 The MemMoRF database for recognizing disordered protein regions interacting with cellular membranes
The MemMoRF database for recognizing disordered protein regions interacting with cellular membranes
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

- Altmetric
Protein and lipid membrane interactions play fundamental roles in a large number of cellular processes (e.g. signalling, vesicle trafficking, or viral invasion). A growing number of examples indicate that such interactions can also rely on intrinsically disordered protein regions (IDRs), which can form specific reversible interactions not only with proteins but also with lipids. We named IDRs involved in such membrane lipid-induced disorder-to-order transition as MemMoRFs, in an analogy to IDRs exhibiting disorder-to-order transition upon interaction with protein partners termed Molecular Recognition Features (MoRFs). Currently, both the experimental detection and computational characterization of MemMoRFs are challenging, and information about these regions are scattered in the literature. To facilitate the related investigations we generated a comprehensive database of experimentally validated MemMoRFs based on manual curation of literature and structural data. To characterize the dynamics of MemMoRFs, secondary structure propensity and flexibility calculated from nuclear magnetic resonance chemical shifts were incorporated into the database. These data were supplemented by inclusion of sentences from papers, functional data and disease-related information. The MemMoRF database can be accessed via a user-friendly interface at https://memmorf.hegelab.org, potentially providing a central resource for the characterization of disordered regions in transmembrane and membrane-associated proteins.
INTRODUCTION
Many proteins contain intrinsically disordered regions (IDRs) that do not fold into well-defined structures in isolation and are best represented by conformational ensembles (1,2). The flexibility enables IDRs to participate in highly specific and reversible interactions, which form a common molecular basis for a wide range of physiological processes, including signalling, gene regulation, cell cycle regulation, scaffolding, or chaperoning and pathological conditions (3–5). Such segments, which show disorder-to-order transition upon forming protein–protein interactions, are called molecular recognition features (MoRFs) (6–8). Recent evidence suggests that IDRs are also important components of membrane-associated proteins (MAPs) and transmembrane proteins (TMPs). In these proteins disordered regions of variable sizes can be located in loops between transmembrane segments or in N- and C-terminal regions (9). Many of these disordered regions possess a common feature, namely, they exhibit lipid induced alteration of ordered/disordered status. Here we suggest to term the segments involved in protein–lipid interactions as MemMoRFs, since they represent a distinct category of context-dependent behaviour when compared to MoRFs.
The fundamental role of MemMoRFs in various cellular functions is demonstrated by their ubiquitous presence in proteins associated with a wide range of cellular processes. A set of proteins with MemMoRFs participates in the regulation of cell cycle, apoptosis and phagocytosis (e.g. NOTCH1 (10) and interferon alpha-inducible protein 27-like protein 1 (11)). Other MemMoRF containing proteins play important roles in cellular trafficking and shaping the cell membrane (e.g. Myc box-dependent-interacting protein 1 (12), α-synuclein (13) and pro-neuregulin-1 (14)). MemMoRFs are expected to be abundant in signalling proteins and were detected in both transmembrane receptors (e.g. integrin β3 (15), PTEN tumor suppressor (16) and receptor tyrosine kinases, such as EGFR (17) and erbB-2 (18)) or membrane-associated proteins (e.g. tyrosine-protein kinase Src (19)). MemMoRFs also play important roles in pathological processes. An important MemMoRF group is associated with neurodegenerative diseases and includes α-synuclein, amyloid-β precursor protein, and prion protein (20). In addition, MemMoRFs have been identified in several viral proteins, such as capsid proteins of HIV1 and HCV (21,22), and TMPs, including a potassium channel of the Influenza A virus (23). Most likely, these viral proteins and various toxins, such as melittin (24) and the GPCR impairing toxin of Gila monster (25), require MemMoRF for engaging with or penetrating through membrane bilayers.
The specific conformational properties of these IDRs are shaped by the chemical composition and physical properties of the membrane, and the relevant IDRs can also shape the properties of the membrane (26–28). An important mode to regulate these protein–lipid interactions is phosphorylation. For example, the intracellular domain of the T-cell receptor ζ forms transient helices in the intracellular membrane leaflet (Supplementary Figure S1) (29). One of the transient helices (LID region) possesses an ITAM (immunoreceptor tyrosine-based activation motif), which includes a tyrosine immersed in the membrane bilayer and is a target for phosphorylation (29). When phosphorylated, the ITAM MemMoRF undocks from the membrane and loses its α-helical character. A similar feature can be observed in transporter regulation, involving single pass TMPs possessing MemMoRFs, such as phospholamban (PLN) and FXYD proteins (e.g. phospholemman, FXYD domain-containing ion transport regulator 4 and Na+/K+ ATPase subunit γ) (Supplementary Figure S1) (30–32). The small size of these proteins makes them attractive for drug targeting (e.g. PLN in cardiac diseases) (33).
Although disorder-to-order transition was detected in numerous studies targeting protein–lipid interactions, this phenomenon has not been examined in a generalized manner. As a consequence, information about these protein segments are scattered in the literature, and the detailed structural properties of MemMoRFs are difficult to interpret in the context of additional structural and functional modules. Here, we report a novel, comprehensive database of experimentally validated MemMoRFs, currently containing 131 examples in 96 proteins as a gold standard set. The MemMoRF database complements existing databases of disordered proteins in general (34,35) and also databases of interactions of disordered proteins with ordered or other disordered proteins (36–38). A separate database was built for MoRFs of protein-protein interactions of MAPs and TMPs (38). In contrast, we collected those flexible protein regions into our MemMoRF database, which participate in reversible membrane binding, associated with conformational changes.
IDENTIFICATION AND SYSTEMATIC COLLECTION OF MemMoRFs
The main aim of this database is to facilitate the investigations and targeting of proteins involving MemMoRFs. As a first step, we collected nuclear magnetic resonance (NMR) structures of TMPs and MAPs, since IDRs of these proteins can be located in the close vicinity of biological membranes. We strongly relied on NMR data, since this spectroscopy method is the most widely used approach to determine lipid interaction of proteins at residue level. NMR structures of TMPs were gathered using the Membrane Protein Browser of Protein Data Bank (PDB) (39). MAPs were looked up in the UniProt database (40) using the ‘peripheral membrane protein’ subcellular location keyword (UniProt 2019.01.14). To characterize the transient secondary structures of IDRs, we did not rely solely on the deposited NMR structures, but also incorporated secondary structure propensity (SSPop; using δ2D) (41) and flexibility (1-S2; using Random Coil Index, RCI) (42). We considered a region intrinsically disordered if the residues within the region exhibited ‘coil’ secondary structure population (>0.5) and high flexibility (>0.15). A region was also considered disordered in the case of corresponding disorder annotation in DisProt, DIBS, MFIB or PFAM databases (34,36–37,43). As a next stage, we also explored invisible regions of X-ray and cryo-EM structures, which belong to TMPs or MAPs possessing annotated disordered regions. We subjected the whole protein set to extensive literature analysis to identify IDRs and MemMoRFs or further validate MemMoRFs detected using NMR or other structural data. Thus, proofs of disorder could be derived from calculations, databases, structural information and literature evidence.
A total number of 538 proteins including 206 TMPs and 332 MAPs were screened for segments located in an IDR and exhibiting altered dynamics in a membrane mimetic. A total of 149 membrane interacting regions were identified in 107 proteins. Only 11 regions were derived exclusively from unresolved residues in X-ray or cryo-EM structures. 131 out of the membrane interacting regions are disordered in aqueous solution and become ordered (n = 107) or retain flexibility (n = 19) upon binding to membrane mimetics. Eighteen regions with stable secondary structure both in solution and in membrane bound state were found. Eighty-four out of 149 membrane interacting regions are in TMPs and the remaining 65 are in MAPs. Intracellular, extracellular and periplasmic localizations were observed in 121, 23 and 2 cases, respectively (Figure 1). SSPop and flexibility values were calculated from published chemical shift data with sufficient quality, available for 41% of our protein dataset. Using data from NMR experiments, X-ray and cyro-EM structures and literature, we found 92 regions among identified IDRs, which were not annotated in the DisProt database (Supplementary Tables S1 and S2).

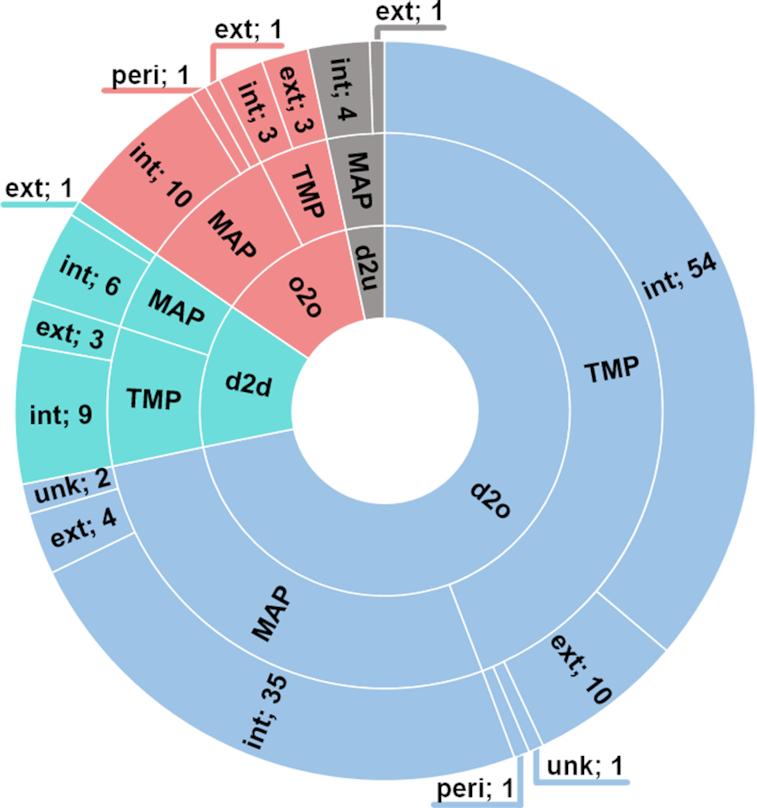
Distribution of MemMoRF types in the database. A total of 149 membrane interacting regions were identified in 107 proteins. One hundred and thirty-one out of these regions are disordered in aqueous solution and become ordered (n = 107) or retain flexibility (n = 19) upon binding to membrane mimetics. Eighteen regions with stable secondary structure both in solution and in membrane bound state were found. d2o: disorder-to-order, d2d: disorder-to-disorder, o2o: order-to-order, d2u: disorder-to-unknown, TMP: transmembrane protein, MAP: membrane associated protein, int: intracellular side, ext: extracellular side, peri: periplasmic side, unk: unknown location.
We categorized the entries depending how their dynamical properties changed upon interaction with the lipid bilayer. In many cases, the corresponding NMR data indicates that the observed transitions resulted in increased α-helical propensity values. In addition, some disordered segments retained their conformational freedom when interacted with membrane mimetics or specific membrane lipids. Although these segments still likely sample a reduced conformational space in the presence of a membrane environment, we labeled their transition as ‘disorder-to-disorder’ to emphasize the dynamics of their membrane bound state. In the case of some of the MemMoRFs derived from X-ray or cryo-EM structures, there is no information on the lipid-bound structure, thus we labeled these entries as ‘disorder-to-unkown’. We also included cases, where the binding helices were stable both in aqueous solution and interacting with the membrane (e.g. helices from kinase suppressor of Ras 1 and BH3-interacting domain death agonist). We labeled these entries as ‘bistable helix’. While they are not classical MemMoRFs, these segments still exhibit a dynamic equilibrium between lipid and water phases and participate in regulatory interactions.
To demonstrate the analysis of various data for IDR and MemMoRF validation and phosphorylation-dependent regulatory disorder-to-order transition, we selected integrin β3, which plays a role in angiogenesis and tumor growth (44,45). Targeting integrin β3 associated signalling was shown to induce apoptosis of endothelial tumor cells (44) and cell permeable peptides derived from integrin β cytoplasmic tails (CT) were developed for angiogenesis inhibition (46). Since these peptides overlap with a MemMoRF, a detailed description of phosphorylation dependent protein and membrane interactions of this region will help to improve the potential therapeutic use of integrin β CT peptides. The complexity of various conformations of the C-terminal integrin β3 MemMoRF under different conditions is shown in Figure 2 and accessible at https://memmorf.hegelab.org/entry/P05106. The C-terminal MemMoRF region (a.a. 770–784) exhibits low helix propensity (<0.5) and high flexibility values (>0.15) in the absence of a membrane mimetic (PDB ID: 2KNC and BMRB ID: 16496) that indicates a disordered state for these residues (47). Although the helical propensity for a.a. 771–776 is lower than the commonly used SSPop threshold (0.5), a small stable helix is present in all of the 20 structures deposited in the 2KNC PDB structure. However, this part of the protein is marked disordered in the DisProt and PFAM databases. Importantly, in the presence of a lipid environment (PDB ID: 2KV9 and BMRB ID: 16771) (48) the decreased flexibility, which approaches the disorder threshold (0.15), confirms the presence of a MemMoRF. At the same time, the helical propensity increases and indicates a stable helix in a smaller part of the sequence (a.a. 776–781). This is in contrast with the long α-helix present in the structural ensemble deposited in the PDB. Moreover, in the paper accompanying these NMR structures (48) the authors noted the presence of transient helical contents and increasing disorder towards the C-terminus based on hydrogen-deuterium exchange experiments. Interestingly, phosphorylation of this MemMoRF promotes disorder (helical propensity <0.5 and flexibility >0.15) in the presence of a membrane mimetic as well (PDB ID: 2LJD and BMRB ID: 17930), thus serves as a conformational switch (49). Again, this NMR ensemble also exhibits a stable small helix between residues 775–780. The potential over-representation of helical content urged us to supplement the entries in the MemMoRF database with supporting statements from papers, and it also cautions database curators that making a decision about disorder should not rely solely on atomistic NMR structures. Phosphorylation-dependent regulation via MemMoRFs is further demonstrated by the T cell receptor ζ and phospholamban (Supplementary Figure S1).
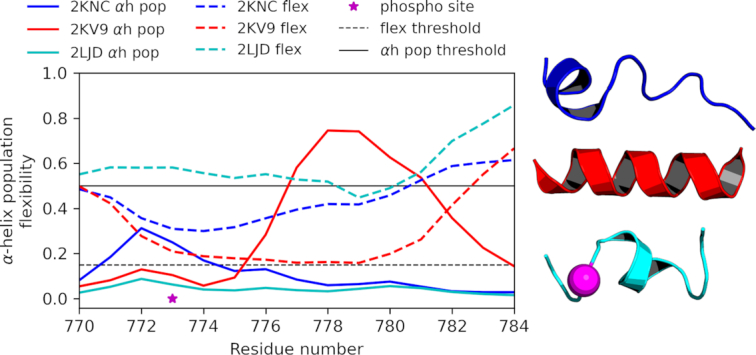
Experimental NMR data provide information on disorder level thus input for MemMoRF identification. Secondary structure population (e.g. helix and coil) and flexibility (1-S2) values were utilized to characterize the per residue disorder-order propensity in NMR ensembles, as exemplified by a MemMoRF from integrin β3. Blue: in organic solvent; red: in DPC; cyan: phosphorylated in DPC; magenta: phosphorylation site; αh pop: α-helix population calculated by δ2D; flex: 1-S2 calculated by RCI, αh pop threshold: 0.5, flex threshold: 0.15.
WEB SERVER AND INTERFACE IMPLEMENTATION
In order to make MemMoRF data accessible and to facilitate research on proteins possessing MemMoRF regions, we developed a web application available at https://memmorf.hegelab.org.
Browsing and searching data
The database provides both browsing and searching functionalities in the ‘Browse’ page, where all entries are listed in a table format. Table columns contain the protein and the gene name, the source organism, the UniProt accession number type, the corresponding PDB and BMRB identifiers and MIM entry, if available, for each protein in our database. The table is sortable and can be filtered by simple queries or using dropdown lists. Searches independent from columns can be performed by typing a query in a text box located at the top of the page.
Entry pages
Each entry starts with the most important, basic information about the protein from UniProt (protein name, accession code, gene name, organism and keywords). Then, MemMoRFs are listed along with their boundaries, type of transition upon membrane binding (e.g. disorder-to-order), localization (e.g. intracellular) and statements supporting disordered and membrane interacting nature of the region.
The SSPop and flexibility calculations, if available, are shown in a graph, which can be saved using the controls next to it. Visibility of corresponding lines can be toggled by clicking on their legend entry. NMR experiments associated with the entry can be selected in a box, right from the graph. This box lists the membrane mimetic used in the NMR experiment, the PDB ID of the calculated NMR ensemble, the BMRB ID and the ‘_Assigned_chem_shift_list.ID’ of chemical shift data used for the calculation. The ‘_Assigned_chem_shift_list.ID’ identifies a specific set of chemical shift values from the BMRB NMR-STAR files, which may contain multiple sets acquired under different experimental conditions.
Sequence specific data, including MemMoRF regions, transmembrane (TM) helices, PFAM domains (43), short linear motifs from ELM (50), post-translational modifications from PhosphoSitePlus (51), IDRs from DisProt, DIBS and MFIB (34,36–37), and segments covered in the PDB are also shown. To further characterize the sequences, Wimley-White hydrophobicity plots and IUPred short predictions were also included into the main graph (52,53). Structures strongly associated with the MemMoRF can be selected for display and manipulation using LiteMol (54). MemMoRFs and TM helices are labelled by default on the structure. In the bottom region of the entry page, additional information is listed to assess the role of MemMoRFs in pathophysiological states. These data include disease phenotypes from MIM (55), disease causing and polymorphic variations from dbSNP (56), and DrugBank records (57). Protein-protein interactions with a link to the IntAct (58) and STRING (59) entries of the protein are followed by an interactive, embedded STRING network graph.
Help and feedback pages
Although the web application is simple and self-explanatory, we provide a comprehensive help page with sections about the following items: i) MemMoRF definition, ii) information resources linked to the memMoRF database, iii) details of our data collection pipeline, iv) search possibilities of the browse page, v) the content of the entry page and vi) links to the statistics and download pages. Users are encouraged to submit comments or questions via a contact form or email located in the feedback page.
Server implementation and database structure
The web application is served via a DJANGO (version 2.1.1) based web interface, fuelled by an SQL database providing fast access to data even in the case of parallel queries. The SQL database contains all the information collected from the literature and various databases (e.g. UniProt, dbSNP, MIM and Drug Bank), organized into multiple tables. Each record in the UniProt table represents a single protein and has the UniProt accession number as a unique key. Other data, including records in the MemMoRF table, are linked to this table by UniProt accession number. In order to provide the best possible user experience on various devices and browsing options for users, the front-end compatibility of MemMoRF is supported by a combination of bootstrap (version 4.3.1) and JQuery (version 2.1.4).
In addition to data access through the web application interface, data can also be downloaded in JSON, XML or TSV formats, or by a RESTful API serving standard JSON format (e.g. https://memmorf.hegelab.org/rest/Q9NR61.json).
CONCLUSION
MemMoRF database delivers data on membrane interacting disordered regions of TMPs and MAPs. These regions exhibit dynamic structural alterations during transition from solvent to a membrane bound state. The collected information has a high potential to contribute to understanding crucial cellular functions and pathological states associated with membrane proteins. Our database provides significant benefits for the broad scientific community because of the following: i) it is a freely accessible, easy-to-use, and organized resource; ii) it includes a gold standard set for determining the sequence requirements for lipid interaction of disordered regions; iii) it represents a high-quality set for developing novel in silico pipelines for MemMoRF identification; and iv) it promotes further experimental investigation and development of drug targeting approaches for MemMoRF containing proteins.
We also provide a new set of disordered protein regions, which are not currently present in other manually maintained databases, based on careful literature curation and NMR-based calculation of secondary structure propensities and flexibility. Our aim is to establish and maintain the MemMoRF database in the long term as a central resource for membrane proteins with lipid bilayer interacting disordered segments.
DATA AVAILABILITY
MemMoRF database and web application are available at http://memmorf.hegelab.org. Since the server is located in the EU, it fully adheres to the General Data Protection Regulation (GDPR).
ACKNOWLEDGEMENTS
We thank Pietro Sormanni and Michele Vendruscolo (Department of Chemistry, University of Cambridge, Cambridge, UK) for providing their scripts for preparing and running δ2D calculations.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research, Development and Innovation Office [NKFIH 127961 to T.H.]; Higher Education Institutional Excellence Programme of the Ministry for Innovation and Technology in Hungary. Funding for open access charge: Semmelweis University, EFOP-3.6.3-VEKOP-16-2017-00009.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.