 Complete minicircle genome of Leptomonas pyrrhocoris reveals sources of its non-canonical mitochondrial RNA editing events
Complete minicircle genome of Leptomonas pyrrhocoris reveals sources of its non-canonical mitochondrial RNA editing events
Nucleic Acids Research



- Altmetric
Uridine insertion/deletion (U-indel) editing of mitochondrial mRNA, unique to the protistan class Kinetoplastea, generates canonical as well as potentially non-productive editing events. While the molecular machinery and the role of the guide (g) RNAs that provide required information for U-indel editing are well understood, little is known about the forces underlying its apparently error-prone nature. Analysis of a gRNA:mRNA pair allows the dissection of editing events in a given position of a given mitochondrial transcript. A complete gRNA dataset, paired with a fully characterized mRNA population that includes non-canonically edited transcripts, would allow such an analysis to be performed globally across the mitochondrial transcriptome. To achieve this, we have assembled 67 minicircles of the insect parasite Leptomonas pyrrhocoris, with each minicircle typically encoding one gRNA located in one of two similar-sized units of different origin. From this relatively narrow set of annotated gRNAs, we have dissected all identified mitochondrial editing events in L. pyrrhocoris, the strains of which dramatically differ in the abundance of individual minicircle classes. Our results support a model in which a multitude of editing events are driven by a limited set of gRNAs, with individual gRNAs possessing an inherent ability to guide canonical and non-canonical editing.
INTRODUCTION
An underappreciated phenomenon in biology is the ubiquity of processing and modification pathways to develop nascent RNA species into their functional forms. The mechanisms for some of these events are simple, such as cleavage of the bacterial two-component ribozyme RNase P to release tRNA from its 5′ leader (1). However, a great deal of RNA processing involves complex molecular machineries and requires fine-tuned recognition of the sites of action and/or regulation of the process itself. RNA processing events, such as splicing that in most eukaryotes produces mature mRNAs from their precursors (2), have been intensely studied despite their daunting complexity (3). Less pursued are unique RNA modification processes that are equally or even more complex yet have much narrower distribution in extant organisms. Such processes and their variations arise with surprising frequency in the mitochondrial-encoded transcripts of certain protists (4). For example, to generate translatable mRNAs, primary mitochondrial transcripts of diplonemids undergo extensive trans-splicing, U-insertions, A/U-appendage, as well as A-to-I, G-to-A and C-to-U editing events (5). Increasing attention given to the mechanisms decoding the uniquely organized mitochondrial genome into functional RNA molecules uncovers their extreme complexity. Recent methodological advances, such as genome editing approaches, complemented by increasingly sophisticated sequence data analyses allow us to tackle the molecular machineries of formerly ‘off-limit’ protistan species.
Obligatory parasitic trypanosomatids belong to the class Kinetoplastea (6). The kinetoplast DNA (kDNA) is a defining feature of this highly diverse and speciose group of unicellular eukaryotes. Easily observable by simple staining and light microscopy, the kDNA network is the inflated, compactly organized DNA of their single mitochondrion (7). The kDNA consists of relaxed and concatenated circular molecules of two types: maxicircles and minicircles. A ‘typical’ single trypanosomatid kDNA is made up of dozens of identical maxicircles and ∼5000 minicircles that are heterogeneous in sequence (8,9). Each maxicircle possesses 9S and 12S rRNA genes along with over a dozen genes that encode subunits of mitochondrial respiratory complexes and the ribosome (7,9). The transcripts of most maxicircle-encoded genes undergo a uridine (U)-insertion/deletion (U-indel) type of RNA editing that creates translatable open reading frames (10).
RNA editing of the U-indel type represents a perfect example of a complicated RNA processing phenomenon confined to the mitochondrion of a particular protist group (11). The specific U insertions and/or deletions within a given mRNA are pinpointed by sequence alignments with small RNAs called guide (g) RNAs, encoded predominantly by the minicircles (12). A gRNA interacts with a nascent transcript forming an mRNA:gRNA duplex that allows a coordinated machinery to insert or remove specific Us (13,14). Some transcripts undergo extensive editing across their whole length (so-called pan-editing), requiring the information inherent in dozens of different gRNAs, while editing of other transcripts is confined to a particular region, requiring only one or very few gRNAs (15).
RNA editing is a sequential process, as it systematically progresses from the 3′ to the 5′ end of the edited mRNA. Once the 3′-most region of a gene is modified as directed by a particular gRNA, it creates a newly edited site that is recognized by the next upstream gRNA, which is then recruited and used consecutively (10). The machinery executing and coordinating these steps is composed of a set of dynamic complexes that in the model species Trypanosoma brucei involve at least 70 dedicated proteins (14). Intriguingly, U-indel editing is complicated by its apparently stochastic, yet ultimately sufficiently precise nature. Only a fraction of U-indels found in recovered reads contribute to what is the consensus (i.e. canonical) translatable sequence for each mitochondrial mRNA. Alternative or supernumerary insertions or deletions are often found in the regions between canonically edited and yet-to-be edited (i.e. pre-edited) sequences of an mRNA undergoing the editing process, termed junction regions (15–17). However, alternative editing or mis-editing events (termed non-canonical) may appear throughout the transcript (18,19). Due to differences in sequence collection obtained in the context of various studies, it is difficult to directly compare non-canonical editing events among trypanosomatid species (15). Moreover, the editing events required for generation of a translatable set of mRNAs significantly differ among species (20), as the extent of editing required for a given transcript is species-specific (21). For example, while ND8 is pan-edited in many trypanosomatid species, it undergoes editing only at its 5′ terminus in Strigomonas oncopelti (22), and is not edited at all in Wallacemonas sp. WSD (23).
Leishmania tarentolae and T. brucei are model systems of two trypanosomatid lineages parasitizing humans and other vertebrates. Recent work has estimated 391 sequence classes in the T. brucei minicircle repertoire. Each minicircle encodes 1–4 gRNAs, and the resulting total number of independently transcribed gRNAs is around 900, correlating with a high editing demand (24). In contrast, the population of L. tarentolae minicircles contains just 114 sequence classes, with each minicircle encoding a single gRNA, yet still meets the requirements for editing in this Leishmania species (25,26). Indeed, the structure, size and organization of kDNA minicircles vary dramatically among trypanosomatids (8,27,28). While there are clearly significant differences in gRNA population and minicircle organization among various trypanosomatids (29), any comparative analysis suffers from a narrow set of species for which a complete and validated minicircle population is available. Furthermore, no study has directly investigated what the impact of gRNA population complexity might be for a species.
In this work, we describe the structure and functional output of the kDNA minicircles of the trypanosomatid Leptomonas pyrrhocoris that is confined to an insect host (19) and is more closely related to L. tarentolae than T. brucei. Here we show that the analyzed gRNA population is rather narrow, yet is associated with abundant non-canonical editing events. We establish a theory of how these phenomena are related via the degree of flexibility in gRNA:mRNA pairing.
Historically, the analysis of gRNA sequences has facilitated major breakthroughs in our understanding of the U-indel RNA editing mechanism (21,30). More recently, the comparison of gRNAs with RNA-seq data has allowed the field to hypothesize about non-canonical U-indels (15). While some data were consistent with editing being guided by alternative or incorrect gRNAs (12,31), an alternative yet mutually non-exclusive explanation was postulated. Possibly, aberrant U additions and deletions were necessary intermediates forcing progressive rearrangements of bonds in the gRNA:mRNA hybrids during editing (16–18,32,33), or were mis-editing events on a subsequently aborted transcripts (19).
Until now, the analyses of hybrids between mRNAs and gRNAs were performed on a case-by-case, site-by-site basis, which limits their applicability. It is desirable that a method establishes mechanisms of the inclusion of non-canonical U-indels for the entire edited transcriptome. Specifically, the determination of the number of non-canonical editing events that result from gRNAs bound to a non-cognate transcript (or an aberrant region of its cognate transcript), relative to those resulting from a single gRNA at a single site directing both non-canonical and canonical patterns is what is sought. Such a method would require interfacing the maxicircle transcriptome with as complete set of gRNAs as possible.
Most canonically edited products of L. tarentolae and T. brucei have been identified, but in neither of these species has the maxicircle transcriptome been sequenced and characterized (including the non-canonical editing events) to the extent of L. pyrrhocoris. Previously, we designed the program suite T-Aligner that reconstructs translatable edited products from maxicircle-derived reads. The T-Aligner toolkit also includes an interface reporting all editing events that happen at each potential location along a transcript where a U could be inserted or deleted. Therefore, T-Aligner output provides quantitative information about both canonical and non-canonical U-indels for every edited mRNA. T-Aligner was developed on L. pyrrhocoris, which seems to exhibit editing on a greater portion of its total mitochondrial transcripts than other trypanosomatids, such as Trypanosoma cruzi and T. brucei (15,19,34). Here, a combination of the maxicircle transcriptome, a rigorously assembled complete set of L. pyrrhocoris minicircles, and an annotated gRNA repertoire has allowed genome-wide editing annotation for the first time.
Here, we have mapped the outcomes of canonical and non-canonical L. pyrrhocoris maxicircle transcriptome editing events and computationally identified gRNAs responsible for directing them. We then established a methodology to quantitatively attribute editing patterns to gRNAs bound to cognate or non-cognate locations across the edited maxicircle transcriptome. Thus, we have developed a system that globally connects all the outcomes of the editing process to the molecules from which the blueprints for U-indel editing emanate. Furthermore, we demonstrate that once annealed to a cognate L. pyrrhocoris transcript, a single gRNA may direct a number of alternative editing events, but that gRNAs annealed in non-cognate locations are responsible for the majority of non-canonical editing events in this species.
MATERIALS AND METHODS
Small RNA extraction
Leptomonas pyrrhocoris H10 cells (35) were grown in BHI media (Sigma-Aldrich, St. Louis, USA), supplemented with 10% heat-inactivated fetal bovine serum (BioSera Europe, Nuaillé, France), 2 μg/ml Hemin (Jena Bioscience, Jena, Germany) and 50 units/ml of Penicillin/Streptomycin (Life Technologies/ Thermo Fisher Scientific, Carlsbad, USA) at 23°C. Mitochondrial vesicles were isolated from 1.5 × 1011 cells on a Percoll gradient as described previously (19). TRIzol reagent (MRC, Cincinnati, USA) was added to the pellet and RNA was isolated according to the manufacturer’s protocol. To remove DNA contamination, the sample was treated with TURBO DNase (Invitrogen/ Thermo Fisher Scientific, Carlsbad, USA). Before proceeding further, the enrichment of mitochondrial transcripts (9S rRNA, ND1 and ND5) was confirmed by RT-qPCR using primers described previously (19) and normalized to cytosolic 18S rRNA. Synthesis of cDNA was performed with random hexanucleotide primers using the Transcriptor First Strand cDNA synthesis kit (Roche, Indianapolis, USA).
Thirteen micrograms of ethanol precipitated mitochondrial-enriched RNA was dephosphorylated using 300 units of Antarctic Phosphatase (New England Biolabs, Ipswich, USA) and purified by TRIzol-chloroform extraction with the Direct-zol RNA miniprep Plus kit (Zymo Research, Irvine, USA). The 5′-phosphorylation was performed by the T4 Polynucleotide Kinase (New England Biolabs), followed by another extraction as described above. A final quantity of 2.5 μg of L. pyrrhocoris mitochondrial RNA was commercially sequenced (Macrogen, Seoul, Korea).
Minicircle assembly
Minicircle sequences were assembled using an algorithm improved from the one described previously (36). It extends the seed sequence by a stepwise search of possible reads that overlap with it at the 3′ end and extend the seed with the most supported k-mer. At each step, from all reads that have an overlap greater than the chosen threshold of l, the read that is longer than z nucleotides (nt) and has the most frequent k-mer taken from read sequence downstream of the overlap was chosen as the most probable extension. k new bases taken from that read were added to the seed sequence. Unlike our previous work (36) using single parameters, here we used different combinations of l (30, 50, 60, 70, 85), z (100, 140, 160, 200) and k (25, 30, 45, 55, 70) parameters to ensure the assembly of all specific classes of minicircles.
Total RNA sequence data (Illumina MiSeq, paired-end, sequencing read length 250 nt, SRA accession numbers SRX2977446 and SRX2977447) were used for the assembly. Trimming for adapter sequences and base quality analysis was done with Trimmomatic v.0.36 (37), merging was performed with BBMerge (38). The initial set of the seeding sequences was prepared by scanning the reads with the ‘grep’ tool from the Linux core utilities, searching for the minicircle conserved sequence blocks (CSB) 1 and CSB3 in the trimmed merged RNA-seq reads, which is again different from the one used previously. Reads that contained both sequences were processed further, extracting ∼100 nt region starting with CSB1 and ending with CSB3, known as the conserved region (CR) (26,27,39). A total of 1601 CRs were used as the initial seeds. For each seed, its number of occurrences in reads was counted with a combination of ‘grep’ and ‘awk’ tools (Linux core utilities). Patterns that occurred in five or more reads were used as seeds (reducing their number to 137). The termination conditions of the algorithm were as follows: the sequence was considered as a complete minicircle if, and only if, after successful extension at step x first z + k nt of growing contig exactly matched a substring from the end of this contig, while the assembly failed if no circularization happened after 100 steps.
Assembled minicircles were positioned to start with CSB1. Identical minicircles present in the assembly (because each minicircle has two CRs) were removed. Extension heuristics of the algorithm prevented assembly of minor sequence subvariants of minicircles (SNVs, i.e. minicircle single nucleotide polymorphisms) in the assembly of 67 major classes that utilized the RNA-seq reads. For the examination of minicircle SNVs, reads were mapped on the two shotgun DNA-seq libraries (SRA accession numbers: SRX1044309 and SRX1044310, which were not used for the assembly) using bowtie2 v.2.3.4.1 (40), followed by pileup generation with SAMtools v. 1.1 (41) and inspection of SNVs in the pileup with a custom Python script. We found SNVs both in the minicircle CRs and variable regions (VRs) with allele frequencies <10%. Those occurring in the low range of allele frequency may primarily be sequencing errors. Those occurring in the range of 2–10% (23 subvariants) were found to be products of mis-mapping due to regions of high localized sequence similarity in the CRs between two minicircles classes. Moreover, most of the 1601 seed sequences that had poor read support were subvariants of one of 134 CRs. They were clustered into 137 groups with an identity level over 95% by the cd-hit v.4.7 program (42). Due to their high identity with their overall group, and the possibility that the variants may be sequencing errors, we did not pursue them further. Properties of the 67 L. pyrrhocoris H10 minicircles assembled are summarized in Supplementary Table S1.
The Leptomonas seymouri minicircles were assembled from the paired-end DNA sequencing reads (Illumina HiSeq, read length: 100 nt) from BioProject PRJNA285179 (43). We did not apply the algorithm used for L. pyrrhocoris assembly to these data, as it requires longer read lengths. Instead, we assembled minicircles using SPAdes v.3.14.0 (44) with default settings, yielding 15 full-length circular molecules and fragments of additional minicircles. A similar approach was used for assembly of minicircles from raw reads for the species listed in Supplementary Table S2.
To verify the assembly completeness, we counted the number of reads containing CSB1 and CSB3 in one RNA-seq and two DNA-seq libraries and compared them with corresponding counts in sequence alignment matrix files produced with bowtie2 v.2.3.4.1 (Supplementary Table S3). To confirm that our approach does not misrepresent essential sequence variants, we used SPAdes v.3.14.0 with the combined RNAseq and DNAseq datasets and obtained the same 67 minicircles, although some molecules remained fragmented.
Minicircle sequence annotation
Motif discovery was done with MEME SUITE v. 5.1.1 (45). Initially, we ran it with the following parameters: ‘any number of repetitions’, ‘motifs to find = 6’, ‘minimal motif width = 6’, ‘maximal motif width = 100’, ‘0-order model for sequences’ to discover motifs de novo. Then, we used the newly discovered 66-mer motif as input for the MAST2/ MEME SUITE v. 5.1.1 to build accurate alignments only for that motif. The repeat finding was performed as described previously (46) with Mreps v.2.6 (47), inverted tool from the EMBOSS package v.6.6.0 (48) and with a dotplot approach using YASS v.1.15 (49) and a Nucmer tool from the Mummer package v.3.23 (50).
Phylogenetic analysis
To construct the phylogenetic tree for minicircle monomeric units, each molecule was split into two units each initiating with CSB1. The tree was built and visualized using the ETE3 toolkit (workflow ‘default_raxml_bootstrap’) (51). This workflow uses Clustal Omega (52) for multiple sequence alignment and RAxML (53) for tree construction. Maxicircle-encoded ND1, ND2, ND4, ND5 and COI (Supplementary Table S2) were used to build the phylogenetic tree of organisms using the tools described above. For most species the genome assemblies were readily available. The maxicircle contig and four above-mentioned genes were identified using the stand-alone version of NCBI-BLAST (54). Transcripts of these genes are not edited, making their identification straightforward. Minicircle contigs were selected using the ‘grep’ tool with the CSB3 sequence as a search pattern. When only raw reads were available (Supplementary Table S2), they were first assembled with SPAdes v.3.14. For Novymonas esmeraldas (55), we extracted the maxicircle and a few minicircle sequences from our own unpublished assembly.
Annotation of gRNAs
The entire bioinformatic workflow for the following sections can be found in Supplementary Figure S1. The canonically edited mRNAs were derived from the mitochondrial transcriptome of L. pyrrhocoris assembled with the latest version of T-Aligner v.3.3.0 (19). Source code for this version is available on GitHub (https://github.com/jalgard/T-Aligner3.3). The poly(A)-enriched RNA-seq dataset (SRX2977446 and SRX2977447), trimmed with Trimmomatic v.0.36 and merged with BBMerge, was used as the input library. T-less cryptogene sequences for ND8, ND9, A6, G3, G4, ND3 and RPS12 were used as references for the extraction of reads with which to perform open reading frame (ORF) reconstructions. The ORF reconstruction tool ‘findorfs’ from the T-Aligner package was used with ‘–orf_tracing_mode coverage’, ‘–orf_search_depth 25’, ‘–orf_filter_esd 5’ (with an exception of A6, where ‘–orf_filter_esd 0’ was used) and ‘–aln_mismatch_max 1’ parameters. A value for the parameter ‘–orf_min_orf_aa’ was chosen close to the length of the homologous protein from the previously analyzed Leishmania amazonensis (56). From all ORFs reconstructed via T-Aligner, we selected as canonical the ORFs that passed through the maximal number of the editing states (number of Us inserted or deleted) that were most supported by mRNA read evidence at each position (19). For partially edited ND7, CYB and MURF2 genes that carry only small edited domains, we used previously published mRNA sequences, GenBank accessions MF409196, MF409189 and MF409190.
Canonical mRNA sequences were aligned to minicircles with an alignment algorithm implemented in C++ as a part of T-Aligner suite. The program implements the longest common substring (LCS) approach similar to that used in (57). This algorithm reports only minicircle:mRNA alignments that lack gaps, allowing G:U pairing and mismatched bases scored greater than a selected threshold. At each position of edited mRNA, the longest (best scoring) alignment with a minicircle is reported. The final thresholds were chosen in a way that only alignments >20 base pairs (bp) with <10 mismatches and an exact match of four or more bp at the end of an alignment (anchor region) were reported as putative gRNA genes. We subsequently ran our algorithm with no restrictions on the anchor region to detect six ‘missing’ gRNAs for G3, ND3, ND9 and ND7. In this case, we aligned only a portion of the edited mRNA where no previously identified gRNAs bound. This degree of stringency that is required to identify gRNAs in L. pyrrhocoris differs from many T. brucei studies; for example, a recent study uses the following stringency: ‘A valid gRNA match is considered if the gRNA alignment is able to align to the edited sequence, has no gaps or mismatches, and the gRNA has an anchoring region of at least six consecutive Watson-Crick base pairs’ (34).
For small RNA sequencing read alignments, reads were trimmed with Trimmomatic v.0.36 and mapped on the minicircle assembly with bowtie2 v.2.3.4.1 with ‘–local, –fast’ options. Resulting alignments were processed with SAMtools v.1.1. For putative gRNA loci identified by small RNA read coverage (but lacking minicircle:mRNA alignments), we repeated the minicircle:mRNA search using all assembled mRNA isoforms.
Alternative gRNA editing analysis
The trimmed merged RNA-seq reads were mapped on the cryptogene reference sequences with T-Aligner v.3.3.0. The program generated dot matrices for each cryptogene, representing observed editing states, which we have previously defined as the number of Us inserted or deleted at any potential site of editing. Each editing site may have multiple edited states that will be variably supported by reads within the transcriptome (19). We then documented all alignments between the collection of 175 annotated minicircle-encoded gRNAs and all reads, mapped with T-Aligner using the approach described above. The resulting gRNA:read alignments were processed with a custom python script, read coordinates were translated onto the coordinates of T-Aligner’s dot matrix, and editing states observed with T-Aligner in the general read population and those supported by the gRNA:read alignments were compared. For each cryptogene, the percent of gRNA:read alignment supporting editing states was calculated. Editing patterns that represent different outcomes of editing for a gRNA were found with a custom Python script (part of the T-Aligner package, available at GitHub), which explores all gRNA:read alignments for each given gRNA, combining the alignments that are mapped on the same cryptogene locus. This procedure results in multiple sequence alignments that compile single gRNA sequences, their cognate cryptogene locus subsequences and one or more edited read sequences per gRNA. The version available at GitHub also performs the event-joining procedure used to generate data summarized in Supplementary Table S5. The event-joining script takes the text file output of ‘editing event mapper’ (Supplementary Figure S1). All read:gRNA alignments are mapped on reference coordinates and overlapping ‘nested’ alignments identified. We join them in ‘event’ if they have exactly the same 3′ end point (e.g. same anchoring sequence at 3′ end of the alignment on the read).
RESULTS
Complete assembly of minicircle classes
Understanding the origins of U-indel editing variation requires a complete and accurate catalogue of minicircles and their gRNAs. Hence, in addition to the 26 available complete kDNA minicircles of L. pyrrhocoris (36), we have assembled an additional 41 minicircles using a modified sequential seed extension algorithm, producing a putatively complete repertoire of 67 minicircles. The structure of a typical L. pyrrhocoris minicircle is shown in Figure 1A, while features of each minicircle are provided in Supplementary Table S1. This complete repertoire was essential to our goals of characterizing the degree of structural and phylogenetic heterogeneity of this kDNA component and allowed for mapping of the entire putative gRNA and small RNA populations.

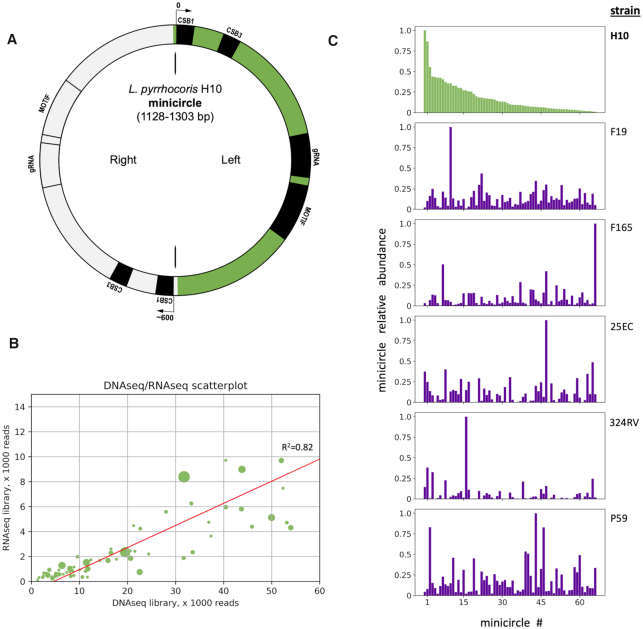
(A) A scheme of a typical L. pyrrhocoris H10 minicircle with two conserved regions (CRs). The zero coordinate is placed at the conserved sequence block 1 (CSB1) within the CR of the left unit. Fifty-five of 67 minicircles are asymmetric in that they possess a left unit encoding a gRNA and a right unit lacking one as shown. The left monomer almost always bears a gRNA and a downstream motif as shown, based on alignments to edited mRNAs and small RNA read coverage. The right monomer is usually inactive (gray) based on small RNA read coverage, but sometimes contains a putative gRNA locus and/or a highly diverged downstream motif (potential locations indicated). Universal minicircle CSB1 and CSB3 are shown. (B) Scatterplot comparing the number of shotgun DNA sequencing reads mapped (a proxy of copy number) to the number of total RNA-seq reads, mapped for each minicircle, and the linear regression line for these values with its R2 value. Each data point on the plot reflects the dimeric minicircle, each dot size is proportional to the level of small RNA sequencing reads mapped on monomeric unit with highest expression (usually the left). (C) Relative individual minicircle class abundances in the kDNA of various L. pyrrhocoris strains, as determined by the number of shotgun DNA sequencing reads mapped on each minicircle. The Y-axis shows read counts for a particular minicircle class divided by the number of reads mapped to the most abundant minicircle class. Each bar shows the coverage of a single minicircle class, with its placement along the X-axis determined by its relative abundance in strain H10 from the highest (left) to the lowest (right). From top to bottom panels show L. pyrrhocoris strains H10, F19, F165, 25EC, 324RV and P59.
Structural organization of trypanosomatid minicircles is species-specific, yet their partition into conserved and variable regions (CRs and VRs) is universal (24–26,58). Consistent with previous results (36), all L. pyrrhocoris minicircles contain two CRs, each encompassing the conserved sequence motifs CSB1 and CSB3 (Figure 1A). Its CSB1 is slightly longer than that in other species (25,59) and has the following sequence: gggtaggggcgttc, while its CSB3 is canonical: ggggttggtgta. A clearly identifiable CSB2, usually found between CSB1 and CSB3, is absent in L. pyrrhocoris minicircles. The two CRs of each minicircle are located in opposing positions, splitting each minicircle into two units of about equal length.
A dot plot of all 67 catenated minicircle sequences (Supplementary Figure S2A) and pairwise alignments of the two closest minicircle pairs (Supplementary Figure S2B,C) demonstrate that minicircles do not have long (over 16 nt) common sequence blocks and there are no pairs of minicircles that are over 90% identical.
Minicircle copy number and expression
We have previously shown that, similarly to L. tarentolae (25), L. pyrrhocoris minicircles are fully transcribed (36). This allowed us to use RNA-seq reads to assemble the minicircles and estimate their abundance, which ranged 275-fold. We also found a 94-fold difference in read coverage using results of DNA shotgun sequencing. Minicircles were subsequently ranked by decreasing read coverage (Supplementary Table S1). Positive correlation between minicircle abundances from RNA-seq and DNA sequencing libraries would suggest a lack of transcriptional regulation at the level of individual minicircles, while divergent patterns would suggest the opposite. Our analysis revealed that the minicircle copy number is strongly correlated with the total RNA-seq reads for that minicircle (R2 = 0.82 for linear correlation) (Figure 1B). Comparing this DNA dataset to a biological replicate DNA dataset yielded an even higher correlation (Supplementary Figure S3). The correlation between RNA- and DNA-derived minicircle abundance was apparent despite a 3-year gap between collection of the DNA and RNA samples, during which the culture had been continuously growing. This strongly suggests that minicircle copy number is stable in L. pyrrhocoris H10 culture.
To determine whether copy number stability extends across different strains of L. pyrrhocoris, we used the raw DNA data from BioProject PRJNA284491 (35). Mapping the minicircle-derived reads of the strains H10, F19, F165, 25EC, 324RV and P59 onto our assembly revealed major copy number disparities between strains (Figure 1C). Even for samples with a relatively high number of mapped reads, there were striking differences in the identities of the predominant minicircle classes. This finding contrasts with the rigorous maintenance of the H10 strain copy number in culture and suggests environmental perturbation as a potential driver of minicircle abundance alterations.
Minicircle phylogeny
Of the closely related species for which minicircle sequences are available, minicircles of Leishmania spp. have a single CR (25), while Crithidia fasciculata, like L. pyrrhocoris described here, possesses minicircles with two CRs (58). To gain insight into the evolution of minicircle architecture within the subfamily Leishmaniinae (60), we assembled (or extracted from available assemblies) minicircles for several key species. Using CSB3 as a CR tag, we determined how many CRs they carry (Supplementary Table S2). We then built a phylogenetic tree and complemented it with the number of CRs in representative minicircles as a marker of their general architecture (Supplementary Figure S4). Minicircles carrying two CRs (dual-CR minicircles) are a unifying feature of the clade ‘II’ represented by the insect-infecting C. fasciculata, L. seymouri, L. pyrrhocoris and Trypanosomatidae sp. LVH60. Of note, the latter species, a close relative of C. fasciculata, was recently isolated from a patient with a fatal visceral leishmaniasis–like disease in Brazil (61). The second CR seems to have emerged after last common ancestor of clades ‘I’ and ‘II’ has branched out from Leishmania and members of the ‘I’ clade preserved mono-CR minicircles. Hence, the minicircle structure of L. pyrrhocoris and other ‘II’ clade members is an exception to the standard minicircles of Leishmaniinae, which are relatively small (∼600–900 nt-long) and carry a single CR and gRNA (62).
As each assembled L. pyrrhocoris minicircle is of the dual-CR type, the question emerges as to the origin of the two separate units. We can hypothesize that this organization either resulted from random catenation of mono-CR circles or is a product of a mono-unit duplication event. The L. pyrrhocoris minicircles range in size from 1128 to 1303 nt, and the contribution of each individual unit to the total length is approximately equal (Supplementary Table S1; compare ‘L’ and ‘R’ length columns).
To test the origin of these units, we split all 67 minicircles into their separate units, each initiating with CSB1, and inferred their phylogenetic relationships. As an outgroup, we used 15 minicircles of the closely related L. seymouri that also possesses dual-CR minicircles. The relationships of all 134 separate units of the L. pyrrhocoris minicircles and 30 separate units of the L. seymouri minicircles are shown in Figure 2. The latter form two distinct clades with high bootstrap support and are stable regardless of the multiple sequence alignment or phylogenetic tree reconstruction algorithms used. Reassuringly, the minicircle units derived from L. pyrrhocoris H10 segregated into clades distinct from those of L. seymouri. They constitute two major clades termed ‘B’ and ‘g’ (named for the background color encompassing the separate clades in Figure 2). Fifty-six of 67 L. pyrrhocoris minicircles possess one unit each from the ‘B’ and ‘g’ clades. A similar pattern occurs also in the L. seymouri minicircles: two units are from different clades. The remaining 11 minicircles possess units that both segregate into the ‘B’ clade (‘2B-type’), whereas there are no ‘2g-type’ minicircles. In summary, the majority of paired minicircle units are of different origin; when from a single clade, they are invariably from the ‘B’ clade.
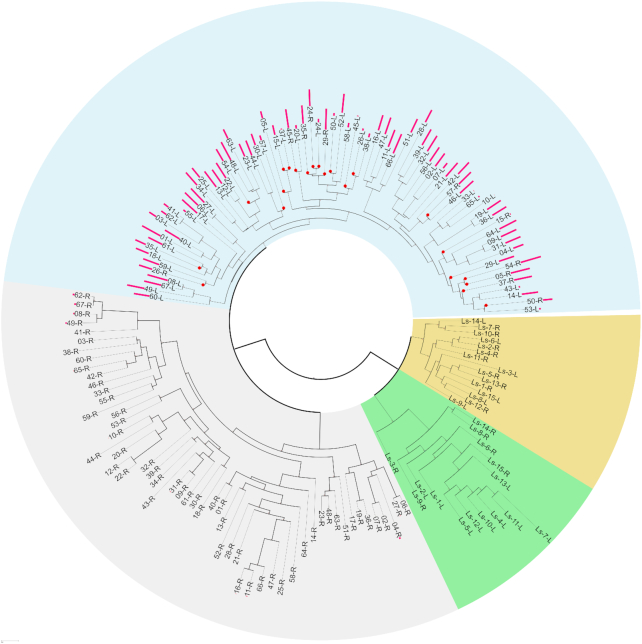
Phylogeny of minicircle monomeric units of Leptomonas pyrrhocoris and Leptomonas seymouri. The background colors encompass clades of monomeric units. The L. seymouri minicircles are composed of one monomer each from the yellow and green clades. The L. pyrrhocoris minicircles are composed either of one monomer each from blue (B) and gray (g) clades (‘Bg- type’), or of both monomers from blue clade (‘2B-type’). Operational taxonomic units that contribute to 2B minicircles are marked with red circles. Pink bar heights for each L. pyrrhocoris monomeric unit are proportional to the negative logarithm of the e-value of finding the 66-mer motif on the respective monomer: higher bars represent higher confidence of motif detection.
Identification of gRNAs directing canonical editing
The primary role of minicircles is to encode the complement of gRNAs required for the essential U-indel editing of the maxicircle-encoded mRNAs. Thus, a full characterization of the minicircle population entails the identification of all gRNAs, which are processed (14) and, thus, likely contain signal sequences driving transcript processing in their vicinity. In T. brucei, the gRNA loci are positioned between 18-mer inverted repeats (24), while in L. tarentolae they are located at a fixed distance from the CSB1 and possess a species-specific motif nearby (25).
Since four different repeat-finding approaches (even with relaxed search settings) failed to identify any inverted repeats in the L. pyrrhocoris and L. seymouri minicircles, these repetitive elements do not seem to comprise their gRNA positional motifs. However, using the MEME SUITE, we found a 66-mer motif ‘tcakdraacgrcygcttrgcgagtatwgagaaccttrctggtmgrktacctgccgaactgtatttt’ in most L. pyrrhocoris minicircle VRs that are the regions in which gRNA loci were anticipated. Sequences of the B-type units fit the consensus sequence much better than those of their g-type counterparts, which seem to lack this motif entirely (Figure 2). Most L. pyrrhocoris minicircles of the type ‘Bg’ constitute a heterogeneous pair of a canonical motif and a divergent unit. Therefore, a sequence consistent with a gRNA positional motif is evident in all L. pyrrhocoris minicircles.
U-indel editing requires the formation, via complementary base pairing, of a gRNA:mRNA duplex. Therefore, we searched for the gRNA loci by finding regions of reverse complementarity between minicircles and edited mRNAs that by T-Aligner were predicted as canonical. First, we searched for the single putative CYB, ND7 and MURF2 gRNAs that are known to be located at specific positions on the maxicircle rather than a minicircle (63,64), in order to test the alignment algorithm’s capacity to predict gRNAs. By aligning these short edited sequences to the L. pyrrhocoris maxicircle (GenBank MN904524), the gRNA loci were identified, provided that mismatches and G:U base pairings within the anchor region were allowed (these alignments are separately listed as the maxicircle-encoded gRNAs in Supplementary Table S4). Small RNA mapping on the maxicircle confirmed that the identified gRNAs are transcribed (described later).
For the canonical ND8, ND9, A6, G3, G4, ND3 and RPS12 transcripts, 170 minicircle:mRNA alignments were found (putative gRNA lengths and other details are listed in Supplementary Table S4). Together, the putative gRNAs derived from these alignments cover most edited positions on the analyzed mRNAs. An attempt was made to identify alignments that would specifically cover positions on edited mRNAs still lacking coverage located within G3, ND3, ND9 and ND7. By relaxing an anchor region parameter in this additional search, five additional putative gRNAs were identified that eliminate all gaps in alignment coverage for the coding regions of all the edited mRNAs. These are highlighted in grey in Supplementary Table S4 and bring the total number of putative minicircle-derived gRNAs to 175.
Many minicircle loci obviously encode more than a single putative gRNA. Seventy-three alignments are secondary, meaning that the same minicircle locus is also involved in a longer alignment within the same set of 175 pairings, while 102 alignments are the longest possible alignment of a minicircle region with any mRNA, and are deemed primary. In the few cases of equally long putative gRNAs within a single minicircle region, the alignment with fewer mismatches was considered primary. Sometimes a primary and secondary gRNA from the same locus overlap in sequence, but usually they are separated by intervening nucleotides. While the same algorithm was used when searching these alignments, the shorter secondary alignments tend to have fewer G:U pairs and more mismatches per nucleotide of alignment length (Supplementary Figure S5), once sorted as such. Start positions of the putative gRNAs corresponding to the primary minicircle:mRNA alignment are listed in Supplementary Table S1.
As expected, these putative gRNA loci are located at specific minicircle regions, which we termed L (on the left unit, coordinates 285–459 bp; always the ‘B-type’) and R (on the right unit, coordinates 855–1100 bp; predominantly, but not exclusively, ‘g-type’). These gRNA loci are positioned at a specific distance from the CSB blocks. They are also consistently 56–62 nt upstream of the 66-mer motif that appears to be analogous to the L. tarentolae ‘bent helical region’, which is thought to be important in nicking the molecule during topological interconversions during replication (58). Thus, the organization of the gRNA-carrying monomer unit of a L. pyrrhocoris minicircle resembles that of a single-CR L. tarentolae minicircle. As the putative gRNA genes are positioned relative to the 66-mer motifs that are best conserved in the B-type units, it is not surprising that from the full complement of 67 L units, invariably of the B-type, there are only six empty (or gRNA-lacking) VRs. In contrast, half of all the R units, which are usually of the g-type, lack putative gRNAs. Finally, 10 out of 11 2B-type minicircles encode two gRNAs. Therefore, g-type monomer units are less likely to contribute information to U-indel editing.
The alignment-finding approach used here identifies putative gRNA loci but does not delineate the exact beginning and end of a functional gRNA. To determine this, we sequenced size-selected small RNAs extracted from L. pyrrhocoris mitochondrial preparations. Mapping these reads on the assembled minicircles identified discrete regions of continuous 10 × or higher read coverage (Supplementary Table S1; ‘small RNA expression’ column), which are defined as the mature gRNA-coding regions. They coincide well with regions that were previously identified via the alignment with edited mRNAs, confirming that while both the L and R regions may contain gRNAs, these are usually present only in the L region of a B-type VR. Interestingly, 17 out of 40 R region minicircle:mRNA alignments were not supported by small RNA library read coverage, again suggesting that they play a lesser role in encoding the information critical for editing. Conversely, two R and three L loci possessed read coverage but were not identified in minicircle:mRNA alignments. These potentially represent five gRNAs that our alignment algorithm failed to detect.
Since functional gRNAs possess at their 3′ ends non-encoded, post-transcriptionally added oligo(U) extensions, the short RNA read population was also used to identify these U addition sites. We extracted all reverse reads initiating with an oligo(A) string of at least 5 nt and trimmed and mapped the read subset onto all 67 minicircles, thus defining the 3′ end positions of the gRNA (Supplementary Table S1; see poly(U) start column). Using the short reads, termini were found for 42 out of the total of 101 mapped gRNAs. Moreover, for many of them, more than one addition site has been identified, indicating variability in the 3′ end trimming of the gRNA prior to oligo(U) addition. Finally, we also mapped identified gRNAs onto the canonical edited mRNA sequences. The position of all gRNAs mapped along the pan-edited RPS12 mRNA is shown in Figure 3, while for other transcripts the same information is provided in Supplementary Figure S6. Complete or even redundant coverage of editing sites on edited mRNAs with this gRNA set was obtained, as long as we included five additional gRNAs identified with relaxed parameters.
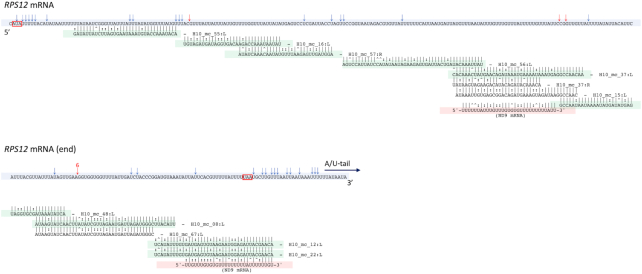
The canonical edited mRNA of the Leptomonas pyrrhocoris RPS12 cryptogene showing the positions of putative gRNA:mRNA alignments. In the alignment string, ‘|’ indicates match; ‘:’ indicates a G:U base pair; ‘∧’ indicates mismatch. Edited RPS12 mRNA sequence is highlighted in blue. Three gRNAs (H10_mc_15:L, H10_mc_12:L and H10_mc_22:L) can also guide the editing of the canonical mRNA of the ND9 cryptogene, alignments to the relevant sequence of ND9 are highlighted in red. Primary gRNAs are highlighted in green. Us that correspond to Ts present at the DNA level are marked with red (deletions) and blue arrows. T-Aligner predicted start and stop codons are boxed in red.
Properties of L. pyrrhocoris gRNAs
As the properties of gRNAs impact their function, we compared the alignment-derived gRNA complement of L. pyrrhocoris (Supplementary Table S4) with that of L. tarentolae and T. brucei that were also determined by implementing the longest common subsequence (LCS) search algorithm (24,25). Guide RNAs of L. pyrrhocoris are shorter, with a median length of 31 nt, while gRNAs of the two-host trypanosomatids have a median length of 43 nt (Figure 4A). However, if regions mapped with small RNA sequencing reads were used instead of the minicircle:mRNA alignment (Figure 4A), the L. pyrrhocoris gRNA length distribution is similar to that of two above-mentioned species. This observation suggests that only an internal portion of a mature L. pyrrhocoris gRNA sequence aligns to a cognate mRNA. With respect to alignment characteristics, the percentage of nucleotide base pairing that includes the allowed weaker G:U pair in the post-editing primary alignments is similar in L. pyrrhocoris and L. tarentolae, but lower than that in T. brucei (Figure 4B). However, the percentage of mismatches excluding G:U base pairing is higher for L. pyrrhocoris relative to the other two species (Figure 4C). To ensure that our parameter set points were not responsible for these differences, we applied the very same methods and allowed mismatch parameters to the publicly available L. tarentolae minicircle and edited mRNA sequences (18). The results recapitulated those published (Figure 4, ‘Lt (tal)’). In summary, gRNA features vary to some extent among the analyzed trypanosomatids, with the L. pyrrhocoris gRNAs being shorter and tolerating more mismatches than those from T. brucei and L. tarentolae.
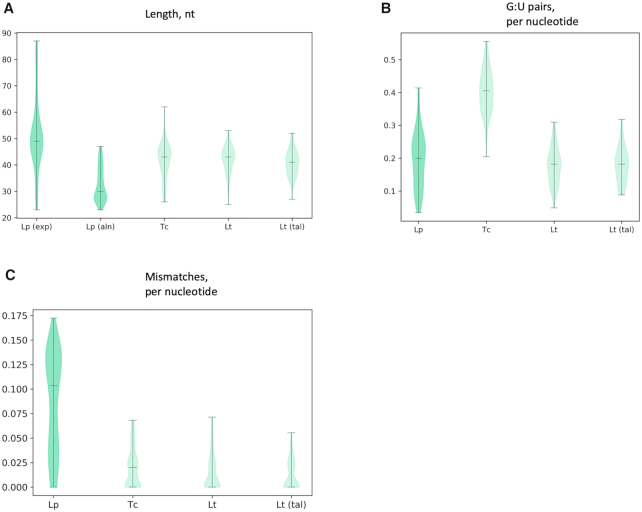
Comparison of gRNA characteristics of three species: Leptomonas pyrrhocoris (L. pyr), Leishmania tarentolae (L. tar; datasets for the analysis were taken from (25)), Trypanosoma brucei (T. bru; datasets for analysis were taken from (24)), and Leishmania tarentolae where characteristics were derived by utilizing our algorithm and parameters on data from (25), noted as ‘Lt(tal)’ (T-aligner derived) violin plot in all subfigures. (A) Distributions of templated gRNA lengths in these species. For L. pyrrhocoris the boxplot on the far left shows the distribution of gRNA length calculated using the lengths as determined by minicircle coverage of short RNA sequencing reads; boxplots 2, 3 and 4 are calculated based on the length of minicircle:mRNA alignments as in (B) and (C). (B) Species-specific distribution of percent of the G:U base pairs of the total pairings per gRNA for all gRNAs. (C) Species-specific distribution of percent of mismatches of the total pairings per gRNA for all gRNAs.
gRNAs determine maxicircle transcriptome diversity
Mitochondrial maxicircle transcriptomes, composed of pre-edited, partially edited and edited mRNAs, are highly complex (15,19). Much of the complexity is due to minor isoforms represented by a wide range of partially and/or alternatively edited molecules. For example, T-Aligner generated 537 isoforms of the L. pyrrhocoris ND8 cryptogene. Most of these isoforms differ in just a few edits and have a very low coverage or are present as a single read and otherwise share 95–99% common sequence. Even cryptogenes with only a small editing domain directed by a single gRNA yield assembled isoforms with edited transcripts differing in a few or even a single position(s). This situation is compatible with a single gRNA generating canonical editing events, and rarely non-canonical ones, or else with alternative sequences resulting from editing directed by a different gRNA annealed to the pre-edited message.
In order to explore the origins of alternative editing, we scanned all cryptogene transcriptome reads (rather than only the canonical ones) with our 175 putative alignment-derived gRNAs, using the same gRNA:mRNA alignment algorithm. The mapped read alignments were grouped by ‘alignment event’, in other words by all pairwise gRNA:mapped read alignments of the same cryptogene site that align to exactly the same nucleotides of the same gRNA. In total, we found 2335 alignment events in which the aligned reads demonstrate possible alternative outcomes. One of 193 such alignment events mapping to RPS12, chosen at random, is shown in Figure 5A. The multiple sequence alignment displays mRNA read-derived possible outcomes of editing of a position on the RPS12 cryptogene with a gRNA encoded by minicircle #48. The anchor region of this gRNA is capable of binding to a sequence present on ∼3700 reads, in which this region is edited by a canonical pathway (Figure 5; boxed). Following its annealing, this particular gRNA appears to direct a series of distinct U insertions, which have a dramatically different read support. From the various editing patterns observed, the major one supported by 3449 reads represents the canonically edited RPS12. However, 16% of reads possess a variety of alternative editing patterns, which differ by the number and positions of inserted Us.
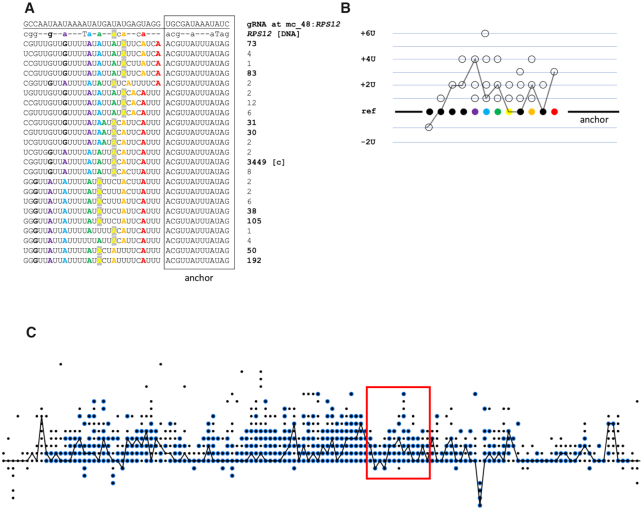
Scenario for multiple alternative editing pathways guided by the same gRNA. (A) U insertion patterns observed in sequences from reads recovered by alignment to a single RPS12 gRNA with an algorithm permitting a degree of mismatch. Read support for each pattern is given in the column on the right. The anchor region is boxed. A rainbow color scheme was used to highlight homologous As between gRNA, pre-sequence, and the edited mRNA reads in each pattern for tracking differences in editing. For example, if the rightmost A (red) pairs with the first U of a gRNA upstream of the anchor, then the AGG nucleotides of the gRNA act as guiding nucleotides, directing the insertion of three Us after this A. However, a group of alternative patterns could have resulted from the rightmost A pairing with the first G upstream of the anchor (which would be considered a mismatch pairing, excluding this G from guiding). (B) Translation of the editing patterns in (A) to the T-Aligner editing state matrix. The X coordinate is equal to the number of the A/G/C cryptogene reference nucleotide, the Y coordinate is the number of inserted or deleted Us after this A/G/C nucleotide (where Y = 0 is the reference level) that were observed in any of the read sequences in (A). Dots representing reference As from (A) are colored in the same way. For example, after the second A (orange), sequences were recovered from the reads in which either 0, 1, 2 or 4 Us were inserted. The canonical editing pattern is shown as lines connecting each respective canonical editing state. (C) The T-Aligner editing state matrix for cryptogene RPS12. The scheme represents the numbers of inserted\deleted Us as dots, located above (insertion) or below (deletion) the reference line, with its X coordinate representing an A/G/C nucleotide position in the cryptogene sequence. Black line shows the path of the canonical edited mRNA for the cryptogene. The editing states observed only in the total RNA sequencing read mapping appear as black dots. The editing states that are also supported by gRNA:read alignments are circled in blue. The red box indicates the region of the RPS12 cryptogene that is edited with mc_48:RPS12 gRNA with alternative outcomes shown in (A).
The alternative G:U alignment patterns observed in the dataset and the allowance of some mismatches appear to be the factors that would permit a single gRNA to drive alternative outcomes. In this scenario, the same nucleotide can act as a guiding nucleotide in one milieu and a non-guiding nucleotide in another one (Figure 5A). Rainbow coloring of As demonstrates the pairing of the same As in DNA (analogous to the pre-edited mRNA) with different gRNA nucleotides in each alignment. If the rightmost A (red) pairs with first U of a gRNA upstream of the anchor, then AGG of the gRNA acts as the guiding nucleotides, directing the insertion of three Us after the A. However, a group of alternative patterns could have resulted from the rightmost A pairing with the first G upstream of the anchor, which would be considered a mismatch pairing, excluding this G from guiding. These complex patterns can be exhibited on T-Aligner’s editing state dot matrix, where the matrix dots depict editing states (the number of inserted and deleted Us) at each editing site observed in the data (Figure 5B). For example, after the second A (orange), sequences were recovered from the reads in which either 0, 1, 2 or 4 Us were inserted.
For each cryptogene, such editing state matrices (corresponding to all reads involved in alternative editing events attributable to 175 gRNAs) were then compared to the editing state matrix produced using all maxicircle-derived reads, regardless of whether they aligned or not to a gRNA. This comparison allows the visualization of the number of editing states observed from raw read mapping that can be attributed to editing with one of the identified gRNAs (Figure 5C for RPS12; see Supplementary Figure S7 for all other cryptogenes). This analysis showed that on average 78% of the editing states are supported by their alignment with the gRNAs that we identified as capable of generating canonically edited sequence in some maxicircle-encoded gene.
Finally, sequential annealing of gRNAs to multiple alternative outputs could compound the transcript diversity resulting from the alternative editing events that are directed by individual gRNAs. To illustrate this, we traced the putative next steps of editing upstream of the RPS12 region detailed in Figure 5A and B. Three possible pathways are shown, including the one directed by the gRNA derived from minicircle #15, resulting in the canonical sequence, and two alternative editing pathways directed by the gRNAs primarily cognate for A6 and ND8 (Figure 6).
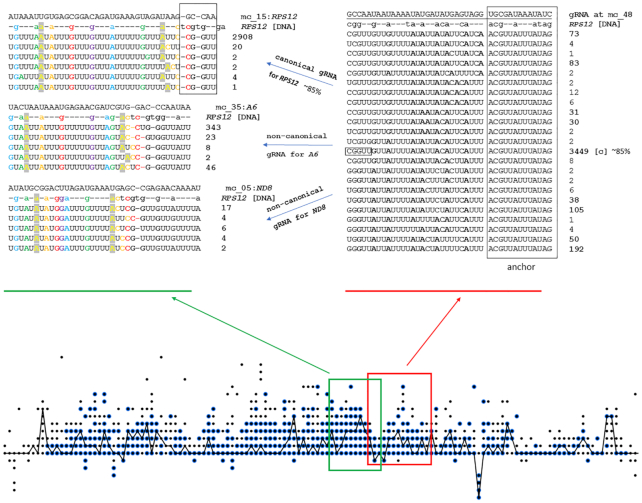
A visualization of proposed canonical and alternative editing. Two sequential editing steps are presented. The RPS12 editing states dot matrix (as in Figure 5C) is shown at the bottom of the figure, depicting the number of editing states observed from raw read mapping that can be attributed to editing with one of our identified gRNAs as blue-circled editing events. Similar dot matrices for additional cryptogenes are found in Supplementary Figure S7. The red square on the dot matrix indicates the region edited with canonical RPS12 gRNA of minicircle #48, and all patterns produced with that gRNA are depicted on the right panel as in Figure 5A. The green square and left panel shows three possible alternatives the next gRNA to bind and guide transcript editing, which was canonically edited with mc_48:L gRNA. Rainbow coloring highlights homologous nucleotides in each pattern. In each case cryptogene reference DNA is aligned with the most supported pattern. Approximately 85% of reads support editing with canonical gRNA coming from minicircle #15 that has primary alignment with RPS12. Alternative patterns are generated with non-canonical gRNAs that have primary alignments with A6 and ND8. Black boxes indicate the anchoring nucleotides used by mc_48:L and mc_15:L gRNAs from canonical RPS12 pathway.
Alignments of each of these alternative upstream gRNAs also incorporate reads with more than one editing pattern (Figure 6). Most non-canonical aligning editing patterns have low read support. Alignments in Figures 5 and 6 reveal a consistent small fraction of reads with editing patterns following the non-canonical pathways. The accumulation of these fractions results in a ‘dissipation ratio’ of editing processivity.
Mechanisms of gRNA usage resulting in non-canonical editing events can be parsed
One thing we did not determine is how many of the alternative editing patterns for the region shown in Figure 5 also aligned to other gRNAs that would typically direct canonical editing at other locations. Is it possible that other (maybe partially overlapping) gRNAs can guide some of the non-cognate editing patterns shown in Figure 5? Multiple technical concerns make it difficult to ascertain whether this could likely occur at any single alignment event.
A related question that is both more universal and can be more easily answered is: do multiple allowable editing events caused by a single canonical gRNA or else binding and action of gRNAs in alternative locations significantly contribute to the non-canonical editing patterns observed in the U-indel edited transcriptomes? Our approach to this question requires defining some terms. We have classified alignment events as being either ‘cognate’ if the gRNA is in a position where it can generate a canonically edited sequence, yet could also guide a non-canonical edit (Figure 5). An alignment event is ‘non-cognate’ if the involved gRNA at a given transcriptome position only aligns to the non-canonical sequence patterns, such as the A6 and ND8 gRNAs aligned to a region of RPS12 in Figure 6. For this analysis, we restricted the editing patterns to only include those supported by at least four reads, in order to reduce noise and possible artifacts from sequencing errors. For simplification, we also utilized an event-joining algorithm to combine alignment events with the exact same gRNA anchor site (some alignment events are simply ‘sequence nested’ versions of other ones). Most (1387) of the 1975 alignment events assembled in this manner are non-cognate, suggesting that at least in L. pyrrhocoris, many non-canonical editing events may be due to the binding of a gRNA that can only direct a non-canonical pattern at that location (Supplemental Table S5). The other 588 alignment events are cognate, in that the involved gRNA aligns with a canonical editing pattern in at least some of the reads of the event.
The cognate and non-cognate alignment events were further classified as being single-pattern (whether canonical or non-canonical) generated by the binding of that particular gRNA to one mRNA pattern only, or multi-pattern as shown in Figures 5 and 6. Very few cognate events (144) were single-pattern. Expectedly, within multi-pattern cognate alignment events (which comprise 588 out of the 729 cognate alignment events total), read support is stronger for alignments of gRNAs to the canonical than non-canonical patterns. A median of 62% of the reads of each cognate gRNA alignment event across the six pan-edited mRNAs displayed the canonical pattern. The remainder of reads were typically multiple alternative patterns of low relative representation. The non-cognate alignment events also usually included one alignment that had a higher read support, but the mean percentage of reads that contained the common pattern was only 50% (Supplementary Table S5). Analysis at the level of individual alignment, previously described in Figures 5 and 6, bears out this collective result. For example, in the canonical alignment event in Figure 5 there are 23 possible outcomes of editing, but a single one with an overwhelming degree of read support. Figure 6 contains two examples of non-canonical alignment events. There are only five possible pattern outcomes for each of the corresponding gRNAs that act in a non-cognate fashion at that site (these gRNAs are capable of guiding canonical patterns in A6 and ND8). In each case there still is a single best-supported pattern, yet it is less overwhelming for these non-cognate alignment events.
Finally, it is possible to discern the relative contributions of cognate gRNA binding and non-cognate gRNA binding in alternative editing. These values can be determined by comparing numbers of alternatively edited reads from two categories. The first category comprises the reads that support all non-canonical editing patterns from 588 cognate multi-pattern alignment events from Supplementary Table S5. The second category is the sum of reads supporting all alignment events for non-cognate gRNAs. The relative abundance of these two categories of reads is a good estimate of the relative contributions of these alternative mechanisms to non-canonical editing patterns. Performing this analysis on our data, a majority of the reads (75%) support non-canonical editing patterns guided by non-cognate gRNAs, while 25% support them guided by cognate gRNAs.
This leads to the question of whether there might be any distinguishing features of alignments of gRNAs bound in cognate arrangements to mRNAs compared to gRNAs our algorithm aligned at non-cognate locations. In fact, in the metrics of alignment length and mismatch rate, the difference between the distributions is statistically significant according to a two-sample Kolmogorov–Smirnov test (P-value < 0.01), while the G:U pair rates do not differ significantly (Supplementary Figure S8).
In summary, our ability to computationally parse cognate and non-cognate gRNA:non-canonical read alignments has led to the following model. When gRNAs bind to transcripts at positions other than their canonical binding location, the resultant editing is restricted in terms of complexity, and these events are individually rare. This is likely caused by weaker binding, as the typical number of mismatches for these alignments is relatively high. In contrast, gRNAs bind to the canonical locations with higher affinity and/or for extended period of time, and these interactions are more complex.
DISCUSSION
Here we present the first complete kDNA minicircle assembly for a monoxenous trypanosomatid flagellate. The putative gRNAs annotated in the assembly span all edited regions of the maxicircle open reading frames. In fact, most editing positions could be canonically edited by more than one, and sometimes up to five gRNA(s). Redundant coverage was also reported for L. tarentolae and T. brucei (24,25,57).
In L. pyrrhocoris, the predominantly observed redundancy is manifested as a gRNA with the ability to anchor to both X and Y mRNAs. For example, the gRNA encoded on the L unit locus of the minicircle #15 has the capacity to direct editing of specific regions of the canonical ND9 and RPS12 mRNAs (Figure 3). This redundancy may reflect the importance of an editing ‘safety net’.
Less impactful according to our calculations, yet possibly more intriguing is the observed potential for a single gRNA to specify different editing patterns for a given transcript region. This is true of both cognate and non-cognate gRNA:transcript interactions (Supplementary Table S5). Computationally, most observed non-canonical editing states can be explained by a combination of these two types of events. By aligning gRNAs with the cryptogene-derived RNA reads, we showed that ∼80% of the editing states observed in the transcriptome can be explained by the gRNA set from 102 identified minicircle loci plus the half dozen located on the maxicircle.
Our analysis leaves ∼20% of known L. pyrrhocoris editing states unsupported by potential cognate gRNAs. It is plausible that all editing states are actually guided by expressed gRNAs, and our failure to detect all of them may be explained by the stringency of our search parameters. For example, we have built the gRNA:raw read alignments with an anchor thresholds of four or more exactly matching bases. To discover five gRNAs that covered gaps in gRNA:mRNA alignments (Supplementary Table S4), we needed to relax these rules. Additionally, in this study we disallowed gaps in the alignments for more rigor, but data obtained with T. brucei suggest that the gRNA:mRNA alignments can tolerate gaps (57). Finally, the alignment was only performed with the gRNA regions that participate in the primary or secondary alignments with the canonical mRNA, but not with the nucleotides external to the aligned regions that were discovered by short RNA sequencing. It is also plausible that at least a portion of these editing states, especially those represented by fewer than four reads, may be sequencing errors or portions of contaminating nuclear genome that mis-align to portions of the maxicircle.
Read support of editing patterns suggests canonical editing to be the most frequent editing outcome following gRNA binding to a cognate position. Still, since some cryptogenes are edited by 6–15 gRNAs, the cumulative degree of non-canonical editing by cognate gRNAs is not inconsequential (Figure 6). First extensively queried between 1990 and 1992 (12,16,21,65,66), the non-canonical editing patterns were presumed to be restricted to junction regions between the edited and pre-edited portions of mRNAs. They were hypothesized to derive from editing by an alternative gRNA, mis-editing by a canonical gRNA for that editing block, or through indiscriminate action of the editing machinery. However, subsequent analyses soon suggested that non-canonical editing patterns may occasionally be incorporated into a translatable mRNA, thus increasing diversity of the mitochondrial proteome (67,68). Indeed, the non-canonical editing patterns have the potential to be recognized as anchors by the non-cognate gRNAs, leading to alternatively edited mRNAs translatable into very different amino acid sequences. Such scenarios have been uncovered at the RNA level for L. pyrrhocoris, T. brucei, Perkinsela sp. and L. tarentolae (19,20,24,25,34,57). However, direct proof for the existence of proteins generated from alternatively edited transcripts is still lacking, leaving doubt as to whether U-indel editing confers a selective advantage in this manner (69). We note that the L. pyrrhocoris gRNA repertoire is almost 10-times smaller than that of T. brucei. The more common mismatches in L. pyrrhocoris gRNA:mRNA alignments that are apparent even in primary gRNA alignments with the canonically edited mRNAs may compensate for its lack of gRNA diversity. In sum, our data suggests that in L. pyrrhocoris, both editing events that originate from anchoring and guiding of a non-cognate gRNA to an mRNA region, and multiple patterns directed by a single gRNA at a single editing loci likely occur, with the former appearing to drive the bulk of non-canonical editing events. Prior to this analysis, there had been no quantitative way to parse these possibilities across the entire edited transcriptome.
Other recent work, utilizing deep sequencing of specific T. brucei mitochondrial mRNAs and their analysis in the context of aligning gRNAs, focuses on discrete questions. The Koslowsky laboratory has focused on identifying alternative functional mRNAs in T. brucei resulting from the utilization of alternative gRNAs (34). Other deep sequencing-based studies allowed for understanding of the initiation and processing of the first gRNA block, or analysis of progression of editing in general with a potential to focus on the junction regions covering a single gRNA editing block (15) and confirmed that it was possible to infer a non-linear modification order for a particular gRNA block from deep sequencing reads (33). It has been hypothesized that the junction regions with the alternative editing patterns do not represent dead-end editing products, but instead necessary intermediates (17,18,32,33). Is it possible that alternative patterns in the L. pyrrhocoris reads that we attribute to differential use of a specific gRNA as either guiding or not guiding, are in fact reads derived from such intermediates? Our results may argue in favor of such a mechanism. The ratio of canonically edited to non-canonically edited reads in multi-pattern cognate alignment events is higher than for the non-cognate ones, despite the overall higher complexity of these patterns in cognate alignment events. This qualitative difference argues for specific features distinguishing the cognate alignment events from the non-cognate ones that are near-certain to result in dead-end products.
The characterization of L. pyrrhocoris minicircles is important for our global perception of gRNA transcription and processing. The only models of trypanosomatid gRNA transcription and processing that are based on empirical studies are developed for T. brucei. It is clear that T. brucei transcription initiates at a fixed distance from a gRNA’s upstream repeat (70). Analogous to T. brucei, L. tarentolae and now L. pyrrhocoris have been shown to lack the inverted repeat sequences but to contain conserved sequence motifs proximal to the gRNA locus, which may instead play a role in initiation of transcription or else processing. We propose that these different regulatory motifs may determine specific mechanisms for gRNA transcription initiation or processing that may impact the length or precision of termini of gRNAs released from their longer precursor molecules. A model for T. brucei gRNA processing posits that expression of the antisense minicircle strand, and its subsequent processing, are responsible for defining the 3′ termini of the mature gRNA trimmed 3′ to 5′ from a much longer precursor (71). However, this model does not provide a role for the T. brucei inverted repeats in processing. Both our RNA-seq and small RNA sequencing reads map to both strands of minicircles, which means that the processing mechanism for L. pyrrhocoris gRNAs could involve antisense RNA. However, in the T. brucei model, U-tailing of both the gRNA and corresponding antisense fragments occurs. Our U-tailed reads are exclusively found on the gRNAs and not on small antisense minicircle products. Presumably, the frequent presence of secondary minicircle:mRNA alignments that we found in L. pyrrhocoris may reflect a potential for variability or flexibility in gRNA processing. While variable gRNA end processing could drive less efficient editing, it might also ultimately increase beneficial transcript diversity.
Our results also demonstrate that understanding the minicircle structure may be important for evolutionary inferences. Within the subfamily Leishmaniinae, L. pyrrhocoris belongs to a clade that invariably possesses dual-CR minicircles. Since most of these species contain two monomeric units of distinct origin, it is unlikely that they arose from the monomeric minicircles via duplication. Their existence can be better explained by a scenario in which an ancestor of the subfamily Leishmaniinae possessed dual-CR minicircles with both units. Subsequently, some of the monomeric units, encoding redundant gRNAs, might have acquired mutations in their 66-mer motif that precluded their processing and stabilization. Although typically only one of each minicircle monomers usually encodes gRNAs and possesses regulatory motifs, in a few cases, both units encode gRNAs. Since products of these loci were not found within the small RNA-seq reads even though all minicircles are fully transcribed, we conclude that their gRNA processing is blocked. Any role for the L. pyrrhocoris minicircle unit that does not engender functional gRNAs has yet to be elucidated.
Finally, in all trypanosomatids investigated thus far, the copy number of the individual minicircle classes drastically varies within a single kDNA network, as observed here as well (Figure 1C). Our finding that the relative abundances of different minicircle classes within a kDNA network vary by strain is also in line with what was observed for two L. tarentolae strains (25,72). However, modeling of relative abundances between several time points assuming random minicircle segregation (73) and empirical measurement of this parameter in the historical L. tarentolae UC laboratory strain suggest that culture samples taken at different times would exhibit alterations in relative abundances of various minicircle classes, leading to a gradual loss of some minicircle populations (72). In L. pyrrhocoris, this appears not to be the case. An underlying assumption with the loss of minicircle classes in the UC strain was that not all minicircle classes were essential for growth in vitro. This was supported by the fact that certain cryptogenes appeared to no longer be completely edited, since in the nutritionally rich medium their protein products were non-essential (72). An increased importance of a diverse minicircle population for L. pyrrhocoris and/or unidentified differences in selective forces between the monoxenous and dixenous species may be the reasons why the H10 strain has achieved an unchanging ratio of different minicircle classes.
To conclude, this description of the complete minicircle genome of L. pyrrhocoris expands our general understanding of minicircle networks, minicircle composition and gRNA features in trypanosomatids, precipitating new conjectures and hypotheses that can be tested empirically as developing tools allow. From the application of our unique bioinformatic tools applied here concurrently to both mRNA and gRNA populations, we demonstrate that the guiding flexibility of individual gRNA at the whole-genome level may be at the root of the alternative editing patterns observed in the U-indel edited transcriptomes. The fact that we can computationally parse gRNA alignment events across the genome is a significant advancement. This approach can now be applied to other species to determine whether the ratio of the non-canonical editing events that can be attributed to the non-canonical gRNA binding versus multiple outcomes of the canonical gRNA binding is similar or different to that of L. pyrrhocoris.
DATA AVAILABILITY
MicroRNA sequencing data can be downloaded from the NCBI SRA under accession SRR12440684. Genbank accession numbers for minicircle sequences are listed in Supplementary Table S1. T-Aligner code is available at GitHub (https://github.com/jalgard/T-Aligner3.3), gRNA:mRNA duplex search program code is a part of T-Aligner 3.3. Minicircles assembly program code, and data processing scripts are available at GitHub (https://github.com/jalgard/scripts-330).
ACKNOWLEDGEMENTS
We thank members of our laboratories for stimulating discussions and Dr. Flegontov (University of Ostrava) for help at the initial stages of this project.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Science Foundation [19-74-10008 to E.S.G.; 19-15-00054 to V.Y.]; ERC CZ [LL1601 to J.L.]; European Regional Developmental Funds [OPVVV16_019/0000759 to V.Y., N.K., J.L.]; University of Ostrava [SGS/2020 to V.Y.]; American Heart Association [16SDG26420019 to S.L.Z.]. Funding for open access charge: European Regional Developmental Funds [OPVVV16_019/0000759].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.