 DOCK7 protects against replication stress by promoting RPA stability on chromatin
DOCK7 protects against replication stress by promoting RPA stability on chromatin
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

- Altmetric
RPA is a critical factor for DNA replication and replication stress response. Surprisingly, we found that chromatin RPA stability is tightly regulated. We report that the GDP/GTP exchange factor DOCK7 acts as a critical replication stress regulator to promote RPA stability on chromatin. DOCK7 is phosphorylated by ATR and then recruited by MDC1 to the chromatin and replication fork during replication stress. DOCK7-mediated Rac1/Cdc42 activation leads to the activation of PAK1, which subsequently phosphorylates RPA1 at S135 and T180 to stabilize chromatin-loaded RPA1 and ensure proper replication stress response. Moreover, DOCK7 is overexpressed in ovarian cancer and depleting DOCK7 sensitizes cancer cells to camptothecin. Taken together, our results highlight a novel role for DOCK7 in regulation of the replication stress response and highlight potential therapeutic targets to overcome chemoresistance in cancer.
INTRODUCTION
DNA replication is a tightly regulated process to ensure accurate duplication and segregation of DNA to daughter cells once per cell cycle. It is particularly vulnerable to various intrinsic and extrinsic DNA damages resulting in replication stress (1,2). Replication stress causes slowed or stalled replication fork progression, and if not properly resolved, fragile replication forks are prone to collapse, which can threaten genome stability (1–3). After replication stress, cell cycle progression is stopped or slowed until replication obstacles are removed or DNA synthesis is restarted to avoid uncontrolled initiation or fork collapse (4,5). Timely and efficient resolution of replication stress is therefore fundamental to counteract these challenges and guarantee genome integrity (4,5).
Many proteins are reported to be involved in replication machineries to prevent excessive nucleolytic degradation of nascent DNA strands and repair replication fork obstacles (6,7). Among them, the replication protein A (RPA) complex, which consists of three subunits, RPA1 (RPA70), RPA2 (RPA32) and RPA3 (RPA14), is essential to protect ssDNA at replication forks and plays a significant role in cancer suppression (8,9). The RPA complex can recruit DNA polymerases α, δ and ϵ for the initiation and elongation steps of DNA replication (10), and ATR kinase via its partner protein ATRIP during DNA replication or replication stress (8,9). The checkpoint kinase, CHK1, acts downstream of ATR to phosphorylate various effectors to induce the DNA damage checkpoint, stabilize stalled forks, suppress new origin firing and repair collapsed forks (11). However, due to the complexity of the RPA interaction and regulation network in different DNA damage conditions, the molecular mechanisms that orchestrate RPA to precisely participate in replication stress response are not well known and need further investigation.
DOCK7, a member of the DOCK180 family, which consists of eleven guanine nucleotide exchange factors (GEFs), regulates the activation of the small GTP-binding proteins Rac1 and Cdc42 by exchanging bound GDP for free GTP (12–16). DOCK7 contains a catalytic DOCK homology region (DHR)-2 and a DHR1 domain that binds phospholipids in the plasma membrane (15). DOCK7 is expressed in all major regions of the human brain during early stages of development and regulates axon development and neuronal polarization (14,16). DOCK7 also acts as an intracellular substrate for ErbB2 to promote Schwann cell migration via its GEF activity (17). In addition, two rare heterozygous variants of DOCK7 were observed in unrelated patients with a phenotype of encephalopathy and eortical blindness (17), further highlighting the important role of DOCK7 in regulating neurogenesis. However, whether DOCK7 has unrecognized roles in addition to neuronal development is still unclear and deserves further investigation.
It has recently been shown that Rho GEFs and Rho GTPases such as Net1, Rgf1p, Rac1 and RhoA could be transported into the nucleus and interfere with the regulation of the DNA damage response and DNA replication (18–20). DOCK2, another DOCK180 family member, also participates in the DNA damage response and affects chemotherapeutic agent sensitivity through indirectly regulating key MMR and DDR factors mRNA levels (21). Previous large-scale proteomic analysis of proteins phosphorylated by ATM and ATR following DNA damage found that DOCK7 is a putative ATM/ATR substrate (22), implying that DOCK7 has a physiological role in regulating the DNA damage response. Herein, we identified DOCK7 as a novel replication stress regulator involved in stabilizing chromatin-loaded RPA1 to ensure proper replication fork restart. Our work uncovers a novel function of DOCK7 in replication stress response and implicates a signaling cascade-dependent mechanism modulating RPA1 phosphorylation and stability in chromatin.
MATERIALS AND METHODS
Cell culture
HEK293T, U2OS and OVCAR8 cell lines were purchased from ATCC. All cell lines have been tested and confirmed by the Mayo Clinic Medical Genome Facility. HEK293T and OVCAR8 cells were maintained in DMEM and U2OS cells were cultured with McCoy’s 5A with 10% FBS. All cell lines were kept in a humidified 37°C 5% CO2 incubator.
Plasmids, reagents and antibodies
FLAG-DOCK7 and FLAG-DOCK7ΔDHR2 were generously provided by Dr. Linda Van Aelst (Cold Spring Harbor Laboratory, NY). FLAG-RPA1 and Myc-RPA1 were generously provided by Dr. Jun Huang (Zhejiang University, Zhejiang, China). GST-PAK1 was generously provided by Dr. Guiling Wang (China Medical University, Liaoning, China). FLAG-Rac1 and Cdc42 were generated by subcloning the cDNA for each into the pCI2-FLAG mammalian expression vector and subsequent subcloning into the pLV3-FLAG lentiviral vector. FLAG-PAK1 was amplified by PCR and inserted into pLV3-FLAG for expression. FLAG-DOCK7 S1438A and V2022A, as well as FLAG-RPA1 S135A, T180A, T590A and ST/A mutants were generated by site-directed mutagenesis (Stratagene). The MDC1 and HA-ATRIP plasmids have been described previously (23). pCMV6M-Pak1 and pCMV6M-Pak1 L107F were purchased from Addgene.
MG132, cycloheximide (CHX), anti-FLAG agarose, anti-Myc agarose, 3xFLAG peptide, hydroxyurea (HU), camptothecin (CPT), 5-ethynyl-2′-deoxyuridine (Edu), cisplatin, biotin, chlorodeoxyuridine (Cidu), 5-iodo-2′-deoxyuridine (Idu) and inhibitors VX-970 (ATR inhibitor), NVS-PAK1–1(PAK1 ihhibitor), R-ketorolac (Rac1/Cdc42 inhibtor) and CK-666 (Arp2/3 Complex Inhibitor) were purchased from Sigma Aldrich. LY2603618 (CHK1 inhibitor) was purchased from MedChemExpress.
Anti-DOCK6 (amino acids 2026–2047; 1:1000) and anti-DOCK7 (amino acids 2110–2132; 1:1000) rabbit polyclonal antibodies were generated at Cocalico Biologicals Inc. (Reamstown, PA) using the indicated KLH-conjugates peptides; Anti-RPA1 was purchased (A300–241A, 1:5000) from Bethyl Laboratories. Anti-RPA32 (sc-56770, 1:2000), anti-Ub (sc-8017, 1:2000) and anti-HA (sc805, 1:2000) were purchased from Santa Cruz; anti-FLAG (F1804, 1:2000) was purchased from Sigma; anti-pS345 Chk1 (2348, 1:1000), anti-pT68 Chk2 (2661,1:1000), anti-phospho-PAK1 (Thr423) (2601, 1:1000), anti-phospho-PAK1 (Ser144) (2606, 1:1000), anti-ATR (2790, 1:1000), anti-Myc Tag (2276, 1:2000), anti-histone H3 (4499, 1:2000) and anti-SQ/TQ motif (9607, 1:1000) were purchased from CST; anti-GAPDH (60004–1-lg, 1:2000), anti-Cdc42 (10155–1-AP, 1:2000), anti-Rac1 (24072–1-AP, 1:1000), anti-RFWD3 (19893–1-AP, 1:1000), anti-MCM7 (11225–1-AP, 1:1000) and anti-PAK1 (21401–1-AP, 1:1000) were purchased from Proteintech Group.
RNA interference
The following shRNAs from Sigma were used in this study: DOCK7 shRNA-1: 5′-ACGTTCTCTAAAGACTATATT-3′, DOCK7 shRNA-2: 5′-ATGACTCAAAGG TACACTATA-3′; PAK1 shRNA-1: 5′-CTTCTCCCATTTCCTGATCTA-3′, PAK1 shRNA-2: 5′-CCAAGAAAGAGCTGATTATTA-3′; RPA1 shRNA: 5′-GCGGCTA CAAAGCGTTTCTTT-3′, Rac1 shRNA: 5′-GAAGATTATGACAGATTAC-3′; Cdc42 shRNA: 5′-TCTTCATTTGAAAACGTGA-3′. The MDC1 and ATR shRNAs have been described previously (23,24).
iPOND (isolation of proteins on nascent DNA)
iPOND assay was performed according to protocol. Briefly, HEK293T cells were labeled with 10 μM EdU for 20 min and then washed with PBS for three times. Cells were then treated with 4 mM HU for 2 h before fixed with 1% formaldehyde for 20 min at room temperature, which subsequently was quenched with 1.25 M glycine. Cells were then permeabilized with 0.25% Triton for 1 h before incubated with click reaction buffer (1 mM biotin azide, 100 mM CuSO4, 20 mg ml-1 sodium L-ascorbate in PBS) for 2 h. After click reaction, cell pellets were resuspended in lysis buffer (1% SDS in 50 mM Tris-HCl, pH 8.0) and subjected to sonication. Cell lysates were then incubated with streptavidin beads overnight at 4 °C before being washed twice with cold lysis buffer, once with 1 M NaCl, and twice with cold lysis buffer. Beads were incubated in 2 × Laemmli buffer and heated at 95 °C in for 10 min before loaded onto SDS-PAGE and immunoblotted with the indicated antibodies.
DNA fiber assay
Briefly, U2OS cells were labeled with 25 μM ldU for 30 min and then washed twice with media before treated with 4 mM HU for 4 h, washed with media again and then added with 200 μM CldU for another 30 min, cells were then washed twice and harvested. Cell pellets were resuspended in 10-fold volume of lysis buffer (200 mM Tris-HCl, pH 7.4, 50 mM EDTA and 0.5% SDS) for 10 min before slowly flow down along the slide which tilted at 15° to horizontal. The slides were fixed in 3:1 methanol/acetic acid and then air-dried overnight. The slides were then treated with 2.5 M HCl for 1 h before neutralized with 0.1 M Na2B4O7, pH 8.5, and rinsed three times in PBST (PBS with 0.1% Tween-20). The slides were blocked with 2% BSA and then incubated with anti-BrdU antibodies (BD Bioscience: 347580, Abcam: ab6326) overnight 4°C. After washing, the slides were incubated with secondary antibodies at room temperature for 1 before washed once with low-salt TBST (36 mM Tris–HCl pH8.0, 0.5 M NaCl, 0.5% Tween-20) and three times with PBST. Slides were mounted with anti-fade solution and visualized using a Nikon eclipse 80i Fluorescence microscope. All fiber lengths were measured using ImageJ.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed according to manufacturer's instruction. Briefly, ER-AsiSI U2OS cells transfected with indicated constructs were fixed by 1% formaldehyde for 10 min at room temperature and then quenched with glycine for 5 min at room temperature. Harvested cells were then digested by Micrococcal Nuclease for 20 min at 37°C to digest DNA to length of approximately 150–900 bp. Nuclear pellets were resuspended with 1x ChIP buffer and then sonicated before centrifugation. The supernatant was immunoprecipitated using 2 μg of FLAG antibody overnight at 4°C, and then added with ChIP-Grade Protein G Magnetic Beads for 2 h 4°C. Elution of chromatin from antibody/beads and then reversal of cross-links by adding NaCl (200 mM) and proteinase K (0.25 mg/ml) for >2 h at 65°C. DNA was thereafter purified for qPCR assay that has been described previously (25).
Western blot and immunoprecipitation
Harvested cells were lysed with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40 with 10 mM NaF, and 1 mg per ml each of pepstatin A and aprotinin for 30 min before centrifugation. Supernatant was immunoprecipitated by indicated agarose beads for 2 h at 4°C. the immunoprecipitates were washed with NETN for three times and then immunoprecipitates were added with 50 μl 1× Laemmli buffer subjected to SDS–PAGE separation. Immunoblotting was performed following standard procedures as previous described (26).
Chromatin fractionation
Chromatin fractionation was performed as described previously (27). In brief, cells were harvested and resuspended in low salt buffer (10 mM Tris-HCl, pH 7.4, 0.2 mM MgCl2, 50 0 mM b-glycerophosphate, 10 mM NaF and 1 mg/ml each of pepstatin A and aprotinin) containing 1% Triton X-100 on ice for 30 min. After centrifugation, the supernatant contained the soluble proteins were collected, and then the pellet contained the chromatin-bound proteins were resuspended in 0.2 N HCl on ice for 20 min before sonicated. After centrifugation, the supernatants were neutralized with 1 M Tris-HCl, pH 8.0. The amounts of each fraction were analyzed by western blotting analysis.
Colony formation assay
1000 U2OS or OVCAR8 cells stably expressing indicated constructs were plated into each well of six-well plates for 24 h before treated with indicated dosage of DNA damage agents for 10–14 days. Colonies were then stained with 5% GIEMSA and counted. Results were normalized to plating efficiencies.
Immunofluorescence
U2OS or OVCAR8 cells transfected with the indicated shRNAs or constructs were seeded on coverslips for 24 h before being treated with the indicated DNA damage agents. Cells were then fixed using 3% paraformaldehyde for 20 min at room temperature, washed three times in 1× PBS, and then permeabilized in 0.1% Triton-X solution for 10 min at room temperature. Cells were blocked with 5% goat serum for 30 min prior to incubate with primary antibodies at 4°C overnight. Subsequently, cells were washed three times using 1× PBS and incubated with Alexa Fluor 488- or Alexa Fluor 594-conjugated second primary antibodies for 1 h at room temperature. Cells were then stained with DAPI for 5 min to visualize nuclear DNA. The coverslips were mounted onto glass slides with anti-fade solution and visualized using a Nikon eclipse 80i fluorescence microscope. More than 200 cells were counted per experiment.
Tumor xenograft
Experiments were performed under the approval of the Institutional Animal Care and Use Committee at Mayo Clinic (Rochester, MN). Control or DOCK7-depleted OVCAR8 cells were injected subcutaneously into the flanks of 6-week-old female athymic nude Ncr nu/nu (National Cancer Institute/National Institutes of Health) mice using 18-gauge needles. Each mouse was injected a 100 μl mixture of 2 × 106 cells with 30% growth factor reduced Matrigel (BD Biosciences). Mice bearing tumors of about 100 mm3 were divided into groups by stratified randomization: control group (saline) and CPT group (10 mg kg−1). Mice were intraperitoneally injected three times per week. Tumor volume was measured every 7 days using calipers, and tumor volume was calculated using the formula length × width2. Mice were sacrificed for tumor dissection on day 28 after the start of treatment. Data were analyzed using Student’s t test.
Statistics
Data in bar and line graphs are presented as mean ± S.E.M of three independent experiments. Statistical analyses were performed with the Student’s t-test. Statistical significance is represented in figures by: * p<0.05; **p<0.01. ***p<0.001.
RESULTS
DOCK7 is involved in the DNA damage response
To test whether DOCK7 is involved in the DNA damage response, we examined the viability of DOCK7-depleted cells versus control cells in response to DNA damage agents including ionizing radiation (IR), cisplatin, hydroxyurea (HU) and camptothecin (CPT), and observed significantly less survival of DOCK7-depleted cells than control cells in response to these DNA damage agents (Figure 1A, C, E and Supplementary Figure S1A). IR and cisplatin first cause DNA double-strand breaks (DSB) and then single-stranded DNA (ssDNA), whereas HU and CPT directly induce replication stress and generate ssDNA. We next performed immunoblot analysis to determine the effect of DOCK7 on ATM-CHK2 and ATR-CHK1 signaling pathways, and found that the phosphorylation of CHK1 but not CHK2 was significantly attenuated in DOCK7-depleted U2OS and HCT116 cells after chemotherapeutic treatment (Figure 1B, D, F and Supplementary Figure S1B–D). On the other hand, DOCK6, which is an orthologous member of the DOCK-C family, which contains both DOCK7 and DOCK8, had no effect on the activation of either CHK1 or CHK2 in response to IR (Supplementary Figure S1E). We next examined the role of DOCK7 in the inhibition of replication stress induced by ATR or Chk1 inhibition. As shown in Supplementary Figure S1F and G, DOCK7 depletion was not able to further increase ATRi and Chk1i-mediated cytotoxicity, indicating that the effect of DOCK7 in replication stress is epistatic with the activation of ATR-Chk1 pathway. These results indicate that DOCK7 participates in the ATR-mediated Chk1 activation and initiation of the DNA damage response.

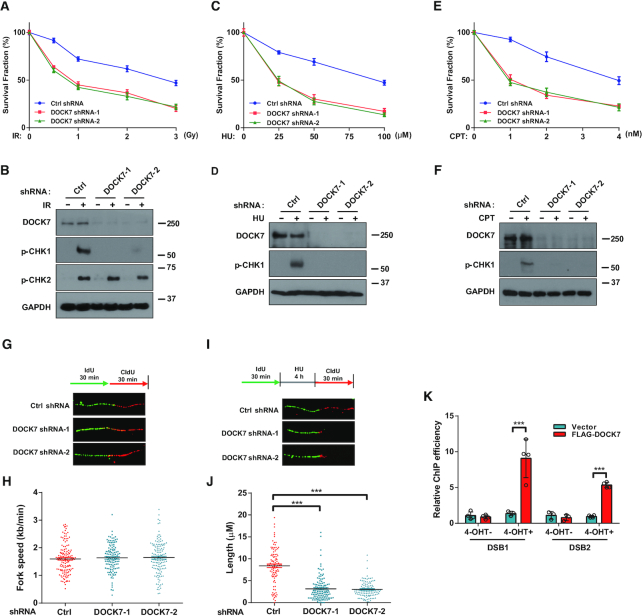
DOCK7 is required for replication stress response. (A, C, E) Survival assays of control and DOCK7-depleted U2OS cells treated with indicated doses of IR, HU or CPT. Data are represented as the mean ± SEM of n = 3 independent experiments. (B, D, F) Phosphorylation of CHK1 and CHK2 were determined by immunoblotting in control and DOCK7-depleted U2OS cells treated with 10 Gy IR, 10 mM HU or 1 μM CPT for 2 h. (G-J) U2OS cells were labeled with 50 μM IdU, and then treated with or without HU, thereafter incubated with 200 μM CldU for indicated time, the fork speed and the length of CIdU track in control and DOCK7-depleted cells were determined by measuring the length of CIdU track panels (H and J), representative pictures of fibers are shown in panels (G and I). The graphs represent mean ± S.D., two-tailed, unpaired t-test. ***p<0.001. (K) ER-AsiSI U2OS cells were transfected with vector control or FLAG-DOCK7 for 36 h before being treated with or without 1 μM 4-OHT for 4 h. After cells were harvested, ChIP experiments were performed using FLAG antibody. Error bars represent SEM from three independent experiments. ***p<0.001.
ATR is a master regulator of cellular responses to DNA replication stress (11); thus, we next determined whether DOCK7 is also required for regulation of DNA replication dynamics. To this end, we utilized the DNA fiber analysis to examine the effect of DOCK7 depletion on replication fork speed and the recovery of stalled forks. As shown in Figure 1G–J, DOCK7-depleted cells showed similar replication fork speed as control cells in the absence of replication stress; however, the recovery of stalled replication forks were decreased in DOCK7-depleted cells in the presence of HU, indicating that DOCK7 is important for replication fork recovery. This is consistent with a role of DOCK7 in ATR signaling. In addition, we found that DOCK7 was recruited to a site-specific DSB in ER-AsiSI U2OS cells as detected by ChIP analysis (25) (Figure 1K). Taken together, these results indicate that DOCK7 is important for DNA replication stress response.
DOCK7 is recruited to chromatin by MDC1 in response to replication stress
While DOCK7 is considered to be a cytoplasmic GEF, our data above suggests that DOCK7 is able to participate in the DNA damage response in the nucleus. Interestingly, we found that DOCK7 is chromatin-associated and its association with chromatin significantly increased as early as 30 min (Supplementary Figure S2A), indicating that DOCK7 is recruited to the chromatin to engage in the DNA damage response upon replication stress. In addition, we further investigated whether DOCK7 is recruited to nascent chromatin upon replication stress using the iPOND (isolation of proteins on nascent DNA) assay and immunofluorescence. As shown in Supplementary Figure S2B and C, the iPOND experiment showed that ectopic DOCK7 was able to be attached on replication forks, and the recruitment was augmented after HU treatment (B); in addition, the percentage of co-localization of EdU and FLAG-DOCK7 was also significantly increased upon replication stress (C), indicating that DOCK7 was recruited to nascent ssDNA to participate in the replication stress response.
We next explored the mechanism of DOCK7 recruitment to the chromatin. MDC1 is an upstream mediator protein, which binds and recruits multiple DNA repair proteins upon variety of DNA damage types including replication stress (28,29). In addition, Srivastava et al. used unbiased PCNA centered approaches and identified that MDC1 is also a potential component of replisome complexes by LC-MS/MS (30). To confirm whether MDC1 is directly involved in replication stress response, we performed the IPOND assay and identified that MDC1 could be recruited to the replication fork (Figure 2A). We found that the chromatin recruitment of DOCK7 was increased when MDC1 was overexpressed (Supplementary Figure S2D), while depletion of MDC1 effectively abolished HU-induced DOCK7 chromatin recruitment (Figure 2B), suggesting that MDC1 is indispensable for the recruitment of DOCK7 to the chromatin. We next investigated whether MDC1 affects the recruitment of DOCK7 to stalled replication forks. As shown in Supplementary Figure S2E, the accumulation of FLAG-DOCK7 at replication forks after HU treatment was significantly decreased when MDC1 was depleted, indicating that MDC1 is indispensable for the replication fork recruitment of DOCK7. In addition, co-immunoprecipitation (co-IP) experiments showed that MDC1 and DOCK7 interaction was significantly increased after HU treatment, and this interaction was dependent on the BRCT, but not the FHA domain of MDC1 (Figure 2C). Because the BRCT domain recognizes phosphor-Ser/Thr (31), this result implies that the interaction between MDC1 and DOCK7 is phosphorylation-dependent. ATR plays central roles in the regulation of replication fork stability and genomic stability maintenance through phosphorylating and activating numerous substrates which contain SQ or TQ motifs (11). We found that DOCK7 was phosphorylated at the SQ/TQ sites in an ATR-dependent manner (Figure 2D), indicating that DOCK7 is a substrate of ATR. In addition, knocking down ATR or inhibiting the activity of ATR could significantly attenuate HU-induced DOCK7 chromatin recruitment (Figure 2E and Supplementary Figure S2F).
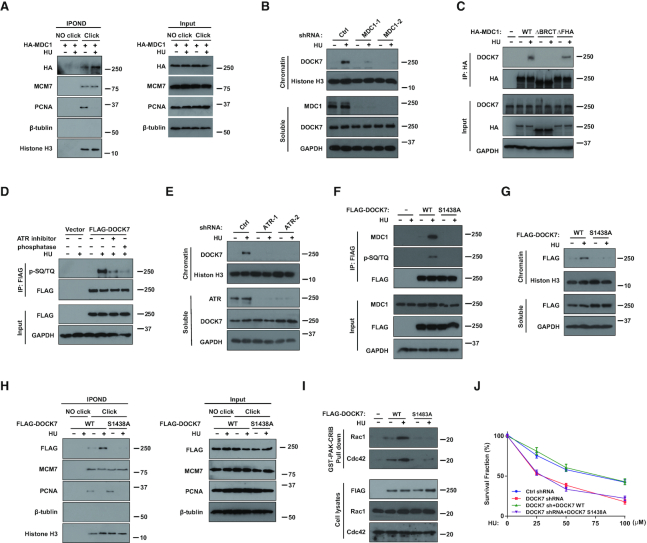
DOCK7 is phosphorylated by ATR and then recruited to chromatin by MDC1 to regulate replication stress response. (AandH) HEK293T cells stably transfected with HA-MDC1 (A), FLAG-DOCK7 WT or S1438A mutant (H) were incubated with 10 μM EdU for 20 min before treated or untreated with HU. Replication fork recruited proteins were isolated by iPOND and blotted with indicated antibodies. (B andE) Control and MDC1-depleted or ATR-depleted U2OS cells were treated with 10 mM HU for 1 h, cells were then harvested and separated into chromatin and soluble fractions, the protein level of DOCK7 in each fraction was detected by immunoblotting assay. (C) HEK293T cells were transfected with indicated MDC1 constructs for 24 h, co-immunoprecipitation (co-IP) assay were performed using anti-HA agarose beads and then blotted with indicated antibodies. (D) HEK293T cells transfected with FLAG-DOCK7 were pre-treated with DMSO or 50 nM VX-970 for 2 h then treated 10 mM HU for 2 h, cell lysates were immunoprecipitated with anti-FLAG agarose beads, and left untreated or were treated with phosphatase, cell lysates were blotted with the indicated antibodies. (F) HEK293T cells transfected with WT or S1438A mutant of DOCK7 were incubated with 10 mM HU for 1 h, cell lysates were then immunoprecipitated with anti-FLAG agarose beads and blotted with indicated antibodies. (G) The protein levels of FLAG-DOCK7 in chromatin and soluble fractions of HEK293T cells transfected with WT or S1438A mutant of FLAG-DOCK7 before or after HU treatment were detected by immunoblotting assay. (I) Lysates from HEK293T cells transfected with WT or S1438A mutant of DOCK7 with or without HU treatment were used in a PAK-CRIB pull-down assay. The immunoprecipitates were subjected to immunoblotting with the indicated antibodies. (J) The survival rate of control or DOCK7-depleted U2OS cells transfected vector control or indicated DOCK7 constructs were assessed by colony formation assay. Error bars represent SEM from three independent experiments.
Previous mass spectrometry analysis studies suggested that Ser-1438 is a potential SQ/TQ site of DOCK7 (22). Thus, we mutated this putative phosphorylation site and found WT but not the S1438A mutant of DOCK7 was phosphorylated under HU treatment (Figure 2F), suggesting that S1438 is a key ATR phosphorylation site of DOCK7. In addition, the interaction between DOCK7 and MDC1 in response to HU was also significantly decreased when Ser-1438 of DOCK7 was mutated (Figure 2F). Consistent with this observation, chromatin and replication fork recruitment of DOCK7 after HU treatment were also both attenuated when the Ser-1438 site of DOCK7 was mutated (Figure 2G and H). In addition, ChIP assays indicated that the recruitment of DOCK7 to DNA damage sites following 4-OHT treatment was significantly decreased when MDC1 or ATR was depleted (Supplementary Figure S2G), indicating that the phosphorylation of DOCK7 by ATR is indispensable for its interaction with MDC1 and recruitment to damage sites upon replication stress.
To explore whether the GEF activity of DOCK7 could be affected by its phosphorylation, we used GST-PAK1 affinity precipitation assays to measure the levels of GTP-loaded Rac1 and Cdc42 from cells expressing WT or the S1438A mutant of DOCK7 (32). As shown in Figure 2I, increased GTP-loaded Rac1 and Cdc42 were observed in cells overexpressing WT but not S1438A mutant of DOCK7 in response to HU, indicating that ATR-dependent phosphorylation of DOCK7 not only regulates its chromatin recruitment, but also promotes the GEF activity of DOCK7.
Next, we inquired whether the phosphorylation of DOCK7 affects its role in DNA damage repair. As shown in Supplementary Figure S2H, HU-induced CHK1 phosphorylation was rescued by the expression of WT but not the S1438A mutant of DOCK7 in DOCK7-depleted cells. In addition, reconstitution of DOCK7-deficient cells with WT but not S1438A mutant effectively reversed the HU hypersensitivity seen in DOCK7-depleted cells (Figure 2J). Combined, these data identify that the phosphorylation and chromatin recruitment of DOCK7 are important for its role in the replication stress response.
DOCK7 regulates the chromatin loading of RPA1 by inhibiting its protein degradation under replication stress
Since the RPA complex is an upstream factor in the ATR pathway and RPA1 exhibits the highest ssDNA-binding activity, we next tested whether DOCK7 modulates RPA1 location and function in response to replication stress. As shown in Figure 3A, the chromatin accumulation of RPA1 and RPA2 were both significantly decreased in DOCK7-depleted cells compared to control cells. In addition, HU treatment resulted in a dramatic increase of RPA foci in both total and EdU-positive cells, which were significantly attenuated when DOCK7 was depleted (Figure 3A and B; Supplementary Figure S3A and B), indicating that deficiency of DOCK7 impairs the chromatin loading of RPA1. We next performed the IPOND assay to examine the recruitment of RPA to the replication fork. As shown in Figure 3C, there was much less RPA1 and RPA2 accumulation on replication forks when DOCK7 was depleted. Significantly, DOCK7 depletion had no effect on the interaction between RPA1 and RPA2, or RPA1 and ATRIP (Supplementary Figure S3C and D). In addition, end resection at DSBs and cell cycle progression were not affected when DOCK7 was depleted (Supplementary Figure S3E and F). We next compared the ssDNA-binding affinity of WT RPA1 and RPA1 ST/A mutant by EMSA. As shown in Supplementary Figure S3G, we incubated increasing amounts of WT or ST/A mutant of RPA1 with Cy5-labeled 31-nt ssDNA and found there was no difference in the binding efficiency of WT and ST/A mutant of RPA1 with ssDNA, indicating that the ssDNA-binding affinity of RPA1 ST/A mutant is the same as that of WT RPA1. Therefore, we inferred that decreased RPA1 chromatin loading in DOCK7-depleted cells might be attributed to an increase in protein degradation rather than decreased affinity. Consistent with our hypothesis, we observed a significant increase of RPA1 protein levels in nuclear chromatin but not the soluble components when MG132 was used to blocked ubiquitin-mediated protein degradation (Figure 3E), indicating that chromatin-loaded RPA1 undergoes degradation, possibly to maintain RPA homeostasis under normal physiological states. We next measured the effect of DOCK7 on chromatin RPA1 protein stability to substantiate their connection. As expected, DOCK7 depletion significantly accelerated the degradation rate of chromatin-loaded RPA1, without affecting soluble RPA (Figure 3F). Moreover, the ubiquitination level of chromatin-loaded RPA1 was markedly decreased when treated with HU, and this phenomenon was effectively blocked in DOCK7-depleted cells (Figure 3G), indicating that DOCK7 regulates chromatin-loaded RPA1 protein accumulation through an ubiquitin–proteasome pathway.
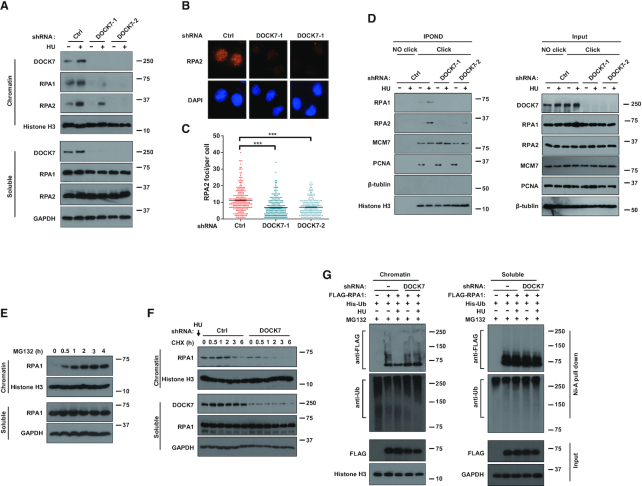
DOCK7 increases the protein stability of RPA1 in chromatin and replication fork. (A) The distribution of indicated proteins in the chromatin and soluble fractions of control or DOCK7-depleted U2OS cells after treated with 10 mM HU for 2 h were determined by immunoblotting assay. (B andC) Representative images (B) and quantification (C) of RPA2 foci. More than 200 cells were counted in each experiment. Error bars represent SEM from three independent experiments. ***p<0.001. (D) Control and DOCK7-depleted HEK293T cells were incubated with 10 μM EdU for 20 min before or after HU treatment. Replication fork recruited proteins were isolated by iPOND and blotted with indicated antibodies. (E) Chromatin and soluble fraction of cell lysates separated from MG132-treated HEK293T were blotted to measure the expression level of RPA1. (F) The protein contents of RPA1 in the chromatin and soluble fraction of control or DOCK7-depleted HEK293T cells treated with 20 μM CHX for different time points were detected by immunoblotting assay. (G) Control or DOCK7-depleted HEK293T cells were transfected with FLAG-RPA1 and His-Ub for 24 h before being treated with 10 mM HU for 1 h, the chromatin and soluble fractions of harvested cell lysates were then immunoprecipitated with nickel (His) beads and blots were probed with indicated antibodies.
DOCK7 regulates RPA1 chromatin loading and replication stress response by activating Rac1/Cdc42-PAK1 signaling pathway
The DHR2 domain of DOCK7 is required for its GEF activity toward Rac1 and Cdc42 (15). Therefore, we asked whether the DHR2 domain of DOCK7 also regulates RPA1 chromatin loading and replication stress response. To this end, DOCK7 GEF mutants in which the DHR2 domain was deleted (DOCK7ΔDHR2) or inactivated through a specific point mutation (V2022A) (33) were used to determine the role of DOCK7 GEF activity in the replication stress response. As shown in Figure 4A and Supplementary Figure S4A–C, expression of WT DOCK7 but not the DOCK7ΔDHR2 or V2022A mutants in DOCK7-depleted cells rescued RPA1 and RPA2 chromatin loading and CHK1 phosphorylation after HU treatment. In addition, the decrease in RPA2 foci, HU hypersensitivity and GTP-loading Rac1 and Cdc42 were all effectively rescued by WT DOCK7, but not DOCK7ΔDHR2 (Figure 4B–E), indicating that the GEF activity of DOCK7 is indispensable to its role in the replication stress response.
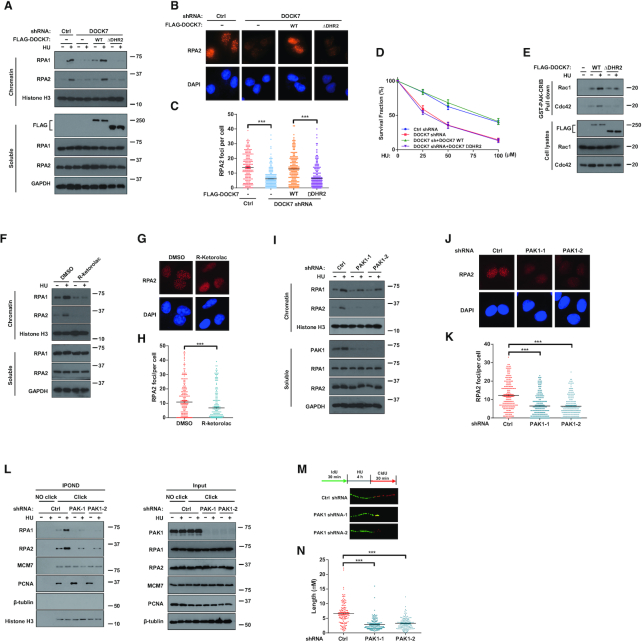
DOCK7-Rac1/Cdc42-PAK1 pathway is critical for replication stress response through regulating the chromatin recruitment of RPA1. (A-C) Control or DOCK7-depleted U2OS cells were transfected with vector control, WT DOCK7 or DOCK7ΔDHR2 truncates for 24 h before treated with 10 mM HU for 1 h. The expression levels of RPA1 and RPA2 in the chromatin and soluble fraction of harvested cells were determined by immunoblotting assay (A); RPA2 foci formation was detected by immunofluorescence (B) and quantified (C). More than 200 cells were counted in each experiment. Error bars represent SEM from three independent experiments. ***p<0.001. (D) Survival assays of control or DOCK7-depleted U2OS cells transfected with vector control, WT DOCK7 or DOCK7ΔDHR2 in response to HU. Error bars represent SEM from three independent experiments. (E) HEK293T cells were transfected with vector control, WT DOCK7 or DOCK7ΔDHR2 for 24 h before treatment with 10 mM HU for 2 h, cells lysates were then used in a GST-PAK-CRIB pull-down assay to detect the activation of Rac1 and Cdc42. (F and I) Chromatin and soluble fractions of cell lysates derived from DMSO or 1 μM R-ketorolac pretreatment (F) and control or PAK1 knockdown (I) U2OS cells were subjected to immunoblot with the indicated antibodies. (G, H, J andK) Representative images (G and J) and quantification (H and K) of RPA2 foci in DMSO or R-ketorolac pretreatment (G and H) and control or PAK1 knockdown (J and K) U2OS cells after HU treatment were detected and analyzed by immunofluorescence. (L) RPA1 and RPA2 proteins isolated by iPOND from control or PAK1 depletion HEK293T cells were detected by immunoblotting assay. (M andN) DNA fiber assay was performed to detect the length of CIdU track after HU was removed in control or PAK1-depleted U2OS cells (N), representative pictures of fibers are shown in (M). The graphs represent mean ± S.D., two-tailed, unpaired t-test. ***p<0.001.
To determine whether the effect of DOCK7 on replication stress regulation is mediated by Rac1 and Cdc42, we used the Rac1/Cdc42 inhibitor R-ketorolac (34) to confirm the role of Rac1 and Cdc42 in the replication stress response. As shown in Figure 4F–H, R-ketorolac pretreatment significantly inhibited HU-induced RPA1 and RPA2 chromatin loading and RPA2 focus formation. In addition, inhibition of Rac1/Cdc42 also caused a significant decrease of CHK1 phosphorylation (Supplementary Figure S4D). Importantly, depleting Rac1 and Cdc42 in DOCK7 knockdown cells did not further reduce RPA1 and RPA2 chromatin loading or CHK1 phosphorylation (Supplementary Figure S4E and F), indicating an epistatic relationship. Taken together, these results suggest that DOCK7 regulates the replication stress response through its GEF activity toward Rac1 and Cdc42.
PAK1 is a serine/threonine protein kinase that serves as an important mediator of Rac1 and Cdc42 GTPase function in cytoskeletal reorganization (35,36); thus, we hypothesized that PAK1 might also be required for DOCK7-associated replication stress regulation. As shown in Supplementary Figure S4G and H, the phosphorylation of PAK1 was significantly increased in response to HU and this phenomenon was attenuated when DOCK7 was depleted, whereas inhibiting the activity of the Arp2/3 complex by CK-666, another downstream effector of Rac1/Cdc42, had no observable effect on HU-induced CHK1 phosphorylation, indicating that PAK1 is the downstream effector of DOCK7-Rac1/Cdc42 pathway under replication stress. We further explored the biological role of PAK1 in replication stress and found that depletion of PAK1 resulted in decreased RPA1 and RPA2 chromatin loading and RPA2 foci formation (Figure 4I–K). In addition, RPA1 and RPA2 accumulation on replication forks were decreased while the stalled replication fork recovery was decreased when PAK1 was depleted (Figure 4L–N), indicating that PAK1 is important for the protection from HU-induced replication stress. Furthermore, PAK1 depletion reduced CHK1 phosphorylation, and sensitized cells to HU-induced colony formation inhibition (Supplementary Figure S4I and J), indicating that PAK1 contributes to the regulation of replication stress-induced DNA damage response. Moreover, PAK1 together with Rac1 and Cdc42 were recruited to DNA damage sites following DSB induction as determined by ChIP assay (Supplementary Figure S4L), indicating that the Rac1/Cdc42-PAK1 complex is a component of the DNA damage response. In addition, we performed iPOND to detect whether MDC1 affects the recruitment of the Rac1/Cdc42-PAK1 complex on stalled replication forks. As shown in Supplementary Figure S2E, the accumulation of Rac1, Cdc42 and PAK1 on replication forks was all significantly decreased upon MDC1 depletion, indicating that MDC1 is also necessary for the recruitment of Rac1/Cdc42-PAK1 complexes to stalled replication forks.
To confirm DOCK7 and PAK1 act in the same pathway required for proper replication stress response, we used a PAK1 inhibitor to block the activation of PAK1 and observed their combined effects. As shown in Supplementary Figure S4M and N, the PAK1 inhibitor effectively impaired HU-induced CHK1 phosphorylation, PAK1 autophosphorylation (37) and RPA2 foci formation. Importantly, PAK1 inhibition did not further impact RPA1 and RPA2 chromatin loading or CHK1 phosphorylation in DOCK7-depleted cells, suggesting that PAK1 is the critical downstream effector of the DOCK7 targets Rac1 and Cdc42.
Phosphorylation of RPA1 at residues S135 and T180 by PAK1 is critical for RPA1-mediated replication stress response
PAK1 phosphorylates a variety of substrates on their serine/threonine residues; thus, we used phospho-tag electrophoresis assay to examine whether RPA1 is the downstream target of PAK1 during replication stress. As shown in Figure 5A and B, a significant shift of FLAG-RPA1 was observed in cells treated with HU by phospho-tag gel analysis, and the phosphorylation pattern of RPA1 was nearly abolished when the activity of PAK1 or Rac1/Cdc42 was inhibited, or DOCK7 was depleted, indicating that DOCK7-Rac1/Cdc42-PAK1 signaling contributes to HU-induced RPA1 phosphorylation. Previous MALDI-TOF mass spectrometry data showed that RPA1 could be hyperphosphorylated in vitro within peptides containing amino acids 112–157 and 569–600 (38). Thus, we inferred that residues S135 and T590 were the potential phosphorylation sites of RPA1 by PAK1. As shown in Supplementary Figure S5A, we found that the T590A mutant of RPA1 was phosphorylated to the same extent as WT RPA1, whereas the S135A mutant partially abolished HU-induced RPA1 phosphorylation, indicating that other sites of RPA1 adjacent to S135 might also be phosphorylated. Consistent with our hypothesis, the T180A mutant was also partially attenuated HU-induced RPA1 phosphorylation, and the S135A/T180A double mutant nearly abolished the phosphorylation of RPA1 (Figure 5C), indicating that PAK1 phosphorylates RPA1 on S135 and T180. We further examined whether PAK1 is physically associated with RPA1 and is able to phosphorylate RPA1 directly. As shown in Supplementary Figure S5B, FLAG-RPA1 was effectively pulled down by purified GST-PAK1 but not GST protein, indicating that PAK1 directly interacts with PAK1. In addition, in vitro kinase assay results showed that 32P labeled ATP was readily incorporated into WT but not the ST/A mutant of RPA1 when co-incubated with activate PAK1 (Figure 5D), thus demonstrating that RPA1 is a direct substrate of PAK1.
![RPA1 phosphorylated by PAK1 at Ser-135 and Thr-180 is critical for its role in replication stress response. (A–C) Control or DOCK7-depleted HEK293T cells were transfected with indicated FLAG-RPA1 constructs for 24 h. Cells were then pretreated with or without inhibitors (R-ketorolac or PAK1 inhibitor) before or after HU treatment. FLAG-RPA1 was coimmunoprecipitated from cell lysates and loaded on both normal and Phospho-tag gel, thereafter blotted with indicated antibodies. (D) Purified WT and ST/A mutant of RPA1 were incubated with or without constitutively active PAK1 (50 aa-150 aa) and incubated with γ- [32P] ATP for 30 min at 30°C before subjected to autoradiography. (E–G) Control or RPA1-depleted U2OS cells were transfected with WT or ST/A mutant of FLAG-RPA1 for 24 h before treatment with 10 mM HU for 1 h, and then the distribution of RPA1 and RPA2 in the chromatin and soluble fractions of cells were determined by immunoblotting (E). Representative images (F) and quantification (G) of RPA2 foci were analyzed by immunofluorescence. More than 200 cells were counted in each experiment. Error bars represent SEM from three independent experiments. ***p<0.001. (H–J) RPA1-depleted cells were transfected with WT or ST/A mutant of FLAG-RPA1 for 24 h before HU treatment, and then IPOND assay in HEK293T cells was performed to detect the distribution of the WT or ST/A mutant of RPA1 on the replication fork (H). DNA fiber assay in U2OS cells was performed to detect the length of CIdU track after HU was removed (J), and the representative pictures are shown in panel (I). The graphs represent mean ± S.D., two-tailed, unpaired t-test. ***p<0.001. (K) Control or RPA1-depleted U2OS cells were transfected with vector control, WT or ST/A mutant of FLAG-RPA1 for 24 h, treated with the indicated concentrations of HU and survival was measured using the colony formation assay. Error bars represent SEM from three independent experiments. (L) The protein stability of WT or ST/A mutant of RPA1 in the chromatin and soluble fraction of RPA1-depleted HEK293T cells under CHX treatment were analyzed by immunoblotting assay. (M) RPA1-depleted HEK293T cells were transfected with WT or ST/A mutant of FLAG-RPA1 and His-Ub for 24 h before HU treatment. Chromatin and soluble fractions derived from harvested cells were immunoprecipitated with nickel (His) beads and then blotted with indicated antibodies](/dataresources/secured/content-1766009657840-f5875c94-99e8-47bd-b865-fd23317547b2/assets/gkab134fig5.jpg)
RPA1 phosphorylated by PAK1 at Ser-135 and Thr-180 is critical for its role in replication stress response. (A–C) Control or DOCK7-depleted HEK293T cells were transfected with indicated FLAG-RPA1 constructs for 24 h. Cells were then pretreated with or without inhibitors (R-ketorolac or PAK1 inhibitor) before or after HU treatment. FLAG-RPA1 was coimmunoprecipitated from cell lysates and loaded on both normal and Phospho-tag gel, thereafter blotted with indicated antibodies. (D) Purified WT and ST/A mutant of RPA1 were incubated with or without constitutively active PAK1 (50 aa-150 aa) and incubated with γ- [32P] ATP for 30 min at 30°C before subjected to autoradiography. (E–G) Control or RPA1-depleted U2OS cells were transfected with WT or ST/A mutant of FLAG-RPA1 for 24 h before treatment with 10 mM HU for 1 h, and then the distribution of RPA1 and RPA2 in the chromatin and soluble fractions of cells were determined by immunoblotting (E). Representative images (F) and quantification (G) of RPA2 foci were analyzed by immunofluorescence. More than 200 cells were counted in each experiment. Error bars represent SEM from three independent experiments. ***p<0.001. (H–J) RPA1-depleted cells were transfected with WT or ST/A mutant of FLAG-RPA1 for 24 h before HU treatment, and then IPOND assay in HEK293T cells was performed to detect the distribution of the WT or ST/A mutant of RPA1 on the replication fork (H). DNA fiber assay in U2OS cells was performed to detect the length of CIdU track after HU was removed (J), and the representative pictures are shown in panel (I). The graphs represent mean ± S.D., two-tailed, unpaired t-test. ***p<0.001. (K) Control or RPA1-depleted U2OS cells were transfected with vector control, WT or ST/A mutant of FLAG-RPA1 for 24 h, treated with the indicated concentrations of HU and survival was measured using the colony formation assay. Error bars represent SEM from three independent experiments. (L) The protein stability of WT or ST/A mutant of RPA1 in the chromatin and soluble fraction of RPA1-depleted HEK293T cells under CHX treatment were analyzed by immunoblotting assay. (M) RPA1-depleted HEK293T cells were transfected with WT or ST/A mutant of FLAG-RPA1 and His-Ub for 24 h before HU treatment. Chromatin and soluble fractions derived from harvested cells were immunoprecipitated with nickel (His) beads and then blotted with indicated antibodies
To further characterize the biological impact of RPA1 phosphorylation, we first examined whether phosphorylation of RPA1 affected its chromatin accumulation. As shown in Figure 5E and Supplementary Figure S5C, HU treatment resulted in overt increases of chromatin accumulation of WT RPA1 in RPA1-depleted cells, whereas only a slight increase was observed with RPA1 ST/A mutant. In addition, re-expression of WT RPA1, but not the ST/A mutant in RPA1-depleted cells rescued RPA2 foci formation (Figure 5F and G). Moreover, RPA1 and RPA2 accumulation on nascent ssDNA and replication fork restart was reduced in ST/A mutant expressing cells (Figure 5H and J), indicating that HU-induced RPA1 phosphorylation promotes the accumulation of RPA1 protein on nascent ssDNA and promotes replication fork recovery. Furthermore, as shown in Figure 5K and Supplementary Figure S5D, reduced CHK1 phosphorylation and increased HU hypersensitivity caused by RPA1 depletion could be effectively reversed by WT but not the ST/A mutant of RPA1, indicating that RPA1 phosphorylation is critical for protecting from replication stress-induced DNA damage. To further examine whether the phosphorylation of RPA1 affects its protein stability in chromatin, we compared the degradation rates and the ubiquitination of chromatin RPA1 in WT and ST/A mutant of RPA1 overexpressed cells. As shown in Figure 5L and M, chromatin ST/A mutant of RPA1 was more unstable than WT RPA1, and continued to undergo ubiquitin-mediated protein degradation when treated with HU, indicating that RPA1 phosphorylation promotes chromatin RPA1 protein stability through attenuating its ubiquitination in response to replication stress.
Because several previous studies showed that the E3 ubiquitin ligase RFWD3 is able to ubiquitinate RPA and facilitate its degradation upon replication stress (39–41), we examined whether the interaction between RPA1 and RFWD3 was affected by the DOCK7/PAK1 signaling axis. As shown in Supplementary Figure S5E and F, the interaction between RPA1 and RFWD3 was not affected when DOCK7 or PAK1 was depleted. In addition, there was no difference between the interaction of RFWD3 with WT RPA1 or the ST/A mutant, indicating that PAK1-mediated RPA1 phosphorylation doesn’t affect its interaction with RFWD3.
DOCK7 depletion enhances chemotherapy of ovarian cancer cells by affecting RPA1 phosphorylation
DOCK7 pathway is critical for replication stress regulation and targeting replication stress is an important therapeutic strategy in ovarian cancer; we, therefore, evaluated the therapeutic potential of targeting the DOCK7 pathway in ovarian cancer. As shown in Supplementary Figure S6A, the total alteration frequency of DOCK7 was about 11% (detected in 22 of 201 samples) for ovarian cancer patients in TCGA from cBio Cancer Genomics Portal [PMID: 22588877]. Most of them were mRNA high (15 samples), followed by amplification (5 samples) and deep deletion (1 sample). These data suggested that DOCK7 expression is upregulated in patients with ovarian cancer. In addition, the DRUGSURV tool [PMID: 24503543] was used to evaluate the effects of DOCK7 expression on clinical outcome in ovarian cancer patients from the GSE13876 [PMID: 19192944]. As shown in Supplementary Figure S6B, the upregulation of DOCK7 expression was significantly related to shorter overall survival (P = 0.0003), indicating that DOCK7 is a marker and a potential target for ovarian cancer therapy. Thus, we next used the ovarian cancer cell line OVCAR8 to investigate the role of DOCK7 upon replication stress. Consistent with our results in other cell lines, DOCK7 depletion significantly reduced CPT-induced RPA2 foci formation in OVCAR8 cells (Supplementary Figure S6C and D).
We next examined the role of DOCK7 in the chemotherapeutic response in ovarian cancer. As shown in Figure 6A, compared with WT ovarian cancer cells, cells with DOCK7 depletion were more sensitive to CPT treatment. We next explored the role of DOCK7 in response to chemotherapy in vivo. As shown in Figure 6B–D, DOCK7 knockdown in OVCAR8 cells did not affect cell growth, but conferred hypersensitivity to CPT treatment in a Xenograft model. Taken together, our results suggested that the DOCK7 pathway may be a potential therapeutic target for sensitizing ovarian cancer cells to chemotherapy.
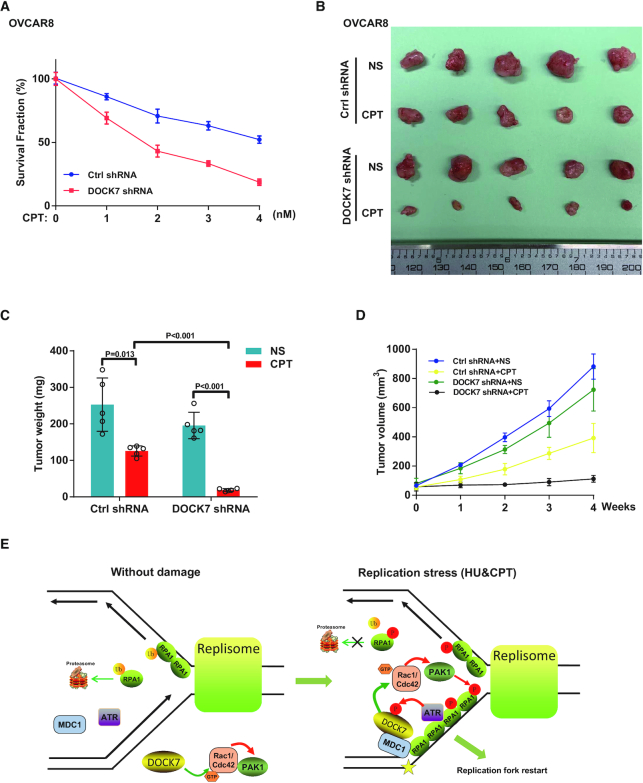
DOCK7 depletion enhances chemotherapy of ovarian cancer cells in vitro and in vivo. (A) Cell survival rate after CPT treatment was determined by colony formation assay in control or DOCK7-depleted OVCAR8 cells. Error bars represent SEM from three independent experiments. (B–D) Control or DOCK7-depleted OVCAR8 cells were subcutaneously injected into the flank of NOD-SCID mice. Mice were treated with saline or CPT (10 mg/kg i.p. 3 days × 8 times). Tumor images were shown in (B), and tumor weight (C) and volume (D) were assessed. Data points (mean ± SEM) are shown from five biologically independent samples by two-sided unpaired t test. (E) Working model of RPA1 phosphorylation regulated by DOCK7 signaling. DOCK7 is phosphorylated by ATR during replication stress and then recruited to the chromatin and DNA damage sites by MDC1. Thereafter, DOCK7 facilitates the GTP-loading of Rac1/Cdc42, which in turn activate PAK1 to phosphorylate and stabilize chromatin-loaded RPA1 to stabilize and enable replication fork restart.
DISCUSSION
The DNA replication machinery ensures the timely and precise completion of genome duplication and safeguards genome integrity by successfully removing replication obstacles caused by exogenous and endogenous replication stress (3,7). The replication stress response pathway consists of a network of DNA repair and checkpoint proteins that are required for efficiently monitoring and resolving replication stress-induced damage (1,42). Here, we identified DOCK7 as a key regulator of replication fork restart in response to replication stress through Rac1/Cdc42-PAK1 signaling-dependent RPA1 phosphorylation and protein stability. This study elucidates a novel player and related mechanism governing the response to replication stress and increases the understanding of how replication stress signaling is dynamically regulated.
Previous studies showed that DOCK7 functions as a cytoplasmic activator to promote axon formation and Schwann cell migration mainly via its downstream targets Rac1/Cdc42 (15,16). Recent evidence suggests that Rac1 and Cdc42 also translocate to the nucleus to play additional roles including DNA damage response (43,44). However, whether subcellular compartmentalization also plays a critical role in regulating DOCK7 signaling and biological outcome is still unknown. Herein, we found that DOCK7 is phosphorylated by ATR and then recruited to the chromatin and DNA damage sites by MDC1 to participate in the replication stress response. ATR is one of the central replication stress response kinases that phosphorylates hundreds of substrates containing SQ/TQ motifs to help cells survive and complete DNA replication under replication stress (11). DOCK7 has a conserved SQ/TQ site at ser-1438 that could be phosphorylated by ATR, which is in turn recognized and recruited by MDC1, an important modular phosphoprotein scaffold in DNA damage response, to chromatin and DNA damage sites. Mutation of this site abolished DOCK7 chromatin accumulation thereby confirming that DOCK7 is a novel substrate of ATR and binding partner of MDC1 to protect against replication stress. In addition, the GEF activity of DOCK7 toward Rac1/Cdc42 was also enhanced by replication stress in WT but not S1438A mutant expressing cells, indicating that the GEF activity of DOCK7 was regulated by ATR phosphorylation to increase the local accumulation of GTP-loaded Rac1 and Cdc42 to quickly deal with replication stress-induced DNA lesions. Whether ATR-phosphorylation releases some DOCK7 autoinhibitory mechanism remains to be determined.
The RPA complex is the central eukaryotic single-strand DNA-binding protein complex that participates in DNA replication and DNA repair (9,45). Among the members of this complex, RPA1 contains four DNA-binding domains (DBDs) and has highest affinity for ssDNA, which protects exposed ssDNA stretches (9). RPA1 also has the ability to specifically interact with numerous DNA repair proteins to facilitate diverse DNA metabolic pathways (8,45). In this article, we show that DOCK7 promotes the chromatin and replication fork accumulation of RPA1 under replication stress. It was reported that several proteins such as SLFN11, G9a and Cdc45 directly associated with RPA1 and affected its ssDNA-binding capacity and chromatin loading (46–48), and PTEN interacts with RPA1 to promote the replication fork recruitment of RPA1 by recruiting OTUB1 to deubiquitinate RPA1 (49). Our results showed that DOCK7 activation of a Rac1/Cdc42-PAK1 signaling node is indispensable for RPA1 phosphorylation and replication fork accumulation under replication stress. In fact, we show that DOCK7 signaling is important for the homeostasis of the chromatin-loaded RPA complex by countering RPA1 ubiquitination and degradation. In addition, RPA complex-coated ssDNA serves as a key platform for ATR recruitment and activation through the interaction with ATRIP and TopBP1 (11). Thus, a positive feedback loop is in place in which ATR-dependent DOCK7 phosphorylation increases chromatin-loaded RPA complexes, which results in the further recruitment and activation of ATR to protect cells against replication stress and maintain genomic stability.
Our results show that DOCK7 depletion or ectopic expression could affect endogenous RPA chromatin loading but with no change in fork dynamics under nondamaging conditions. The reason maybe that endogenous replication stress sources such as cell cycle progression, replication–transcription conflicts and ribonucleotides mis-incorporation consistently exist and cause the obstacles in replication fork progression (42,50). RPA is one of the first responders to coordinate DNA replication and therefore a considerable amount of RPA coats the exposed ssDNA even under nondamaged conditions (9,50). The dynamic RPA1 chromatin loading and subsequently recruited DNA repair proteins are tightly and accurately regulated to orchestrate proper replication stress response and maintain genomic stability during cell growth and division (9). Our results show that the GEF activity of DOCK7 regulates the phosphorylation of RPA1 on the replication fork through the activation of a Rac1/Cdc42-PAK1 pathway. Therefore, it is possible that under nondamaging conditions, DOCK7 depletion, Rac1/Cdc42-PAK1 pathway inhibition, or inhibition of PAK1 kinase activity, contributes to altered RPA phosphorylation level and its chromatin accumulation. However, besides RPA, replication fork dynamics are spatio-temporally regulated by a complex DNA repair network (51). Therefore, under nondamaging conditions, DOCK7-Rac1/Cdc42-PAK1 pathway is insufficient to alter fork dynamics without the coordination of other activated replication stress regulators.
In recent years, emerging evidence shows that post-translational modifications of the RPA complex are critical for its biological functions. For example, the SUMOylation of RPA1 increases its interaction with RAD51 and enhances DNA repair through HR (52). The acetylation of RPA1 by PCAF/GCN5 is critical for UV-induced nucleotide excision repair; while the lysine crotonylation (Kcr) of RPA1 regulated by CDYL promotes the interaction between RPA1 and ssDNA in response to CPT treatment (53). The ubiquitination of RPA2 by PRP19 promotes the recruitment and activation of ATR-ATRIP to DNA damage sites (54). The most studied modification is phosphorylation. RPA2 is phosphorylated by CDK2, ATM, ATR and DNA-PK at different serine/threonine residues sites, which are implicated in DNA repair and checkpoint response by regulating its association with the replication machinery and other proteins (55). Previous mass spectrometry results also suggested that RPA1 is phosphorylated in response to DNA damage (38), although the functional significance was not clear. In this article, we found that Ser-135 and Thr-180 of RPA1 are directly phosphorylated by PAK1 in a DOCK7-Rac1/Cdc42-dependent manner. Mutation of these two sites attenuates chromatin and replication fork-loaded RPA1 stability, and inhibits the ability of PRA1 in mediating replication fork restart under replication stress. PAK1 is one of the best characterized downstream effectors of Rac1/Cdc42 and has been implicated in the DNA damage response through regulating MORC2 phosphorylation as well as regulating the expression of DNA-repair related genes (56,57). It is still unclear how phosphorylation of RPA1 decreases its ubiquitination. Previous data showed that RPA1 and RPA2 could be ubiquitinated by the E3 ligase RFWD3, and thereafter undergo degradation to facilitate timely removal of RPA and progression of late-phase HR in replication stress response or DNA interstrand crosslink repair (39,41). Our data showed that the ubiquitination of chromatin RPA1 was decreased upon treatment with HU (1h), which helps to maintain sufficient RPA1 on chromatin and protect exposed ssDNA. It is possible that the ubiquitination of RPA1 plays a critical role in regulating the dynamic chromatin recruitment of RPA complexes at different stages. When replication stress occurs, more ssDNA is generated and cells need more RPA protein recruitment on the chromatin to protect the integrity of replication fork, at this time, DOCK7-associated pathway inhibits the ubiquitination of RPA1 in a phosphorylation-dependent manner; whereas during resolution of the stalled replication forks, RPA complexes need to be ubiquitinated and removed to allow HR progression. In addition, our results showed that RPA1 phosphorylation didn’t affect the interaction between RPA1 and RFWD3. Therefore, it is possible that phosphorylated RPA1 recruits other ubiquitination regulators such as deubiquitinases to remove ubiquitin from chromatin RPA1 at the early stage of replication stress response, which coordinately with RFWD3 to dynamically regulate the ubiquitination pattern of RPA1, finally promoting fork restart after replication stress. Future studies, aimed at identifying the unknown regulators and their mechanisms of action will illustrate the complicated and dynamic ubiquitination process of RPA1 during the replication stress response.
RPA1, RPA2 and RPA3 exist as a very stable heterotrimeric complex to regulate DNA replication and repair. Though RPA1 knockdown doesn’t affect the protein level of RPA2 (58), the association of RPA2 and RPA3 with DNA and RPA2 foci is significantly diminished after RPA1 knockdown (59). Structurally, there are four DNA-binding domains (DBD) located on RPA1 and only one on RPA2, which affect ssDNA binding affinity. The DBDs of RPA1 are indispensable for initial RPA-ssDNA complex formation in the low-affinity binding mode and sequentially RPA1 and RPA2 bind ssDNA in the high-affinity binding mode (9). Our results showed that RPA1 phosphorylation upon replication stress decreases the ubiquitination of chromatin-loaded RPA1, leading to an accumulation of RPA1 on stalled replication forks. This helps the DBD domain of RPA2 binding with RPA1-coated ssDNA, thereafter contributing to increased RPA2 binding stability
Finally, we found that DOCK7 is highly expressed in a cohort of patients with ovarian cancer and the expression level of DOCK7 is significantly correlated with shorter overall survival. In addition, DOCK7 depletion sensitized CPT-induced tumor growth inhibition in vivo, indicating that DOCK7 signaling pathways are a novel target for ovarian cancer therapy used by replication stress-inducing agents, and combination treatment with PAK1 or Rac1/Cdc42 inhibitors might be a promising strategy to overcome chemoresistance in ovarian cancer.
In conclusion, our findings reveal a novel mechanism linking DOCK7 signaling and replication stress response. We identify the role and regulatory mechanism of RPA1 phosphorylation in controlling RPA stability in chromatin, which is important for DNA replication fork restart. Moreover, our study suggests that targeting this pathway could be a promising strategy to enhance the efficacy of DNA damaging agents.
DATA AVAILABILITY
All correspondence and material requests should be addressed to M.D., D.D.B. and Z.L.
ACKNOWLEDGEMENTS
This research was supported by funding from the Mayo Clinic Center for Biomedical Discovery (Z.L., D.D.B. and J.W.) and the Fraternal Order of Eagles Cancer Research Fund (to M.D.), grant number FP00089365.
Author Contributions: M.G. and G.G designed and conducted experiments, analyzed data and wrote the manuscript; Z.L. and D.D.B. edited the manuscript; J.H., X.H., H.H., W.K, F.Z., X.T., Q.Z, C.Z, Q.Z., J.L., Y.Y., Z.X., P.Y., K.L. and J.W. provided technical and data analysis assistance. P.Y. provided reagents for experiments. M.D., D.D.B. and Z.L. conceived and supervised the project, designed experiments and analyzed data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Mayo Clinic. Funding for open access charge: Mayo Clinic.
Conflict of interest statement. None declared.
REFERENCE
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.