and proceed to translocate, unwind and degrade the DNA at 1000 bp s. Within 5 s, it will be modified by an appropriately oriented χ-site and initiate loading of RecA which can locate homologous targets in 30–120 s. Once the D-loop has formed, PriA will bind to the 3′-end of the invading ssDNA and direct reloading of DnaB on to the template lagging strand. Therefore, for this pathway, the best-case scenario is that restart can initiate as quickly as 35 s but is likely to be slower due to the presence of other binding proteins on the DNA impeding the overall reaction progress. Collectively, the current knowledge base indicates that fork restart catalyzed by two independent pathways, dictated by the nature of the stalled fork structure, is a rapid process, consistent with the requirement for the repair of forks and resumption of DNA replication being essential to the viability of the cell (8).
FUNDING
Work in the Bianco laboratory is supported by National Institutes of Health Grant [GM100156 to PRB].
Conflict of interest statement. None declared.
Notes
Present address: Yue Lu, Department of Physics, Emory University, Atlanta, GA, 30322-2430, USA.
REFERENCES
1.
2.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
250.
251.
252.
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.