 Human METTL18 is a histidine-specific methyltransferase that targets RPL3 and affects ribosome biogenesis and function
Human METTL18 is a histidine-specific methyltransferase that targets RPL3 and affects ribosome biogenesis and function
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the second and third authors should be regarded as Joint Second Authors.

- Altmetric
Protein methylation occurs primarily on lysine and arginine, but also on some other residues, such as histidine. METTL18 is the last uncharacterized member of a group of human methyltransferases (MTases) that mainly exert lysine methylation, and here we set out to elucidate its function. We found METTL18 to be a nuclear protein that contains a functional nuclear localization signal and accumulates in nucleoli. Recombinant METTL18 methylated a single protein in nuclear extracts and in isolated ribosomes from METTL18 knockout (KO) cells, identified as 60S ribosomal protein L3 (RPL3). We also performed an RPL3 interactomics screen and identified METTL18 as the most significantly enriched MTase. We found that His-245 in RPL3 carries a 3-methylhistidine (3MH; τ-methylhistidine) modification, which was absent in METTL18 KO cells. In addition, both recombinant and endogenous METTL18 were found to be automethylated at His-154, thus further corroborating METTL18 as a histidine-specific MTase. Finally, METTL18 KO cells displayed altered pre-rRNA processing, decreased polysome formation and codon-specific changes in mRNA translation, indicating that METTL18-mediated methylation of RPL3 is important for optimal ribosome biogenesis and function. In conclusion, we have here established METTL18 as the second human histidine-specific protein MTase, and demonstrated its functional relevance.
INTRODUCTION
The transfer of a methyl group from the methyl donor S-adenosylmethionine (AdoMet) to a biomolecule is a common biochemical reaction, catalyzed by a number of different methyltransferases (MTases) (1). Bioinformatics analysis has indicated that the human genome encodes more than 200 AdoMet-dependent MTases (2). Many of these enzymes still remain uncharacterized, although the biochemical function of several human MTases has been uncovered in recent years. The so-called seven β-strand (7BS) MTases, which have a characteristic core fold consisting of seven β-strands, also referred to as the ‘Rossmann-fold’, constitute the largest group of MTases in humans (2,3). These enzymes methylate a wide range of different substrates, including metabolites, lipids, nucleic acids and proteins. The SET-domain proteins represent the second largest group of MTases in humans, and they mainly target lysines in proteins (4).
Proteins are subject to a wide range of post-translational modifications (PTMs). Methylation is a PTM occurring mainly at lysines and arginines, but other residues, such as glutamine or histidine, can also be methylated (5–11). In the case of histidine methylation, which is a relatively poorly studied PTM, the two nitrogen atoms of the imidazole ring of a histidine can be monomethylated, yielding 1-methylhistidine or 3-methylhistidine (1MH or 3MH; also referred to as π- or τ-methylhistidine, respectively). Both forms exist in mammals, but only a few methylhistidine-modified proteins have been firmly established (12–16). However, recent high-throughput studies have identified hundreds of candidate histidine-methylated proteins in mammals, indicating that this modification is widespread (11,17). Only two histidine-specific protein MTases have so far been described in eukaryotes. First, the MTase encoded by open reading frame (ORF) YIL110W in Saccharomyces cerevisiae was found to mediate the introduction of 3MH at His-243 in the 60S ribosomal protein L3 (Rpl3), and therefore denoted Hpm1 (histidine protein methyltransferase 1) (10). Second, the human SET-protein SETD3 was recently identified as the long-sought MTase responsible for introducing 3MH at His-73 in actin (8,11).
When yeast Hpm1 was discovered in 2010, the orphan MTase METTL18 (also known as C1orf156) was pinpointed as its closest human sequence homolog, and also suggested as a possible functional ortholog (10). Nevertheless, the function of METTL18 remains obscure a decade later. METTL18 is one of the 11 human members of the so-called MTase Family 16 (MTF16) (18–20), and we and others proposed MTF16 as a family of lysine (K)-specific MTases (KMTs) (18,19). Indeed, KMT activity has now been reported for nearly all human MTF16 members, but no studies have focused on METTL18. Furthermore, yeast Hpm1 and human METTL18 only exhibit a modest sequence similarity (31% identity; 50% similarity), comparable to the similarity between two other MTF16 members, yeast Efm6 and human METTL21A, which target different substrates, and are thus not orthologs (21,22). Moreover, attempts to complement phenotypes of Hpm1-deficient yeast by the expression of human METTL18 were unsuccessful (23). On the other hand, human RPL3 was reported as a main METTL18 interactant in pull-down experiments, suggesting that RPL3 may indeed be the target of METTL18 (18). In conclusion, the function of METTL18 has remained unresolved, requiring further investigation.
The ribosome represents a universally conserved molecular machine that translates the genetic information in mRNA into a protein sequence. The human ribosome (80S) consists of four ribosomal RNAs (rRNAs) and 80 proteins, which are distributed between the small (40S; contains 18S rRNA) and large (60S; contains 28S, 5.8S and 5S rRNAs) subunits (24). While 5S rRNA is transcribed from a separate gene, the remaining rRNAs are transcribed from a joint gene in the form of one, long primary transcript (47S pre-rRNA), which is processed through cleavage and trimming by several specific ribonucleases to yield the mature 18S, 28S and 5.8S rRNAs (25). Both rRNA and ribosomal proteins contain several different modifications, including numerous methylations (26–28). The assembly and maturation of the ribosome, occurring primarily within the nucleoli, is a highly orchestrated process, where timely processing of precursor rRNAs and insertion of specific rRNA and protein modifications is essential (29). To fully understand the functional significance of these modifications in the context of ribosome biogenesis and function, it is necessary to identify the various enzymes responsible for their introduction.
Since the function of METTL18 has remained unresolved, we set out to elucidate various functional aspects of this enigmatic MTase. We found METTL18 to be a nuclear protein, accumulating in nucleoli and containing a nuclear localization signal (NLS). Recombinant METTL18 methylated a single protein present in nuclear extract of human METTL18 KO cells, which was identified by mass spectrometry (MS) as the ribosomal protein RPL3. Using both biochemical and cellular approaches, we demonstrated that METTL18 introduces 3MH at position 245 in RPL3. Moreover, processing of pre-rRNA and mRNA translation were both affected by METTL18 ablation.
MATERIALS AND METHODS
Gene cloning and mutagenesis
The plasmid constructs and the strategy to generate them are described in detail in Supplementary Table S1. In brief, ORF were amplified by polymerase chain reaction (PCR), and cloned into indicated vectors by standard ligation-dependent cloning, or by ligation-independent cloning using In-Fusion® HD Cloning Plus kit (Takara). Mutations within ORFs were introduced by site-directed mutagenesis using PCR splicing by overhang extension (PCR SOEing) as previously described (30). Sanger sequencing was used to verify all cloned constructs.
Bioinformatics analysis
Sequence alignments were generated using algorithms embedded in the JalView interface (http://www.jalview.org/) (31).
Transient transfection of HeLa cells and fluorescence microscopy
HeLa cells were grown in RPMI 1640 + GlutaMAX™ medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco) and 100 U/ml penicillin-streptomycin (P/S) (Lonza). HeLa cells were transiently transfected, using the Lipofectamine® 3000 transfection kit (Invitrogen), with pEGFP-N1-derived plasmids (Clontech) encoding human full-length (FL) METTL18 or its variant consisting only of amino acids 233–265 that contains a putative nuclear localization sequence (NLS), all fused to the N-terminus of enhanced green fluorescent protein (EGFP). The FL and NLS METTL18 constructs were either WT or K250G/R251G-mutated. When indicated, cells were co-transfected with pMRFP-C1-RPL3 plasmid encoding human RPL3 fused to the C-terminus of monomeric red fluorescent protein (MRFP) (32). After overnight incubation cells were stained with 1 μg/ml DAPI (Sigma-Aldrich) to visualize the nuclei. Live cells were imaged using an Olympus FluoView 1000 (I × 81) inverted confocal fluorescence microscopy system, equipped with a UPLSAPO 60× NA 1.35 oil objective (Olympus). The different fluorophores were excited at 405 nm (DAPI), 488 nm (EGFP) and 559 nm (MRFP), and Kalman averaging (n = 3, sequential) was used to record multichannel images. The fluorescent signals emitted from EGFP, MRFP and DAPI were acquired through green, red and blue channels, respectively, and merged.
Generation of HEK293-derived cell lines for ectopic expression of ORFs
ORFs encoding GFP alone, or GFP fused to human RPL3, either WT or R246A/R249A-mutated (RPL3 fused to N-terminus of GFP) were cloned into the pcDNA5/FRT/TO plasmid (Thermo Fisher Scientific) under the control of a doxycycline (Dox)-inducible promoter, similarly as described in ref. (32). HEK293-derived, Flp-In™ T-REx™ 293 cells (Thermo Fisher Scientific) were co-transfected with pOG44 and pcDNA5/FRT/TO-derived plasmids, according to manufacturer's instructions, and selected with 200 μg/ml Hygromycin B (Thermo Fisher Scientific). The pool of surviving cells was maintained in high-glucose DMEM + GlutaMAX™ medium (Gibco) supplemented with 10% FBS and P/S. To test ORF expression, cells were left untreated or induced with 1 μg/ml Dox for 24 h and then fixed in cold acetone for 10 min. Cells were visualized by fluorescence microscopy after incubation with 1 μg/ml Hoechst 33258 (Sigma-Aldrich) to stain the nuclei (λex = 405 nm), similarly as described in the above section.
Generation of HAP1-derived stable cell lines
HAP1-derived cells were grown in IMDM + GlutaMAX™ medium (Gibco) supplemented with 10% FBS and P/S. HAP1 METTL18 knock-out (KO) cells were generated as a custom (non-exclusive) project by Horizon Discovery (Austria). The METTL18 gene was disrupted in haploid HAP1 parental cells using CRISPR-Cas9, with guide RNA designed to target part of the exon located immediately downstream of the Post I motif, which is required for enzymatic activity of 7BS MTases. Individual clones were selected by limiting dilution, and frame-shifting events within the targeted gene were determined by sequencing of genomic DNA. The METTL18-deficient cell line used in this study (Horizon Discovery HZGHC000541c002) contains a 2-bp insertion within exon 2, thus generating a version of the METTL18 protein consisting of the initial 230 amino acids of METTL18 (i.e. truncated 13 amino acids downstream of the Post I motif), followed by two residues of out-of-frame sequence and a premature stop-codon. Complementation of METTL18 KO cells was performed similarly as described previously (33) by transfection with p3×FLAG-CMV-14-derived plasmids encoding either WT or D217A-mutated human METTL18, all bearing a C-terminal 3×FLAG-tag. Transfected cells were selected and expanded in medium containing 1 mg/ml geneticin (Gibco). Individual clones of complemented cells were screened by western blot for the presence of the FLAG-tag and GAPDH (as loading control), using appropriate antibodies (see western blot analysis section).
Cell fractionation and Western blot analysis
Cells were grown in 10-cm diameter plates until 50–80% confluent. After washing with phosphate-buffered saline (PBS), cells were scraped in PBS and centrifuged (5 min, 500 × g). Cell lysis and fractionation into cytoplasmic and nuclear fractions was performed at 4°C, using a modified rapid, efficient and practical method (34). Cell pellets were lysed on ice, for 2 min, in 300 μl Fractionation Buffer (PBS, supplemented with 0.1% NP-40 and 1% protease inhibitors cocktail (P8340, Sigma)). The lysate was vortexed and 100 μl of whole cell extract (WCE) was transferred to another tube, which was kept on ice. The remaining lysate was centrifuged (45 s, 16 100 × g) to sediment the nuclei, and 100 μl of the supernatant, designated cytoplasmic fraction (Cyt), was transferred to a new tube, which was kept on ice. The rest of the supernatant was discarded, and the nuclear pellet was washed twice in 500 μl Fractionation Buffer and centrifuged (45 s, 16 100 × g). After the final centrifugation, the supernatant was discarded, and the nuclear pellet was resuspended in 200 μl Fractionation Buffer, and designated nuclear fraction (Nucl). WCE, Cyt and Nucl fractions were adjusted to contain 5 mM CaCl2 and 1 U/μl micrococcal nuclease (Thermo Fisher Scientific), then incubated at room temperature for 6 min, with intensive vortexing every minute, and finally placed on ice. Protein concentration in WCE and fractions was determined using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Protein (50 μg) from WCE, or from Cyt and Nucl fractions (corresponding to the same amount of cells), was resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a PVDF Immobilon®-FL transfer membrane (Merck), which was stained with Ponceau S and blocked using Odyssey® Blocking Buffer (TBS) (LI-COR) diluted 1:1 (v/v) in TBS. The membrane was then incubated with primary antibodies: mouse anti-FLAG (F1804, Sigma-Aldrich), rabbit anti-GAPDH (ab9485, Abcam), or rabbit anti-H3 (ab1791, Abcam), diluted in Odyssey® Blocking Buffer mixed 1:1 (v/v) with TBS, containing 0.05% Tween-20. The primary antibodies were detected with LI-COR secondary antibodies coupled with infrared fluorescent dyes, either goat anti-mouse IRDye® 680RD, or goat anti-rabbit IRDye® 800CW, according to the manufacturer's instructions and visualized using LI-COR Odyssey CLx Imaging System. When necessary, the same membrane was re-probed with different primary and secondary antibodies, without stripping. Precision Plus Protein Dual Color Standards (Bio-Rad), or Cameleon Duo Pre-stained Protein Ladder (LI-COR), were used to evaluate the size of polypeptides visualized by western blot. The signal intensities of the individual proteins were quantified using the software package of the Odyssey Imaging System. The METTL18-FLAG signal from the Cyt or Nucl fractions was calculated as the percentage of the total (Cyt + Nucl) signal.
Expression and purification of recombinant His-tagged METTL18
The ORF encoding human METTL18, or a METTL18 variant with its initial 94 amino acids deleted (Δ94 METTL18), either WT, H154A- or D217A-mutated, was cloned into the pET28a plasmid (Novagen) to contain N-terminal 6×His-tag. Plasmids were transformed into Escherichia coli strain BL21-CodonPlus (DE3)-RIPL (Agilent) and protein expression was induced overnight with 0.1 mM IPTG at 18°C. Isolation of 6×His-tagged proteins was performed at 4°C. Bacteria were lysed in Lysis Buffer 1 (50 mM Tris–HCl pH 7.4, 500 mM NaCl, 1% Triton X-100, 5% glycerol, 25 mM imidazole) supplemented with 1× Complete™ (EDTA-free) protease inhibitor cocktail (Roche) and 10 U/ml Benzonase nuclease (Sigma-Aldrich), and lysates were clarified by centrifugation (30 min, 45 000 × g). Cleared lysates were applied to Ni-NTA-agarose (Qiagen) equilibrated in Lysis Buffer 1. The resin was washed sequentially with Wash Buffer (50 mM Tris–HCl pH 7.4, 5% glycerol, 25 mM imidazole) supplemented with 0.5 M NaCl, then with Wash Buffer supplemented with 2.0 M NaCl, and finally again with Wash Buffer supplemented with 0.5 M NaCl. His-tagged proteins were eluted using Wash Buffer supplemented with 0.5 M NaCl and 200 mM imidazole, and buffer-exchanged to Storage Buffer (50 mM Tris–HCl pH 7.4, 100 mM NaCl, 10% glycerol) using centrifugal concentrators (Sartorius, 50 kDa cut-off). Proteins were aliquoted and stored at −20°C. Proteins were >90% pure as assessed by SDS-PAGE and Coomassie Blue staining. Protein concentration was determined using Pierce BCA Protein Assay Kit.
In vitro methyltransferase assay using [3H]AdoMet
To test MTase activity of recombinant METTL18 on cellular material, 10 μl reactions were assembled on ice containing: 1× Storage Buffer, ∼1 μg of recombinant Δ94 METTL18 (WT, H154A- or D217A-mutated), 0.5 μCi of [3H]AdoMet (Perkin-Elmer) ([AdoMet]total = 0.64 μM, specific activity = 78.2 Ci/mmol) and ∼50 μg protein from WCE, Cyt or Nucl fractions. Reactions were incubated at 30°C for 1 h, and then analyzed by SDS-PAGE and fluorography, similarly as previously described (35). When METTL18 activity was tested on ribosomes, ∼12 μg protein from isolated ribosomes was added and after 1 h incubation at 30°C, the 10 μl reactions were supplemented with 2 μg RNase A, and incubated at room temperature for further 15 min, before SDS-PAGE analysis. Typically, all fluorography experiments were performed three times, with similar results, and data from a representative experiment are shown.
Isolation of crude ribosome fraction
Cells were grown in 15-cm diameter plates until ∼80% confluent. After washing with PBS, cells were scraped in PBS, centrifuged (5 min, 500 × g) and frozen at −80°C until needed. Ribosome isolation was performed at 4°C, using a previously published protocol (36), with some modifications. Typically, cells from three plates were briefly thawed on ice, and lysed on ice, for 10 min, in 600 μl Ribosome Lysis Buffer (50 mM Tris–HCl, pH 7.4, 250 mM KCl, 5 mM MgCl2, 250 mM sucrose, 1% Triton X-100, 1% protease inhibitor cocktail (P8340, Sigma-Aldrich)). The lysate was centrifuged (10 min, 1000 × g) to sediment the nuclei, and then the supernatant was further clarified by centrifugation (10 min, 16 100 × g). The clarified supernatant was adjusted to the final concentration of 0.5 M KCl, using 2 M KCl added in small portions. The adjusted suspension, containing ribosomes, was subsequently carefully layered on top of 1.2 mL Sucrose Cushion (50 mM Tris-HCl, pH 7.4, 500 mM KCl, 5 mM MgCl2, 1 M sucrose) prepared in polycarbonate centrifuge tubes (13 × 51 mm, Beckman-Coulter), and centrifuged at 4°C (4 h, 250 000 × g), in an Optima Max ultracentrifuge using a MLS-50 rotor (Beckman-Coulter). The supernatant and the sucrose cushion were carefully removed, the ribosomal pellet was briefly rinsed with 250 μl cold Ribosome Wash Buffer (50 mM Tris–HCl, pH 7.4, 500 mM KCl, 5 mM MgCl2) and finally resuspended by intensive pipetting in 200–400 μl Ribosome Storage Buffer (50 mM Tris–HCl, pH 7.4, 25 mM KCl, 5 mM MgCl2). Ribosome solution was then cleared from undissolved particles by centrifugation (10 min, 16 100 × g), aliquoted and frozen at −80°C. Typically, isolated ribosomes had an A260/A280 ratio of ∼1.8. Ribosomes were quantified by RNA content, assuming that OD260 = 14 corresponds to 1 mg/ml of ribosomes, i.e. ∼0.5 mg/ml of ribosomal proteins (36).
GFP-trap immunoprecipitation
Flp-In™ T-REx™ 293-derived cells engineered to ectopically express RPL3-GFP or GFP, under Dox-induced promoter, were incubated with 1 μg/ml Dox for 24 h, harvested and lysed at 4°C, for 15 min, in RIPA buffer (Sigma-Aldrich), supplemented with 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and 1× Complete™ (EDTA-free) protease inhibitor cocktail. The lysate was cleared at 4°C by centrifugation (10 min, 16 100 × g), diluted in Dilution Buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA) and subjected to immunoprecipitation using GFP-Trap® agarose beads (Chromotek) at 4°C, for 2 h. After washing the beads with the Dilution Buffer, samples were further processed for MS analysis.
MS identification of proteins interacting with GFP-fusion proteins
Proteins isolated by GFP-Trap® immunoprecipitation, using RPL3-GFP (bait) or GFP only (control), were eluted and digested by adding 25 μl of 2 M urea, supplemented with 1 mM dithiothreitol (DTT) and 5 ng/μl trypsin. The resulting peptides were alkylated by adding 5 mM iodoacetamide, then desalted using StageTips (37) and analyzed using EASY-nLC 1200 system coupled to a QExactive HF-X mass spectrometer (Thermo Fisher Scientific) operating in a data-dependent acquisition mode (38). MS files were analyzed with MaxQuant (v1.6.0.1) (39), using the default settings except for the following variable modifications: mono-methylation (Lys, Arg, His), di-methylation (Lys, Arg) and tri-methylation (Lys). Proteins were quantified using the MaxLFQ algorithm (40). All analysis of proteomics data was performed using Perseus (version 1.6.5.0) (41).
MS analysis of proteins from subcellular fractions
Proteins present in Nucl/Cyt fractions and isolated ribosomes, or R246A/R249A-mutated RPL3-GFP isolated by GFP-Trap® immunoprecipitation, were resolved by SDS-PAGE, stained with Coomassie, and the portion of gel containing protein of interest was excised and subjected to in-gel proteolytic digestion using trypsin (Promega), chymotrypsin (Roche), Lys-C (Roche) or proteinase K (Sigma-Aldrich). Peptides resulting from proteolytic digestion were alkylated by adding 5 mM iodoacetamide, desalted using ZipTips, and analyzed by liquid chromatography system coupled to a QExactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific), similarly as described previously (35).
MS data were analyzed using in-house maintained human protein sequence databases using SEQUEST™ and Proteome Discoverer™ (Thermo Fisher Scientific), similarly as described previously (42). MS/MS spectra of peptides corresponding to methylated RPL3, were manually searched by Qual Browser (v2.0.7).
Amino acid analysis by LC-MS/MS
Cell pellets (from ∼80% confluent 15-cm diameter plates) or dried ribosomes (an equivalent of ∼0.6 mg of ribosomal proteins) were dissolved in 0.6–0.8 ml 6 M ultrapure HCl (Thermo Fisher Scientific) and incubated under vacuum at 120°C, for 24 h, using 1 ml vacuum hydrolysis tubes (Thermo Fisher Scientific). Alternatively, ribosomes were incubated at room temperature, for 15 min, with 0.2 mg/ml RNase A, then resolved by SDS-PAGE, and then the region of Coomassie Blue-stained gel corresponding to RPL3 was cut out from gel, chopped, vacuum-dried and finally transferred to 1 ml vacuum hydrolysis tubes for HCl-hydrolysis, as described above. After 24 h of HCl-hydrolysis, the resulting solutions were transferred to plastic tubes and evaporated overnight, at 65°C. Dry pellets were then dissolved in 2 ml ultrapure H2O, the solutions were filtered (0.45 μm) and again evaporated at 65°C. The remaining dry material was then analyzed by Bevital (Bergen, Norway) using LC-MS/MS (analysis platform C), to detect and quantify histidine, 1MH and 3MH.
Total RNA extraction and analysis by northern blot
Total RNA was extracted using TRI Reagent (Sigma-Aldrich). The aqueous phase was further extracted with phenol-chloroform-isoamylic alcohol (25:24:1; Sigma-Aldrich), and then with chloroform. Total RNA from the aqueous phase was recovered after precipitation with 2-propanol, and then rinsed twice in 70% ethanol. Total RNA was denatured for 10 min at 70°C, and analyzed by electrophoresis using 1.2% agarose gels, containing 1.2% formaldehyde and Tri/Tri buffer (30 mM triethanolamine, 30 mM tricine, pH 7.9) (3 μg total RNA/lane). RNA was then passively transferred to a Hybond N+ nylon membrane (GE Healthcare). To analyze small RNAs, total RNA was separated by electrophoresis using 6% polyacrylamide gels (acrylamide:bisacrylamide 19:1), containing 7 M urea and Tris-borate EDTA buffer and electro-transferred to a nylon membrane. After UV crosslinking, nylon membranes were pre-hybridized for 1–2 h, at 45–48°C, in 6× SSC, 5× Denhardt's solution, 0.5% SDS and 0.9 μg/ml of tRNA. Membranes were incubated overnight with 5’-radiolabeled oligonucleotide probes, whose sequences are indicated in Supplementary Table S2. After hybridization, membranes were washed twice, for 10 min, in 2× SSC, 0.1% SDS and once in 1× SSC, 0.1% SDS and then exposed to PhosphorImager Screens (Fuji), which were then scanned using the Typhoon Trio Imager System (GE Healthcare), and quantified using the MultiGauge software.
Measurement of polysome profiles
Cells were incubated for 10 min in Dulbecco's-modified Eagle's medium (DMEM) containing 100 μg/ml cycloheximide (CHX) (Sigma-Aldrich). All subsequent steps were performed in the presence of 100 μg/ml CHX. Cells were collected by centrifugation at 4°C (5 min, 300 × g) after treatment with trypsin and CHX. Cell pellets were mechanically disrupted with a Dounce homogenizer in 800 μl of buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT) containing CHX. The lysate was cleared from nuclei by centrifugation at 4°C (10 min, 1000 × g), and the upper cytoplasmic fraction was further clarified by two successive centrifugations at 10 000 × g, 5 min each. An aliquot containing 1 mg protein was loaded on a 10–50% (w/w) sucrose gradient prepared in buffer A, which was generated with a Gradient Master former (BioComp Instruments). The gradient was centrifuged at 4°C (2 h 15 min, 36 000 rpm), using a SW41Ti rotor (Optima L100XP ultracentrifuge; Beckman-Coulter), and the fractions were collected with a Density Gradient Fractionation System (Teledyne Isco) with continuous reading of OD254nm.
Ribosome profiling
Ribosome profiling libraries of HAP1 cells were generated in duplicates (WT cells) or triplicates (METTL18 KO cells). Cells were washed with ice-cold PBS, flash frozen in liquid nitrogen and transferred on ice. A total of 400 μl Lysis Buffer 2 (10 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 0.5 mM DTT, 0.5% sodium deoxycholate (w/v), 100 μg/ml CHX) was dripped onto the cells, which were scraped after lysis buffer had thawed. The lysate was cleared from nuclei by centrifugation at 4°C (5 min, 10 000 × g). Subsequently, ribosome protected fragments were generated by digesting 10 A260 units of the cleared lysate with 900 U RNase I (Thermo Fisher) at 22°C, for 1 h. The reaction was stopped by addition of 15 μl SUPERase In RNase inhibitor (Thermo Fisher) and loaded on a 10–50% (w/v) sucrose gradient prepared in Gradient Buffer (50 mM Tris–HCl pH 7.5, 50 mM NH4Cl, 12 mM MgCl2, 0.5 mM DTT, 100 μg/ml CHX), and centrifuged at 4°C (3 h, 35 000 rpm), using a TH-641 rotor (Thermo Scientific). The 80S ribosome fraction was collected and total RNA isolated. Subsequently, RNA was separated by electrophoresis using a 15% polyacrylamide gel containing 8 M urea (1 × TBE buffer), and 28–32 nt ribosome footprints were extracted from the gel. Finally, libraries were generated as described (43), but using adenylated 3′-adapters that contained four randomized nucleotides to reduce ligation biases (44). Libraries were mapped to the hg38 transcriptome (UCSC_knownGene), and subsequent analysis was performed using a custom script. A-site codon occupancy was determined as previously described (45), while excluding the first 15 nt of each ORF to reduce position-specific biases. About 29–31 nt long reads starting in the 0-frame were used, and the A-site defined as positions 15–17 nt from the start of the read. Occupancy was normalized to the adjacent codons in the +1, +2 and +3 positions. Ribosome occupancy at the +1 position (18–20 nt from the start of the read) was normalized by occupancy in the +2, +3 and +4 positions. DESeq2 using a p-adjusted threshold of 0.05, and a log2 fold change threshold of 0.5, was used for differential gene translation analysis of ribosome protected fragments (46). Gene ontology (GO) term analysis was performed using Gorilla (47).
Statistical analysis
The independent two-sample Student's t-test was used to evaluate the probability (p-value) that the means of two populations are not different.
RESULTS
Human METTL18 is an evolutionarily conserved 7BS MTase containing hallmark motifs
The function of METTL18 has hitherto remained elusive, and it has been unclear whether METTL18 is an ortholog of yeast Hpm1. Thus, we have taken an unbiased approach to elucidate the role of this enzyme. The crystal structure of human METTL18 in complex with S-adenosylhomocysteine (PDB ID: 4RFQ) shows the presence of seven β-strands forming a twisted β-sheet, characteristic of 7BS MTases. Protein sequence searches indicated that putative METTL18 orthologs are present in all eukaryotic organisms, including animals, fungi and plants (Supplementary Figure S1). An alignment of METTL18 sequences from various species revealed conserved motifs that are hallmarks of 7BS MTases (Motif I, Post I and Motif II), as well as the so-called ‘DXXY-motif‘ (consensus D/E-X-X-Y/F) shared by MTF16 members (19). While the MTase domain of METTL18 is well conserved, its N-terminal region is not, and can vary in size, being particularly short in some organisms, such as Caenorhabditis elegans (Supplementary Figure S1), indicating that it may not be strictly required for enzymatic activity.
METTL18 contains a non-classical NLS and localizes to nucleoli
To elucidate the function of METTL18, we studied its intracellular localization. For this, HeLa cells were transfected to transiently express a METTL18-GFP fusion protein. Live cell imaging revealed that METTL18-GFP was present both in the cytoplasm and the nucleus, but accumulated in the nucleus, particularly in nuclear foci reminiscent of nucleoli. Indeed, these foci co-localized with RPL3, which is known to accumulate in nucleoli and has been used as a marker for this organelle (Figure 1A and B) (32,48). Nuclear proteins often contain a positively charged NLS, which confers their nuclear localization. We found that the sequence PDVKRCR (amino acids 247–253 in METTL18), which is localized to a region of undetermined three-dimensional structure, matched the consensus of Class 2 NLSs (P/R-X-X-K-R-(ˆDE)-K/R; X, any amino acid; ˆDE, any amino acid except D or E) (49), suggesting it may represent a putative NLS. This sequence is highly conserved in vertebrates, but not in other animals, fungi or plants (Supplementary Figure S1) and to test whether it represents a functional NLS, two positively charged residues (K250 and R251) were replaced by glycine (K250G/R251G) in a METTL18-GFP construct. Mutant METTL18-GFP did not show accumulation in the nucleus, in contrast to the WT protein (Figure 1A). Moreover, a fusion protein between GFP and the NLS-containing portion of METTL18 (amino acids 233–265) accumulated preferentially in the nucleus, but was excluded from the nucleoli (Figure 1A and B), whereas the corresponding K250G/R251G-mutated protein showed uniform distribution between the nucleus and the cytoplasm (Figure 1A).

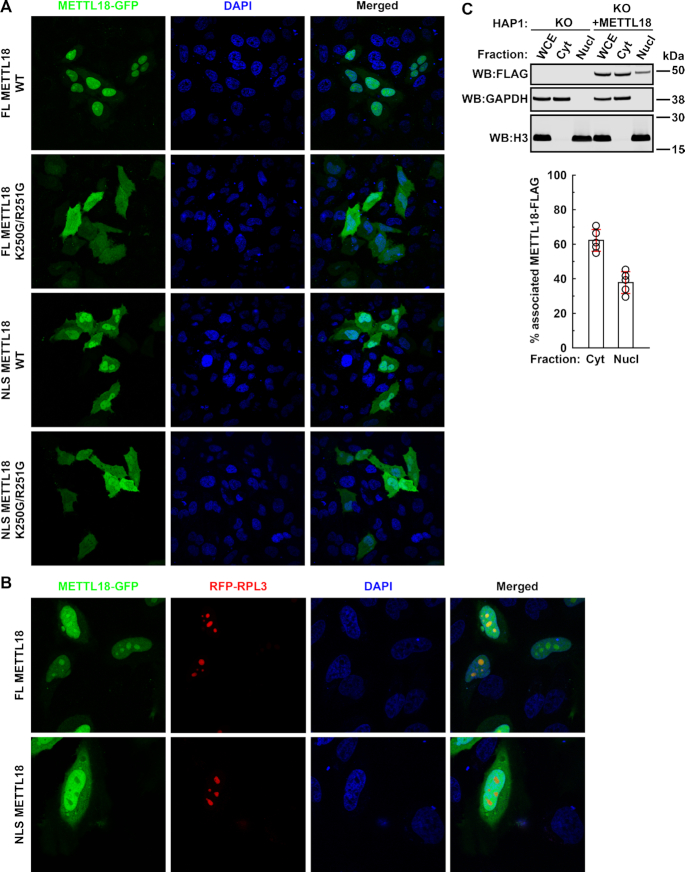
Human METTL18 contains a functional NLS and accumulates in nucleoli. (A) Intracellular localization of METTL18-GFP fusion proteins. HeLa cells were transiently transfected with plasmids encoding variants of METTL18-GFP (METTL18 fused to the N-terminus of EGFP), either METTL18 full length (FL) or a fragment consisting of amino acids 233–265 (NLS), and either WT or K250G/R251G-mutated. Shown are representative live cell images of GFP (green) and nuclear (DAPI; blue) staining. (B) Intracellular localization of METTL18-GFP and RFP-RPL3 fusion proteins. HeLa cells were transiently co-transfected with plasmids encoding RFP-RPL3 and METTL18-GFP variants, either FL or NLS. Shown are representative live cell images of GFP (green), RFP (red) and nuclear (DAPI; blue) staining. (C) Subcellular distribution of METTL18-FLAG. METTL18 KO HAP1 cells were complemented with METTL18 by stable transfection with a plasmid encoding METTL18-FLAG (for details see Supplementary Figure S2). Cells were lysed, and the WCE was fractionated into cytoplasmic (Cyt) and nuclear (Nucl) fractions, which were analyzed by SDS-PAGE and western blotting (WB:), and probed with anti-FLAG antibody. Expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and histone H3 is shown as loading and specificity control for Cyt and Nucl fractions, respectively. Shown are results from a representative experiment (top), and the quantitation of METTL18-FLAG signal associated with Cyt and Nucl fractions, based on results from five independent experiments (bottom). Shown are average values, with error bars (red) representing standard deviation.
To independently confirm the nuclear localization of METTL18 observed by microscopy, HAP1 cells with METTL18 gene KO were complemented with ectopically expressed METTL18-FLAG (Supplementary Figure S2), and then fractionated into cytoplasmic and nuclear fractions. Immunoblotting, using anti-FLAG antibodies, revealed that ∼60% of METTL18-FLAG was associated with the cytosolic fraction, while ∼40% was associated with the nuclear fraction (Figure 1C). In summary, our results show that human METTL18 contains a Class 2 NLS that confers its localization to the nucleus, where it accumulates in nucleoli.
METTL18 is a protein methyltransferase
Several human MTases can, in the recombinant form, directly methylate their respective substrates in a cell extract. Previously, we have successfully used extracts from MTase KO cells, where the relevant substrates are found in the unmethylated form (33,50–52). Thus, we set out to investigate recombinant METTL18, containing an N-terminal 6×His-tag, for such activity. We were unable to express full-length METTL18 in E. coli. However, a variant lacking the initial 94 residues (Δ94 METTL18), thereby matching the size of the METTL18 homolog from C. elegans, was efficiently expressed and purified. This recombinant, truncated METTL18 protein was tested for MTase activity using a cell extract from HAP1 METTL18 KO cells in the presence of [3H]AdoMet, allowing the detection of [3H]-methylated proteins by fluorography as distinct bands in SDS-PAGE. Also, we performed such experiments with the isolated cytoplasmic or nuclear fractions from these cells (Figure 2A). In these experiments, we observed several [3H]-labeled proteins but their methylation was typically METTL18-independent, likely originating from human MTases present in the extract. The exception was a ∼36 kDa protein representing automethylated recombinant METTL18, and a ∼46 kDa protein, whose methylation was dependent on the added recombinant METTL18 (Figure 2A). The [3H]-labeled ∼46 kDa protein was observed both in the cytoplasmic and the nuclear fractions, but could not be detected above the strong METTL18-independent background in the WCE (Figure 2A). We also noticed that the ∼46 kDa protein was present in the insoluble part of the nuclear fraction, while the ∼36 kDa band, i.e. auto-methylated METTL18, was soluble (Supplementary Figure S3). Reassuringly, the ∼46 kDa protein was not methylated by the putatively inactive D217A-mutated METTL18 (Figure 2B and Supplementary Figure S1). (Note: Asp-217, which is part of motif Post I, is putatively necessary for AdoMet-binding, and the corresponding residue was shown to be required for activity of similar 7BS MTases (19,21,30,33,51,53)). This indicates that methylation of the ∼46 kDa protein is directly catalyzed by METTL18, rather than by a contaminating E. coli–derived MTase. Finally, the ∼46 kDa protein did not become methylated when a nuclear fraction from HAP1 WT, rather than METTL18 KO cells, was used (Figure 2C), indicating that it represents a bona fide METTL18 substrate that is fully methylated in cells expressing endogenous METTL18.
![Recombinant METTL18 methylates a nuclear-associated protein in vitro. (A) METTL18-dependent protein methylation in HAP1 METTL18 KO cell extract. Cells were lysed and the WCE was fractionated into cytoplasmic (Cyt) and nuclear (Nucl) fractions. A total of 50 μg of protein from WCE, or the corresponding amount from the indicated fractions, was incubated with [3H]AdoMet (0.5 μCi) and Δ94 METTL18 (3 μM), and then analyzed by SDS-PAGE and transferred to a membrane. Incorporation of [3H]-methyl into proteins was visualized by fluorography (top) of a Ponceau S-stained membrane (middle), which was additionally probed for expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and histone H3 (bottom) as loading and specificity controls for Cyt and Nucl fractions, respectively. Red arrow indicates METTL18-dependent methylation of ∼46 kDa protein present in Nucl and Cyt fractions. (B) D217A mutation of METTL18 abrogates its enzymatic activity. A total of 40 μg of protein from nuclear fraction of KO cells was incubated with [3H]AdoMet and Δ94 METTL18, either WT or D217A-mutated and [3H]-methyl incorporation into proteins was visualized by fluorography (top) as in (A). Ponceau S-stained membrane (bottom) is shown as loading control. (C) The ∼46 kDa protein present in nuclear fraction is nearly fully methylated in HAP1 WT cells. A total of 40 μg of protein from nuclear fraction of WT or KO cells was incubated with [3H]AdoMet and Δ94 METTL18, and [3H]-methyl incorporation into proteins was visualized by fluorography (top) as in (A). Ponceau S-stained membrane (middle) and expression of histone H3 (bottom) are shown as loading controls.](/dataresources/secured/content-1766021126690-3c7c1d7b-e526-4bbf-99bb-443e00260158/assets/gkab088fig2.jpg)
Recombinant METTL18 methylates a nuclear-associated protein in vitro. (A) METTL18-dependent protein methylation in HAP1 METTL18 KO cell extract. Cells were lysed and the WCE was fractionated into cytoplasmic (Cyt) and nuclear (Nucl) fractions. A total of 50 μg of protein from WCE, or the corresponding amount from the indicated fractions, was incubated with [3H]AdoMet (0.5 μCi) and Δ94 METTL18 (3 μM), and then analyzed by SDS-PAGE and transferred to a membrane. Incorporation of [3H]-methyl into proteins was visualized by fluorography (top) of a Ponceau S-stained membrane (middle), which was additionally probed for expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and histone H3 (bottom) as loading and specificity controls for Cyt and Nucl fractions, respectively. Red arrow indicates METTL18-dependent methylation of ∼46 kDa protein present in Nucl and Cyt fractions. (B) D217A mutation of METTL18 abrogates its enzymatic activity. A total of 40 μg of protein from nuclear fraction of KO cells was incubated with [3H]AdoMet and Δ94 METTL18, either WT or D217A-mutated and [3H]-methyl incorporation into proteins was visualized by fluorography (top) as in (A). Ponceau S-stained membrane (bottom) is shown as loading control. (C) The ∼46 kDa protein present in nuclear fraction is nearly fully methylated in HAP1 WT cells. A total of 40 μg of protein from nuclear fraction of WT or KO cells was incubated with [3H]AdoMet and Δ94 METTL18, and [3H]-methyl incorporation into proteins was visualized by fluorography (top) as in (A). Ponceau S-stained membrane (middle) and expression of histone H3 (bottom) are shown as loading controls.
Identification of RPL3 as target of METTL18
To identify the ∼46 kDa protein that exhibited strong, METTL18-dependent methylation, the insoluble part of the nuclear fraction from HAP1 cells was resolved by SDS-PAGE and the region of the gel matching the ∼46 kDa protein was excised, trypsin-digested, and its protein content analyzed by MS. Several nuclear proteins were identified in the relevant region (Supplementary Table S3). Strikingly, one of the top hits was RPL3, the counterpart of yeast Rpl3, the reported target of yeast Hpm1 (10). RPL3 was also found among the top hits found in the ∼46 kDa region of the cytoplasmic fraction (Supplementary Table S4). Taken together, this suggests that RPL3 is a target of human METTL18.
A previous interactomics experiment, using tagged METTL18 as bait, identified RPL3 as a main interactant (18). To further investigate the potential interaction between RPL3 and METTL18, we used a HEK-293-derived cell line to ectopically express a RPL3-GFP fusion protein under the control of a doxycycline-inducible promoter, and observed that RPL3-GFP was enriched in nucleoli (Supplementary Figure S4). We affinity purified RPL3-GFP from these cells using a GFP-affinity resin, and the co-purifying proteins were quantified by MS, and compared to those isolated from a cell line overexpressing GFP only, thereby identifying proteins that specifically interacted with RPL3, and excluding proteins binding to the beads or the GFP affinity tag. Notably, we found that there were two MTases among the 74 proteins that were most strongly (>16-fold) enriched by RPL3, namely METTL18 and the tRNA methyltransferase TARBP1, further corroborating that METTL18 and RPL3 interact (Figure 3A; Supplementary Tables S5 and S6). Since TARBP1 has been reported to bind RNA (54), one may speculate that the observed interaction between RPL3 (which also is an RNA-binding protein) and TARBP1 is indirect and RNA-mediated.
![METTL18 interacts with RPL3 in cells and methylates ribosomal RPL3 in vitro. (A) RPL3 interactome. Flp-In™ T-REx™ 293 cells, expressing GFP or RPL3-GFP under doxycycline-inducible promoter, were treated for 24 h with doxycycline (1 μg/ml). GFP and RPL3-GFP were isolated using GFP-Trap®, trypsin-digested and analyzed by MS to identify co-purifying proteins. Volcano plot represents proteins enriched by RPL3-GFP versus GFP alone. The solid curved line represents the significance cut-off (FDR = 0.01 and s0 = 0.1). The RPL3 (bait), METTL18 and TARBP1 (interactants) are indicated, while other identified proteins (n = 1576) are listed in Supplementary Table S5. Strongly (>16-fold) enriched proteins (n = 74) are indicated by the dashed rectangle, and are listed in Supplementary Table S6. (B andC) Recombinant METTL18 methylates ribosomal RPL3 in vitro. (B) METTL18 WT, but not the inactive mutant (D217A), methylates ribosomal RPL3. A total of 12 μg of ribosomal proteins isolated from HAP1 METTL18 KO cells was incubated with [3H]AdoMet (0.5 μCi) and Δ94 METTL18 (3 μM), either non-mutated or D217A-mutated and then analyzed by SDS-PAGE and transferred to a membrane. Incorporation of [3H]-methyl into proteins was visualized by fluorography (top) of a Ponceau S-stained membrane (bottom) shown as loading control. Arrows show the positions of RPL3, RPL4 and METTL18. (C) Ribosomal RPL3 isolated from WT cells is nearly fully methylated. 12 μg of ribosomal proteins isolated from HAP1 WT or METTL18 KO cells was incubated with [3H]AdoMet and Δ94 METTL18, and analyzed as in (A). Incorporation of [3H]-methyl into proteins was visualized by fluorography (top), and the Ponceau S-stained membrane (bottom) shown as loading control.](/dataresources/secured/content-1766021126690-3c7c1d7b-e526-4bbf-99bb-443e00260158/assets/gkab088fig3.jpg)
METTL18 interacts with RPL3 in cells and methylates ribosomal RPL3 in vitro. (A) RPL3 interactome. Flp-In™ T-REx™ 293 cells, expressing GFP or RPL3-GFP under doxycycline-inducible promoter, were treated for 24 h with doxycycline (1 μg/ml). GFP and RPL3-GFP were isolated using GFP-Trap®, trypsin-digested and analyzed by MS to identify co-purifying proteins. Volcano plot represents proteins enriched by RPL3-GFP versus GFP alone. The solid curved line represents the significance cut-off (FDR = 0.01 and s0 = 0.1). The RPL3 (bait), METTL18 and TARBP1 (interactants) are indicated, while other identified proteins (n = 1576) are listed in Supplementary Table S5. Strongly (>16-fold) enriched proteins (n = 74) are indicated by the dashed rectangle, and are listed in Supplementary Table S6. (B andC) Recombinant METTL18 methylates ribosomal RPL3 in vitro. (B) METTL18 WT, but not the inactive mutant (D217A), methylates ribosomal RPL3. A total of 12 μg of ribosomal proteins isolated from HAP1 METTL18 KO cells was incubated with [3H]AdoMet (0.5 μCi) and Δ94 METTL18 (3 μM), either non-mutated or D217A-mutated and then analyzed by SDS-PAGE and transferred to a membrane. Incorporation of [3H]-methyl into proteins was visualized by fluorography (top) of a Ponceau S-stained membrane (bottom) shown as loading control. Arrows show the positions of RPL3, RPL4 and METTL18. (C) Ribosomal RPL3 isolated from WT cells is nearly fully methylated. 12 μg of ribosomal proteins isolated from HAP1 WT or METTL18 KO cells was incubated with [3H]AdoMet and Δ94 METTL18, and analyzed as in (A). Incorporation of [3H]-methyl into proteins was visualized by fluorography (top), and the Ponceau S-stained membrane (bottom) shown as loading control.
We were unable to show methylation of recombinant RPL3 by recombinant METTL18 in vitro, and to obtain more direct evidence that RPL3 is methylated by METTL18, we isolated ribosomes from HAP1 METTL18 KO cells and used those, together with [3H]AdoMet and recombinant METTL18, as substrate in an in vitro MTase reaction. We found that a single band, identified by MS as RPL3, was indeed [3H]-methylated by METTL18 WT, but not by the catalytically inactive METTL18 mutant, D217A (Figure 3B). Moreover, in vitro methylation of RPL3 was only observed using ribosomes from METTL18 KO cells, and not with WT ribosomes (Figure 3C), suggesting that RPL3 is nearly fully methylated in WT cells. Furthermore, ribosomes isolated from WT and METTL18 KO cells showed similar amounts of RPL3, indicating that lack of METTL18 does not impair RPL3 incorporation into ribosomes (Figure 3C).
Human METTL18 methylates ribosomal RPL3 at His-245
To further investigate METTL18-dependent methylation of RPL3 in cells, we digested ribosomal RPL3 with various proteases, followed by MS analysis in search of methylated RPL3-derived peptides. No methylated peptide could be identified when RPL3 was digested with commonly used proteases, such as trypsin or chymotrypsin, while digestion with Lys-C generated peptide KLPRKTHRGLRK (residues 239–250) that was found monomethylated in HAP1 WT cells, but the exact localization of the methyl group within this peptide could not be resolved by MS/MS analysis (Supplementary Figure S5 and data not shown). However, digestion of RPL3 with proteinase K generated a shorter peptide KLPRKTHRGL (residues 239–248) that was detected as fully monomethylated in WT cells, but completely unmethylated in METTL18 KO cells (Figure 4A). MS/MS analysis of this peptide revealed His-245 as the site of monomethylation (Figure 4B). Importantly, in KO cells complemented with METTL18-FLAG (Supplementary Figure S2), methylation of His-245 was largely restored, while in KO cells complemented with D217A-mutated METTL18-FLAG, His-245 remained completely unmethylated (Figure 4A).
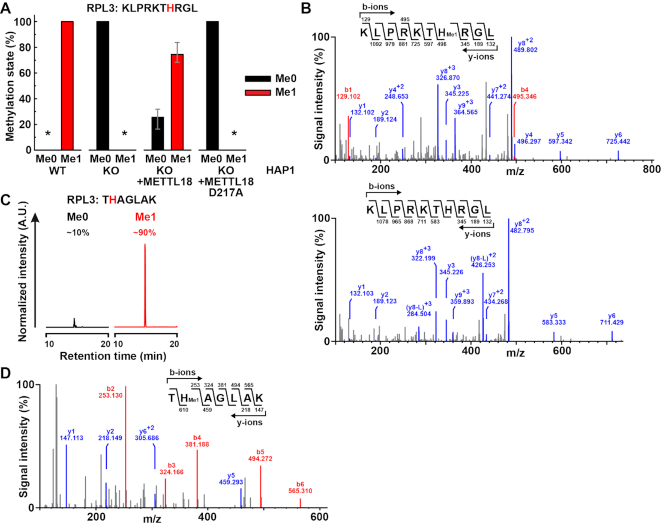
Human RPL3 is monomethylated at His-245 by METTL18 in cells. (A) Complementation of METTL18 KO cells with METTL18 restores monomethylation of RPL3 at His-245. Ribosomes isolated from HAP1 WT, METTL18 KO or KO cells expressing FLAG-tagged METTL18, either non-mutated or D217A-mutated (see Supplementary Figure S2) were resolved by SDS-PAGE. The portion of gel containing RPL3 was proteinase K-digested and analyzed by MS. Shown are the mean relative intensities of MS signals, gates for different methylation states of the indicated, RPL3-derived peptide, with His-245 marked in red. Three independent analyses of each cell line were performed. Error bars represent range of values. An asterisk indicates that the relevant peptide was not detected. (B) MS/MS fragmentation spectra show monomethylation of RPL3 at His-245 in HAP1 WT cells (top) and absence of methylation in METTL18 KO cells (bottom). (C andD) R246A/R249A-mutated RPL3 is monomethylated at His-245 in HEK293-derived cells. Flp-In™ T-REx™ 293 (HEK293) cells expressing R246A/R249A-mutated RPL3-GFP under doxycycline-inducible promoter, were treated for 24 h with doxycycline (1 μg/ml) and the mutated RPL3-GFP fusion protein was isolated using GFP-Trap®, and resolved by SDS-PAGE. The portion of gel corresponding to RPL3-GFP was Lys-C-digested and analyzed by MS. (C) Shown are representative, normalized extracted ion chromatograms, gated for different methylation states of RPL3-derived, Lys-C-generated peptides, encompassing residues 244–250 of R246A/R249A-mutated RPL3, present in cells, with His-245 marked in red. Percentages indicate the relative area under each peak. A.U., arbitrary units. (D) MS/MS fragmentation spectrum shows monomethylation of R246A/R249A-mutated RPL3 at His-245 in HEK293-derived cells.
Our attempts to detect RPL3 methylation by MS were initially (prior to using proteinase K) unsuccessful. We suspected that the high number of Lys and Arg residues in the sequence surrounding the putative methylation site His-245, (corresponding to the Hpm1 target site His-243 in yeast Rpl3) was unfavorable regarding charge state and/or protease digestion. Thus, we generated HEK-293-derived cells that ectopically expressed a R246A/R249A-mutated RPL3-GFP fusion protein from a doxycycline-inducible promoter (Supplementary Figure S4), and then isolated the mutated RPL3-GFP using GFP affinity resin. R246A/R249A-mutated RPL3-GFP was then subjected to digestion with Lys-C, and analyzed by MS. We identified the peptide THAGLAK (residues 244–250 of R246A/R249A-mutated RPL3) and found it to be ∼90% monomethylated at His-245 (Figure 4C and D). This further corroborates His-245 as the METTL18 target site, and also indicates that METTL18 shows some tolerance for amino acid substitutions in the vicinity of this site.
METTL18 mediates 3MH modification of RPL3
Next, we set out to determine which isomer of methylhistidine, 1MH or 3MH, is introduced by METTL18. To this end, we isolated total and ribosomal proteins from WT and METTL18 KO HAP1 cells, and then subjected these to acid hydrolysis into amino acids, which were analyzed and quantitated by LC-MS/MS. For total protein, WT and KO cells showed no significant differences in the content of 1MH and 3MH (Figure 5A and data not shown). For the ribosomes, however, the level of 3MH was dramatically reduced in the KO relative to the WT, while 1MH was not detected (Figure 5A and data not shown). Since we had already identified RPL3 as target of METTL18, we determined the 3MH content specifically in ribosome-associated RPL3. Clearly, the relative content of 3MH was much higher (>10×) in isolated RPL3 compared to bulk ribosomes, directly demonstrating that RPL3 is 3MH-modified. Moreover, 3MH was virtually absent in RPL3 from METTL18 KO cells, and was partially restored by complementation with enzymatically active METTL18, but not by the enzymatically inactive mutant (Figure 5B). In summary, the above results demonstrate that RPL3 contains 3MH, and that this modification depends on the enzymatic activity of METTL18.
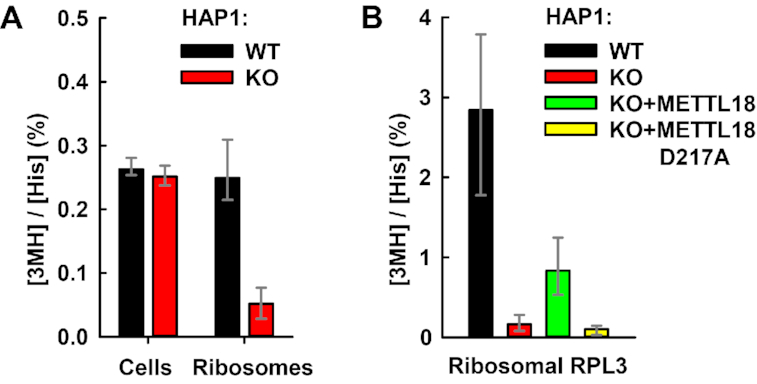
METTL18 introduces 3MH modification in cellular RPL3. (A) METTL18 KO abrogates 3MH modification in ribosomes. Whole cells or ribosomes isolated from HAP1 WT or METTL18 KO cells were subjected to HCl-mediated hydrolysis into amino acids, and the content of His and 3MH was determined by LC-MS/MS. Shown is the average relative amount of 3MH detected in indicated cells and ribosomes, expressed as percentage of total His. Error bars represent range of values from 3 (cells) or 4 (ribosomes) independent analyses. (B) METTL18 enzymatic activity is specifically required for 3MH modification of RPL3. Ribosomes isolated from HAP1 WT, METTL18 KO or KO cells expressing FLAG-tagged METTL18, either non-mutated or D217A-mutated (see Supplementary Figure S2) were resolved by SDS-PAGE. The region of gel containing RPL3 was excised and subjected to amino acid analysis, as in (A). Shown is the average relative amount of 3MH detected in RPL3, expressed as percentage of total His. Error bars represent range of values from four to five independent analyses.
METTL18 is automethylated at His-154
Fluorography experiments showed that recombinant METTL18 is intensely [3H]-labeled when incubated with [3H]AdoMet (Figures 2 and 3), indicating automethylation. To examine whether automethylation also occurs during METTL18 expression in bacteria, and to determine the modified site(s), recombinant METTL18 was analyzed by MS in search for methylated peptides. Indeed, the peptide SFSSHSDLITGVYEGGLK, corresponding to residues 150–167 of METTL18, was found to be almost fully monomethylated in WT recombinant METTL18 (>95%), but not in its D217A-mutated counterpart, where it was completely unmethylated (Figure 6A). MS/MS analysis revealed His-154 of METTL18 as the site of methylation (Figure 6B). Importantly, we found that the endogenous METTL18 that was pulled down by RPL3-GFP during anti-GFP affinity purification (Figure 3A), was exclusively detected as the monomethylated (at His-154) form (Figure 6A and Supplementary Figure S6), demonstrating that this modification occurs in human cells, and not only during expression/purification of recombinant METTL18 from E. coli.
![METTL18 is monomethylated at His-154. (A) Recombinant and cellular METTL18 is monomethylated at His-154. Recombinant Escherichia coli-expressed Δ94 METTL18, either WT or D217A-mutated, was resolved by SDS-PAGE and the portion of gel containing METTL18 was trypsin-digested, and analyzed by MS. Alternatively, Flp-In™ T-REx™ 293 (HEK293) cells, expressing RPL3-GFP under doxycycline-inducible promoter, were treated for 24 h with doxycycline (1 μg/m). RPL3-GFP was isolated using GFP Trap®, trypsin-digested and co-purifying proteins were identified by MS, similarly as in Figure 3A. Endogenous METTL18 was identified as an RPL3 interactant (see Figure 3A). Shown are the relative MS signals corresponding to the unmetylated or monomethylated versions of the indicated, METTL18-derived peptide, with His-154 marked in red. Two (E. coli-derived METTL18) or six (HEK293-derived METTL18) independent analyses were performed. Shown are the mean relative intensities of MS signals, and error bars represent range of values. An asterisk indicates that the relevant peptide was not detected. (B) MS/MS fragmentation spectra show monomethylation of His-154 in recombinant METTL18 WT (top) and absence of methylation in enzyme-inactive D217A-mutated METTL18 (bottom). (C) H154A mutation inhibits METTL18-dependent methylation of RPL3. A total of 40 μg of protein from nuclear fraction of HAP1 METTL18 KO cells was incubated with [3H]AdoMet (0.5 μCi) and Δ94 METTL18 (3 μM), either WT or H154A-mutated, and then analyzed by SDS-PAGE and transferred to a membrane. Incorporation of [3H]-methyl into proteins was visualized by fluorography (top) of Ponceau S-stained membrane (bottom), shown as loading control. Arrows indicate the position of automethylated METTL18 and the ∼46 kDa protein substrate, previously identified as RPL3.](/dataresources/secured/content-1766021126690-3c7c1d7b-e526-4bbf-99bb-443e00260158/assets/gkab088fig6.jpg)
METTL18 is monomethylated at His-154. (A) Recombinant and cellular METTL18 is monomethylated at His-154. Recombinant Escherichia coli-expressed Δ94 METTL18, either WT or D217A-mutated, was resolved by SDS-PAGE and the portion of gel containing METTL18 was trypsin-digested, and analyzed by MS. Alternatively, Flp-In™ T-REx™ 293 (HEK293) cells, expressing RPL3-GFP under doxycycline-inducible promoter, were treated for 24 h with doxycycline (1 μg/m). RPL3-GFP was isolated using GFP Trap®, trypsin-digested and co-purifying proteins were identified by MS, similarly as in Figure 3A. Endogenous METTL18 was identified as an RPL3 interactant (see Figure 3A). Shown are the relative MS signals corresponding to the unmetylated or monomethylated versions of the indicated, METTL18-derived peptide, with His-154 marked in red. Two (E. coli-derived METTL18) or six (HEK293-derived METTL18) independent analyses were performed. Shown are the mean relative intensities of MS signals, and error bars represent range of values. An asterisk indicates that the relevant peptide was not detected. (B) MS/MS fragmentation spectra show monomethylation of His-154 in recombinant METTL18 WT (top) and absence of methylation in enzyme-inactive D217A-mutated METTL18 (bottom). (C) H154A mutation inhibits METTL18-dependent methylation of RPL3. A total of 40 μg of protein from nuclear fraction of HAP1 METTL18 KO cells was incubated with [3H]AdoMet (0.5 μCi) and Δ94 METTL18 (3 μM), either WT or H154A-mutated, and then analyzed by SDS-PAGE and transferred to a membrane. Incorporation of [3H]-methyl into proteins was visualized by fluorography (top) of Ponceau S-stained membrane (bottom), shown as loading control. Arrows indicate the position of automethylated METTL18 and the ∼46 kDa protein substrate, previously identified as RPL3.
Conceivably, the observed automethylation at His-154 may play a role in regulating or optimizing METTL18 activity. To investigate the importance of residue His-154 for METTL18 function, we generated a corresponding mutation, H154A and tested the MTase activity of the mutant protein by the fluorography-based assay, using the nuclear fraction from HAP1 METTL18 KO cells as source of unmethylated RPL3. We observed that H154A-mutated METTL18 was automethylated to a similar extent as the WT enzyme, but the methylation of the ∼46 kDa substrate, i.e. RPL3, was strongly reduced (Figure 6C). This indicates that automethylation of METTL18 can occur at sites other than His-154 (at least in the H154A-mutated protein), but that the integrity of His-154 is required for METTL18-mediated methylation of RPL3. In summary, our results show that METTL18 is stoichiometrically automethylated at His-154, and suggest that such automethylation may modulate METTL18 activity.
METTL18 ablation affects early stages of pre-rRNA processing
METTL18 was found to co-localize with the ribosomal protein RPL3 in nucleoli (Figure 1). This suggested that METTL18-mediated methylation of RPL3 may occur during ribosomal biogenesis in nucleoli, and potentially play an important role in this process. Ribosome biogenesis involves extensive processing of rRNA precursors (pre-rRNAs) into mature rRNA. The 18S, 5.8S and 28S rRNAs originate from the same primary transcript, 47S pre-rRNA, which is cleaved by various endoribonucleases at specific cleavage sites located within external transcribed spacers (5′ETS and 3′ETS) and internal transcribed spacers (ITS1 and ITS2; Figure 7A; reviewed in (25)). Various cleavage products are further trimmed from both ends by exoribonucleases to produce the mature rRNAs (Figure 7A). After rapid cleavage at distal sites A’ and 02, which gives rise to 45S pre-rRNA, various intermediates are formed, determined by the relative efficiency of endonuclease cleavage at various sites (55). We therefore set out to investigate the effect of METTL18 ablation on pre-rRNA processing. To this end, we isolated total RNA from WT and METTL18 KO HAP1 cells, and analyzed it by gel electrophoresis and Northern blotting. Using specific probes, we detected various pre-rRNAs, and compared their steady-state levels by quantifying the observed changes (Figure 7B and C). We found that the amount of 47S and 45S pre-rRNAs were similar in WT and KO cells, indicating that METTL18 ablation does not affect transcription and initial processing at sites A’ and 02. However, we detected a significant decrease in 41S pre-rRNA as well as its precursor 41S+3’, concomitant with an increase in 30S pre-rRNA in KO compared to WT cells, whereas the levels of 32S, 21S and 12S pre-RNAs remained unchanged (Figure 7B and C). These alterations in 30S and 41S rRNA levels suggest that cleavage at sites A0 and 1 is impaired in the KO cells (see ref. 25 for an outline of the pre-rRNA processing pathway in human cells). The observed changes in these pre-rRNA intermediates in METTL18 KO cells were, however, not accompanied by significant changes in the steady-state levels of downstream intermediates (21S, 18S-E) or of mature rRNAs, including 18S, 28S and 5.8S rRNAs, which were similar in WT and KO cells (Figure 7C and D). This indicates that 5'-ETS processing is not blocked, but rather slowed down in the KO cells. In summary, the above results indicate that METTL18 ablation, and consequently lack of RPL3 monomethylation at His-245, affects the dynamics of pre-rRNA processing, but has no apparent effect on the amount of mature rRNAs.
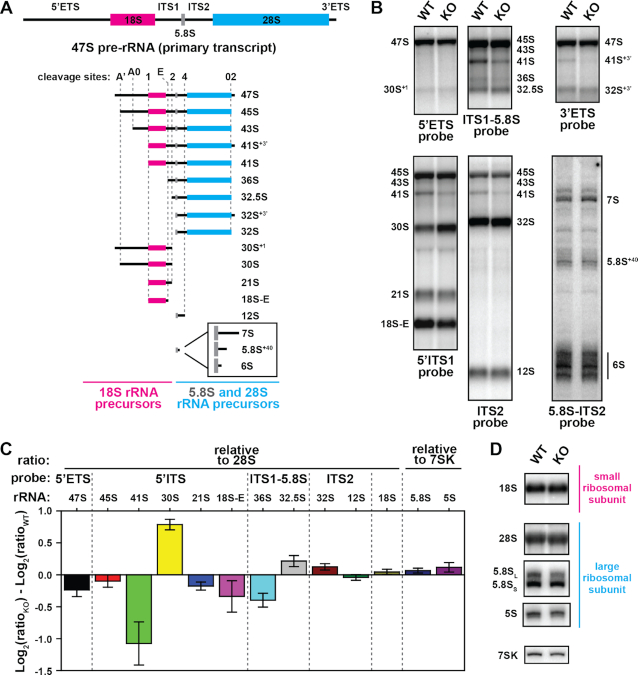
Comparison of pre-rRNA processing in HAP1-derived cells. (A) A schematic view of the human rRNA primary transcript, 47S pre-rRNA (top) and its maturation intermediates (bottom). The sequences corresponding to 18S (magenta), 28S (cyan) and 5.8S (gray) rRNAs are indicated, flanked by external (5’ETS, 3’ETS) and internal (ITS1, ITS2) transcribed spacers indicated by solid black lines. Position of main endoribonuclease cleavage sites is indicated by dotted gray lines. Short pre-5.8S precursors generated by exoribonuclease trimming of 12S pre-rRNA are shown enlarged in the inset. (B–D) Steady-state pre-rRNA processing in WT and METTL18 KO cells. Total RNA was isolated from cells and separated on agarose gel, except for shorter rRNAs (5S, 5.8S) that were separated on polyacrylamide gel and then northern-blotted on membrane, which was probed using indicated DNA probes (for probe sequences, see Supplementary Table S2). (B) Visualization of pre-rRNA intermediates in WT and KO cells, detected as bands using indicated probes. (C) Quantitation of changes in band intensities observed in KO relative to WT cells. Shown are mean values normalized to either 28S rRNA or 7SK RNA (small non-coding RNA, not related to rRNA), with error bars representing S.D. (n = 3). (D) Visualization of 18S rRNA from small ribosomal subunit, and 28S, 5.8S and 5S rRNAs from large ribosomal subunit, present in WT and KO cells. The 7SK is shown as loading control.
METTL18 ablation affects translation dynamics
Methylation modification of human translation factors has been shown to significantly affect mRNA translation (35,50,51,56–58). Since we observed that METTL18 influences pre-rRNA processing, we reasoned that METTL18 may also influence other aspects of ribosome biogenesis and impact on translation. Therefore, we investigated whether the loss of METTL18 function affects mRNA translation. First, we used HAP1 WT and METTL18 KO cells and performed polysome profiling to assess global translation. Polysome profiling uses sucrose-gradient centrifugation of cytoplasmic extracts, to determine the amount of ribosomal subunits, monosomes and polysomes (Figure 8A). We observed that the amount of 80S ribosomes was similar in WT and METTL18 KO cells; however, the amounts of small subunits (40S), large subunits (60S) and polysomes were clearly decreased in the METTL18 KO (Figure 8A and B). This indicates a defect in formation of 40S subunits, and also suggests that KO cells have impaired translation initiation, i.e. loading of new ribosomes on mRNA, and/or increased translation speed.
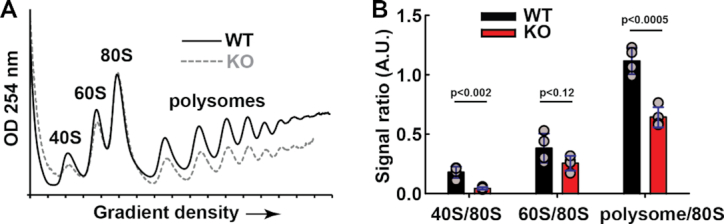
METTL18 ablation results in altered polysome profiles in HAP1-derived cells. (A) Polysome profiles of cytoplasmic fractions from WT and METTL18 KO cells. Cytoplasmic fraction was isolated from indicated cells, centrifuged in 10–50% sucrose gradient and OD254nm was measured across the gradient to show the distribution of small (40S) and large (60S) ribosomal subunits, monosomes (80S) and polysomes. Shown are representative profiles for WT (solid black) and METTL18 KO (dashed gray) cells, out of four independent experiments. (B) Quantitation of relative amounts of 40S, 60S, 80S and polysomes observed in WT and METTL18 KO cells, based on four replicates of polysome profiles obtained as in (A). Blue whiskers represent SD.
Second, to obtain a more detailed picture on translation dynamics, we performed ribosome profiling, a deep-sequencing-based method to analyze transcriptome-wide ribosome occupancy at sub-codon resolution. This allows quantification of ribosomal A-site occupancy of individual codons and determination of the relative translation rates of individual codons between two conditions (43). Also, this method can be used to assess translation at the gene level, and we found cellular processes that are linked to RNA metabolism, the cell cycle and metabolic processes to be upregulated in METTL18 KO relative to WT cells (Figure 9A), while cell signaling-related GO terms were significantly down-regulated (Figure 9B). We found that the relative A-site occupancy of the majority of codons did not differ between METTL18 KO and WT cells (Figure 9C). However, the codon GAA (Glu) showed a substantial increase in occupancy in METTL18 KO cells, i.e. corresponding to a decreased translation rate, whereas several codons showed decreased occupancy, i.e. corresponding to an increased translation rate, most markedly GGA (Gly) and GTT (Val) (Figure 9C). As a control, we also analyzed ribosome occupancy at the codon downstream of the A-site (i.e. a codon that has not yet been translated) and, reassuringly, we did not see any appreciable differences between the METTL18 KO and WT cells (Figure 9C). In summary, we found that METTL18 ablation and lack of RPL3 monomethylation at His-245 led to a global reduction of polysome formation and introduced codon- and gene-specific changes in translation.
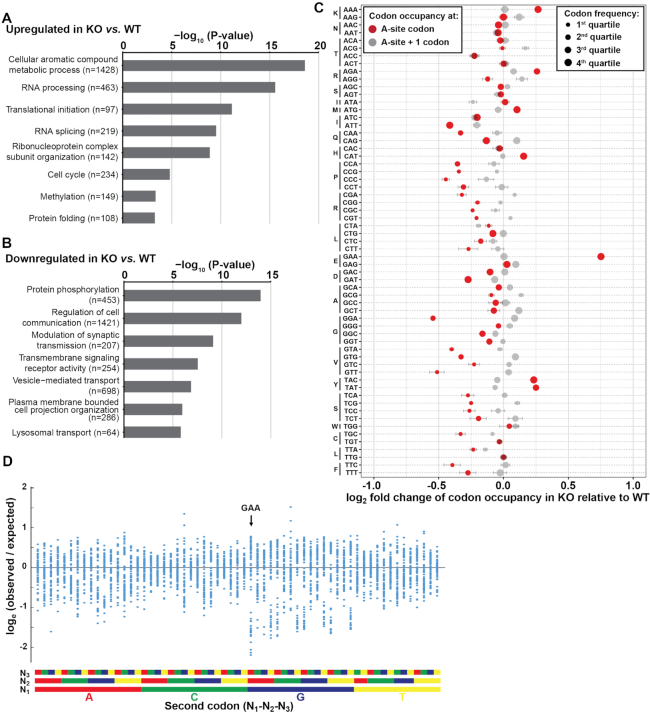
METTL18 ablation results in altered translation in HAP1-derived cells. (A) GO term analysis (Cellular Process) of genes significantly upregulated on the footprint level in METTL18 KO cells relative to WT cells. Number of genes matching the respective GO term category is indicated in brackets. (B) Similar as in (A) but for downregulated genes. (C) The relative ribosome occupancy of mRNA codons in METTL18 KO cells relative to WT cells. The occupancy at acceptor (A-site) is shown in red, and the occupancy at downstream, not yet translated, codon (+1-site) is shown in gray. Spread of data is indicated by horizontal whiskers (mean ± SD, n = 3). Circle size indicates the relative frequency of codons in quartiles (larger is more frequent). (D) GAA is frequently found as the second codon in underrepresented codon pairs. Shown are the observed versus the expected frequencies, expressed as the natural logarithm, of the 3721 (61 × 61) codon pairs present in the human ORFeome. The codon pairs are grouped according to the identity of the second codon. The data were taken from Supplementary Table S1 in Coleman et al. (62).
DISCUSSION
In the present work, we have firmly established that METTL18 is a histidine-specific MTase, and that it catalyzes the introduction of 3MH at His-245 in human RPL3. Consistent with this function, we found that METTL18 localizes to nucleoli, the sites of ribosome maturation, and that METTL18 KO affects rRNA maturation and mRNA translation. Given that METTL18 KO cells are viable, also in long-term culture, we conclude that METTL18 is a non-essential protein, which likely serves a role in optimizing ribosome biogenesis and function.
In general, MTF16 members appear to interact tightly with their substrates, which in several cases were first identified through screens for MTase interactants (19,21,56,59). This appears to be the case also for METTL18, because RPL3 was previously identified as a METTL18 interactant (18). Conversely, METTL18 was one of two annotated MTases, among the top 74 strongly (>16-fold) enriched hits in our RPL3 interactomics screen (Figure 3A and Supplementary Table S6). In agreement with the apparent strong affinity of METTL18 for its substrate, we found that recombinant METTL18 was able to methylate RPL3 in cellular extracts. However, we were unable to detect enzymatic activity of METTL18 on recombinant RPL3, and similar observations were made for yeast Hpm1, as well as for Rkm2, a S. cerevisiae MTase that targets a ribosomal protein (10,60). Interestingly, these MTases recognize their substrates only in the context of a partially or fully assembled ribosome. Given this mode of recognition, it seems less likely that METTL18 has additional substrates. Indeed, we did not detect methylation of proteins other than RPL3 when incubating cellular extracts with METTL18. Nevertheless, it cannot be excluded that additional substrates exist, e.g. low-abundance proteins or substrates masked by the bands resulting from automethylation in the extracts. Also, the observed automethylation of METTL18 at His-154 may suggest that this MTase has the potential to target histidines in sequences other than the (herein) established target site in RPL3. Studies of yeast Hpm1 have suggested the existence of substrates other than Rpl3 (61), and it will be of interest to investigate whether METTL18 has additional substrates as well.
When His-243 in yeast Rpl3 was first reported as the target of Hpm1, the evidence was convincing, but indirect (10). Rpl3 from Hpm1-deficient yeast showed a mass reduction corresponding to the lack of a methyl group, and lacked a methylated amino acid that co-eluted with 3MH in chromatography (10). No specific methylated histidine residue was detected by MS of Rpl3 and its proteolytic fragments, but His-243 was concluded to be the modification site because it was the only histidine residue present in the single segment (amino acids 234–249) that was not covered by the MS analysis (10). This initial study on Hpm1 was followed up by excellent work that further corroborated Hpm1 as a MTase introducing 3MH into Rpl3, but no direct (‘sequence specific’) MS evidence was presented to confirm His-243 as the methylation site (23,61). However, by using an unconventional protease, proteinase K, and the ectopic expression of a more ‘MS-friendly’ RPL3 mutant protein, we were able to directly establish the corresponding residue in human RPL3, His-245, as the target site of METTL18, thus also further corroborating His-243 in Rpl3 as the target of Hpm1 in yeast.
Our results show that ablation of METTL18 yields an accumulation of 30S pre-rRNA (precursor of 18S rRNA of the small subunit), and a concomitant decrease of 41S pre-rRNA (precursor of 5.8S and 28S rRNAs of the large subunit) (Figures 7B and C). Similar changes in pre-rRNA processing have been observed by others, and attributed to defects in early processing of the 5′ ETS at cleavage sites A0 and 1 (Figure 7A, and reviewed in ref. (25)). The observed changes did not, however, affect the relative steady-state levels of mature rRNAs (Figure 7D). This phenotype is similar to that observed in yeast deficient in the METTL18 ortholog Hpm1, which also showed accumulation of pre-rRNAs containing non-cleaved 5′ ETS, but without significant changes in the relative amounts of mature rRNAs. In particular, the Hpm1-deficient yeast strain showed accumulation of 35S (primary transcript) and 23S (precursor of 18S rRNA from small subunit) pre-rRNAs, corresponding to human 47S and 30S pre-rRNAs, respectively (23,61). Thus, ablation of Rpl3 histidine methylation has a similar effect on pre-rRNA processing in human and yeast cells.
However, some features of ribosomal assembly appear to be differently affected by METTL18 and Hpm1 KO. METTL18 KO cells show a reduced 40S/80S subunit ratio, and also a reduced amount of polysomes, while the ratio of 60S/80S is not significantly affected (Figures 8A and B). This suggests a slight deficit of 40S subunits in cells lacking METTL18, which is in line with observed perturbation in 5′ ETS processing of pre-rRNA resulting in accumulation of 30S pre-rRNA (Figures 7B and C). In contrast, Hpm1-deficient yeast displayed reduced levels of 60S subunits, but no significant changes in polysome level (23,61). It was suggested that Hpm1 has substrates other than Rpl3, and concluded that some of the phenotypes of Hpm1 KO are related to methylation of these, yet unidentified, non-Rpl3 substrates (61). Conceivably, differences regarding non-Rpl3 methylation may also underlie some of the differences between phenotypes caused by METTL18 and Hpm1 KO.
In our ribosomal profiling experiments, the codon GAA (Glu) stood out, showing a dramatic increase in occupancy (Figure 9C). Interestingly, we recently performed similar experiments on cells devoid of the 28S rRNA specific N6-adenosine MTase, ZCCHC4, and we also observed the highest increase in occupancy for the GAA codon (32). Moreover, the two codons (GGA and GTT) that showed the strongest decrease in occupancy in the METTL18 KO cells, were also among the ones that were mostly decreased in ZCCHC4 KO cells. One may therefore speculate that certain codon/tRNA interactions are particularly sensitive to perturbations of the ribosome, e.g. through abolishment of methyl modifications. Of note, although we observed codon-specific changes during translation in cells lacking MTases targeting the key translation factor eEF1A, these alterations in codon occupancy were very distinct from those observed in cells lacking METTL18 and ZCCHC4 (32,50,51). Regarding the strong increase in occupancy at GAA, one study highlighted the codon pair GCA-GAA as being strongly underrepresented in humans (62). This study also provided expected versus observed frequencies for all 3721 (61 × 61) codon pairs, and we plotted these data grouped by the last codon of the codon pair. Intriguingly, the GAA-ending codon pairs clearly stood out as encompassing a large fraction of the under-represented codon pairs (Figure 9D). Interestingly, AA-ending codons (AAA, CAA, GAA) have previously been characterized as ‘difficult’ codons, whose translation is particularly dependent on the presence of wobble uridine modifications in tRNA (45,63). Correspondingly, our present (METTL18) and previous (ZCCHC4) data suggest that absence of ribosome modifications can cause stalling at the GAA codon in the A-site, and we further speculate that the strong under-representation of certain GAA-ending codon pairs may reflect that GAA is a ‘challenging’ codon for the translation apparatus. An alternative explanation for the similar codon-specific phenotypes of METTL18 and ZCCHC4 KO cells could be that the abrogation of METTL18-mediated RPL3 methylation somehow affects ZCCHC4-mediated 28S rRNA methylation, or vice versa, but we consider this less likely.
RPL3 is arguably one of the most important and most-studied eukaryotic ribosomal proteins. It is universally present in organisms from all three domains of life, as indicated by its alternative name, ‘uL3’. Furthermore, RPL3 is localized in the vicinity of the peptidyltransferase center (PTC) inside the ribosomes, interacting with the so-called aminoacyl-tRNA accommodation corridor (Figure 10A) (64). Current insights into the role of RPL3 in ribosome function mainly stem from studies in yeast, which suggest that Rpl3 plays an important role in intra-ribosome communication by allosterically transmitting the ribosome's tRNA occupancy status to the elongation factor binding region and the PTC (64). Interestingly, the Hpm1 target site in yeast Rpl3 is localized within the so-called basic thumb, which appears to act as a rocker switch during such communication (61,64). Importantly, yeast Rpl3 and human RPL3 are very similar (66% sequence identity) and the sequence of the basic thumb (residues 234–246 in human RPL3) is identical, while the histidine targeted by METTL18/Hpm1 (His-245/His-243, respectively) shows a virtually identical orientation in the structures of the human and yeast ribosomes (Figure 10B). This agrees well with our observation that ablation of METTL18 gave similar effects on pre-rRNA processing to those observed in Hpm1-deficient yeast cells. Taken together, this indicates that both METTL18-mediated methylation of RPL3 and its functional role are conserved from yeast to humans.
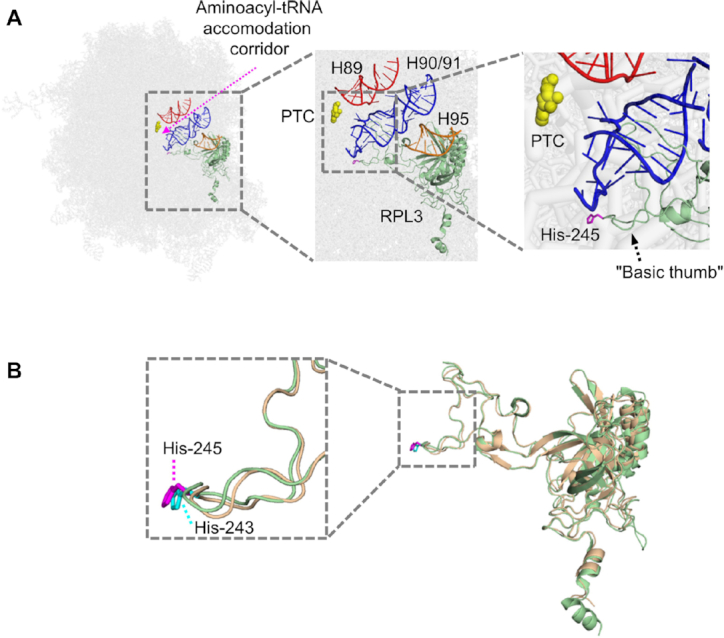
Localization of His-245 in RPL3 in the ribosome structure. (A) His-245 in RPL3 is localized close to the PTC of the human ribosome. Helices 89 (H89; red) and 90/91 (H90/91; blue) in rRNA form the corridor (purple dotted arrow) that channels aminoacyl-tRNA to the PTC, which is indicated by yellow coloring of the key catalytic residue A4397 in 28S rRNA (corresponding to A2451 in Escherichia coli 23S rRNA and to A2820 in Saccharomyces cerevisiae 25S rRNA). His-245 (purple) in RPL3 (green) is localized to the so-called basic thumb, which interacts with H90/91. H95 (orange) represents the ‘sarcin-ricin loop’ (which is subject to cleavage by certain plant toxins). The figure has been adapted from similar figures that were based on the S. cerevisiae ribosome (61,64). (B) His-243 (cyan) in yeast Rpl3 (beige) and His-245 (purple) in human RPL3 (green) show a near-identical orientation. The figure was made using PyMOL Molecular Graphics System, version 1.3 (Schrödinger, LLC), and available structures of the yeast (PDB ID: 4V7R) and human (PDB ID: 4UG0) ribosomes (72,73).
The role of RPL3 in ribosome biogenesis and function has not been extensively investigated in mammals or other metazoans. However, a high-throughput study, which assessed the effects of individual knockdown (by RNAi) of all the 80 human ribosomal proteins, reported that RPL3 knockdown negatively affected the early to intermediate cleavage steps leading to the formation of 28S rRNA (65). Furthermore, an important ‘extra-ribosomal’ role for human RPL3 has been established through a number of studies [reviewed in (66)]. If ribosome biogenesis in nucleoli is disturbed, e.g. by drugs or metabolic stress, ribosome-free RPL3 will be released and will accumulate in the nucleus, where it acts as a transcription factor (67,68). Ribosome-free RPL3 can have a multitude of effects, including the induction of G1 arrest and inhibition of apoptosis (66,67). Moreover, the genomes of humans and other vertebrates encode a RPL3 paralog, denoted RPL3-like (RPL3L), with strong similarity (78% sequence identity) to RPL3, and with a reported ability to repress myotube formation in muscle cells (69,70). In future studies, it will be of interest to investigate how METTL18-mediated histidine methylation influences canonical and extra-ribosomal functions of RPL3 and RPL3L.
Less than a decade ago, none of the human MTF16 members had been functionally characterized. Notably, with the current investigation, enzymatic activities have now been reported for all 11 enzymes, and their collective substrate specificity has, by the present work, been expanded to also encompass histidine methylation. The first human histidine-specific MTase was uncovered only recently, when it was shown that SETD3 was the long-sought MTase responsible for introducing 3MH into His-73 in actin (8,11). Thus, METTL18 represents the second human MTase to be discovered. So far, histidine methylation has been firmly demonstrated for only a handful of mammalian proteins. However, these proteins participate in a wide range of biochemical and cellular processes, and high-throughput proteomic analyses have indicated the presence of histidine methylation in hundreds of proteins (11,17). Thus, protein histidine methylation holds promise as a very interesting and fruitful field of future research, which is likely to yield important insights into how protein function is optimized and regulated by this modification.
ACKNOWLEDGEMENTS
We thank Ingrid F. Kjønstad and Marit Ledsaak for technical assistance. We would like to thank the PRO-MS Danish National Mass Spectrometry Platform for Functional Proteomics and the CPR Mass Spectrometry Platform for instrument support and assistance. We thank Oslo NorMIC Imaging Platform (Department of Biosciences, University of Oslo) for the use of cell imaging equipment.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Norwegian Cancer Society [107744-PR-2007-0132 to P.Ø.F.]; Research Council of Norway [FRIMEDBIO-240009 to P.Ø.F.]; Max Planck Society and DFG Grant [LE 3260/3-1 to S.A.L.]; Lundbeck Foundation [R231–2016-2682 to M.E.J.]; Novo Nordisk Foundation [NNF16OC0022946 to M.E.J., NNF14CC0001 to J.V.O.]. Agence nationale de la recherche [ANR-16-CE11-0029-01 to P.E.G]. Funding for open access charge: Norwegian Cancer Society.
Conflict of interest statement. None declared.
Notes
Present address: Magnus E. Jakobsson, Department of Immunotechnology, Lund University, Medicon Village, Lund, Sweden.
Present address: Rita Pinto, Department of Molecular Oncology, Institute for Cancer Research, Oslo University Hospital, Oslo, Norway.
REFERENCES
1.
2.
3.
4.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.