
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

- Altmetric
RNA structure formation in vivo happens co-transcriptionally while the transcript is being made. The corresponding co-transcriptional folding pathway typically involves transient RNA structure features that are not part of the final, functional RNA structure. These transient features can play important functional roles of their own and also influence the formation of the final RNA structure in vivo. We here present CoBold, a computational method for identifying different functional classes of transient RNA structure features that can either aid or hinder the formation of a known reference RNA structure. Our method takes as input either a single RNA or a corresponding multiple-sequence alignment as well as a known reference RNA secondary structure and identifies different classes of transient RNA structure features that could aid or prevent the formation of the given RNA structure. We make CoBold available via a web-server which includes dedicated data visualisation.
INTRODUCTION
Any RNA transcript in a living cell not only encodes information on its encoded final functional products (e.g. proteins or non-coding RNAs), but also how it is supposed to be regulated in its specific cellular contexts in vivo. While RNA structure has long been acknowledged as a means to assign a final, functional role to a transcript (e.g. highly structured transcripts such as transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs)), there is now mounting evidence that more transient RNA structures are also involved in regulating key events of gene expression on transcriptome level such as alternative splicing (1), RNA editing (1,2), RNA localisation (2–4) and RNA degradation (5).
These transient RNA features appear and dis-appear on time-scales that are similar to key biological processes such as transcription, splicing, A-to-I editing and RNA structure formation. To complicate matters further, these processes are known to happen co-transcriptionally and in the same cellular compartment, i.e. while the transcript is being made in vivo. So far, it has thus been difficult to experimentally investigate and dis-entangle the intricate interplay of these key processes of the transcriptome in vivo which are key to defining the functional products in any living cell. Transcriptome-wide, hypothesis-driven studies in silico can greatly help in that regard. Based on a computational study of several large transcriptome data sets, we managed to show with significant statistical evidence that co-transcriptional A-to-I editing can regulate alternative splicing via induced changes to local RNA structure elements near splice sites (1).
Overall and based on a wide range of experimental and computational evidence, we know by now that RNA structure formation in vivo is highly dependent on the cellular context in vivo as well as the kinetics of RNA structure formation in vivo (6). One and the same RNA transcript can readily encode more than a single functional RNA structure. Similarly to the concept of alternative splicing, these RNA structure features can be differentially expressed depending on the particulars of the transcript at a specific time and location in vivo, a concept we recently introduced as alternative RNA structure expression. Which encoded RNA structure is expressed at any given point of the transcript’s cellular life in vivo depends on subtleties such as trans interaction partners (e.g. proteins, ligands, other RNAs), ion concentrations, the temperature and the kinetics of RNA structure formation.
Most computational RNA secondary structure prediction methods assume the one-sequence-one-structure dogma by predicting a single RNA secondary structure (from now on simply called RNA structure) for any given input RNA; well-known examples include the widely-used methods Mfold and RNAfold (7–12). In reality, however, biologically relevant transcripts can encode between zero and multiple functionally relevant RNA structures which are differentially expressed depending on the particulars of the cellular context in vivo. Most RNA structure prediction methods aim to predict the RNA structure that corresponds to the (pseudo-knot free) RNA secondary structure configuration with the minimum overall free energy (so-called minimum-free energy (MFE) approach) in an in vitro setting. This so-called MFE RNA structure, however, may not be expressed in any biologically setting in vivo.
One conceptual alternative to predicting RNA structure that are functionally relevant in vivo is to try to identify the RNA structure(s) that have been conserved during adequately chosen times of evolution. This approach typically, but not always (9), requires as input a multiple-sequence alignment (MSA) of orthologous or homologous RNAs from several, evolutionarily related species. This strategy has the significant conceptual advantage of not having to understand why RNA structure features have been conserved provided they have been conserved. This so-called comparative approach to RNA structure prediction currently yields the state-of-art in terms of prediction accuracy (13). These methods typically employ mathematically principled approaches such as stochastic context-free grammars (SCFGs) which can explicitly capture different RNA structure formation hypotheses (rather than capturing only the single MFE strategy) and are capable of estimating the reliability of their own predictions (14,15).In this fashion, a recently-developed method know as R-scape (16) employs a G-test statistics in order to assess the statistical significance in terms of E-value for evolutionary-conserved base-pairs within RNA structure containing multiple sequence alignments.
Even these state-of-the-art methods, however, typically ignore the kinetic process of co-transcriptional RNA structure formation. With comparative methods, one can hope that the evolutionarily conserved RNA structure features that are detected and combined into the predicted RNA structures still correspond to the relevant final, functional RNA structure that matter in vivo. When considering non-comparative methods that are guided by the MFE strategy such as Mfold and RNAfold, however, it is less clear how they could be modified to take the overall effects of co-transcriptional RNA structure formation into account. Their predictive performance has long been know to decrease markedly for input sequences longer than 200 nucleotides (nt) for which kinetic effects of co-transcriptional are expected to matter most (17). We showed with the RNA structure prediction program CoFold (18) that it is technically and conceptually possible to incorporate overall effects of co-transcriptional into Mfold, thereby significantly increasing its prediction accuracy, especially for long input sequences. Since then, however, we also found compelling evidence that some transient RNA structure features can be just as highly conserved during evolution as the features of the final, functional RNA structure (19–21). This unfortunately implies that even the state-of-the-art comparative methods may become confused by these highly conserved transient RNA structures features which they may erroneously try to combine into the predicted final, functional RNA structures.
An early computational data study by us from 2013 (19) showed that known, structured RNA genes not only encode these final RNA structures themselves, but also information on how to form them co-transcriptionally in vivo. For this, we proposed a classification of transient helices (so-called clashing helices) based on their compatibility with helices of the known, functionally relevant RNA structure. In that context, we defined helices as consecutive stretches of base-pairs. We here extend the original classification of these clashing, transient helices to the functional classes shown in Figure 1. As the corresponding Table 1 shows, different classes can play different functional roles by either aiding (Figure 2) or preventing (Figure 3) the formation of the know reference RNA structure during co-transcriptional folding in vivo. Our original study from 2004 (21) discovered statistically significant 5′-to-3′ asymmetries by quantifying the overall effect that these clashing, transient helices have on co-transcriptional RNA structure formation in vivo, as the evolutionary prevalence of 3′ clashing helices was much lower than the 5′ one. This asymmetry arises from the different effects that the different types of helix have on the co-transcriptional formation of the final structure. Potentially beneficial helices of type 5′-cis and 3′-trans tend to be more conserved that helices of type 3′-cis and 5′-trans that could prevent the co-transcriptional formation of the known RNA structure.

![Different functional classes of transient helices that can emerge during co-transcriptional folding and interfere with the formation of the final RNA structure: 5′-trans, 5′-mid, 5′-cis, 3′-trans, 3′-mid and 3′-cis. The base-pair between nucleotides i and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{upgreek}
\usepackage{mathrsfs}
\setlength{\oddsidemargin}{-69pt}
\begin{document}
}{}$\overline{i}$\end{document} is part of a helix of the known RNA structure. The base-pair between c and i is part of a transient helix that clashes with the helix involving base-pair i and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{upgreek}
\usepackage{mathrsfs}
\setlength{\oddsidemargin}{-69pt}
\begin{document}
}{}$\overline{i}$\end{document} of the known structure. The nomenclature for the different types of transient helices depends on the location of the competing base-pair with respect to the base-pair with which it competes (trans, mid or cis) and the location of the location of the transient helix w.r.t. the known helix it clashes with (upstream (5′) or downstream (3′)).](/dataresources/secured/content-1765944343956-b58c0487-d7cc-4aa0-b5e5-1dd92f5b5697/assets/gkaa900fig1.jpg)
Different functional classes of transient helices that can emerge during co-transcriptional folding and interfere with the formation of the final RNA structure: 5′-trans, 5′-mid, 5′-cis, 3′-trans, 3′-mid and 3′-cis. The base-pair between nucleotides i and is part of a helix of the known RNA structure. The base-pair between c and i is part of a transient helix that clashes with the helix involving base-pair i and of the known structure. The nomenclature for the different types of transient helices depends on the location of the competing base-pair with respect to the base-pair with which it competes (trans, mid or cis) and the location of the location of the transient helix w.r.t. the known helix it clashes with (upstream (5′) or downstream (3′)).
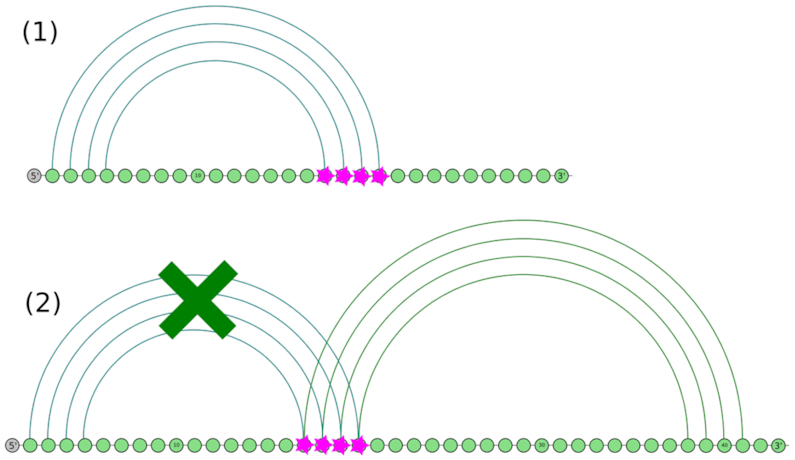
Co-transcriptional role of a transient helix of type 5′-cis which can easily yield to the formation of the helix it is clashing with during co-transcriptional RNA structure formation by temporarily engaging some of its basepairs from its 5′ side and thereby preventing the formation of other structure features from pairing with them.
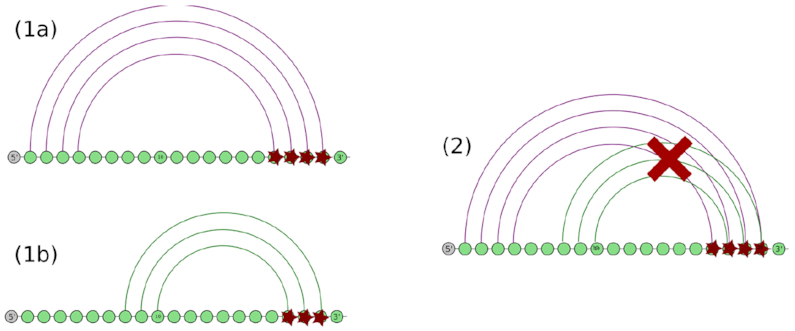
Co-transcriptional role of a transient helix of type 5′-trans. A transient helix of type 5′-trans can easily prevent the co-transcriptional formation of the helix it is clashing with by getting the RNA structure formation kinetically trapped during co-transcriptional folding as it directly competes with the 5′ side of the original helix for the 3′ side at the time of the formation of the helix.

| trans | mid | cis | |
|---|---|---|---|
| 5′ | Disruptive | Neutral | Beneficial |
| 3′ | Beneficial | Neutral | Disruptive |
The experimental identification of transient RNA structure features remains a complex and time-consuming task that is typically strongly tailored to the specific requirements and features of a single transcript of interest. Existing approaches can be subdivided into in vitro and in vivo methods. In vitro strategies allow for the tight control of key features such as the temperature, transcription rate, concentration of certain ions and select interaction partners and are thus often preferred to facilitate reproducibility. These in vitro methods, however, cannot capture some of the (known or unknown) complexities of the cellular environment in vivo that may critically influence the RNA structure formation in vivo (22). In his review paper from 2000 (23), Higgs already noted marked discrepancies in determined RNA structures that are due to differences in how and in which molecular context the molecule of interest was probed experimentally (e.g. in vivo versus in vitro, NMR versus X-ray). It should thus not come as a surprise that these differences will probably extend to the experimental determination of transient RNA structure features which can be expected to be even more sensitive to the particular features of the cellular environment in vivo.
The first experimental protocol for the high-throughput probing of co-transcriptional RNA structure features on a transcriptome-wide scale in vivo and in vitro, called SPET-seq, was published by Incarnato et al. only in 2017 (24). This method is capable of investigating transient RNA structures at single-nucleotide resolution and for individual transcription intermediates due to the fast-acting reagent DMS. SPET-seq’s main limitation is that it primarily enables the RNA structure investigation of the 3′-ends of transcripts. Even with this limitation, however, the research team gained valuable and unique insight into the RNA structure formation of mature cytosolic RNAs in E. coli.
Strobel et al. (25) recently succeeded in extending SHAPE-probing to a high-throughput platform that allows for the simultaneous testing of several variants of the same riboswitch during co-transcriptional structure formation in vitro. The formation of the ZTP riboswitch that they investigate is known to be kinetically controlled and had to be carefully resolved time-wise in order to be able to determine which folding intermediate is involved in binding the key ligand (alarmone ribonucleotide ZMP) during transcriptional elongation. They achieve this by stalling the RNA polymerase at different positions along the DNA, by SHAPE-probing the thus resulting transcripts of different lengths and by combining this with a high-throughput protocol for reading out the resulting transcript-specific sequences of SHAPE-probing signals.
Their method, however, probes a transcript of less than 200 nucleotides length. The influence of folding kinetics during co-transcriptional folding is typically expected to be a major influence for transcript larger than that (26) which constitutes the majority of primary transcripts for many organisms. The experimental determination of co-transcriptional folding intermediates and their functional role(s) in vivo thus remains a real challenge, see for example the long (and alternatively spliced) Xist gene that plays a key role in dosage compensation in mammals and beyond (27).
One orthogonal strategy to the high-throughput, SHAPE-based probing of multiple co-transcriptionally stalled versions of the same emerging transcript is single-molecular (SM) microscopy which investigates one particular molecular at a time, e.g. the recent review (28). This type of microscopy allows for the investigation of even minor effects of ligand binding and its impact on global RNA structure formation.
In same cases, well-established methods such as mutations and genetic selection can be employed to gather evidence for the existence and location of meta-stable structures that are co-transcriptionally formed in vivo in case they have distinct functional roles whose absence or presence can be readily assessed in vivo, e.g. as recently performed by Masachis et al. to uncover a new layer of toxin-antitoxin regulation in Helicobacter pylori (29).
Large-scale experiments of histone transcripts with a mutated and slower version of the nominal polymerase II combined with chemical RNA structure probing of emerging poly-A+ transcripts reveal that the correct transcription speed is key to their correct pre-mRNA maturation (30). More specifically, Saldi et al. show that the slower transcription yields polyadenylated histone mRNAs that extend past the stem-loop processing site, thereby establishing a causal link between transcription speed, co-transcriptional structure formation and correct mRNA maturation for this specific class of genes.
Beyond this particular class of pol II described genes, co-transcriptional events may play a key role in regulating correct splicing, see (31) for a recent review by the Neugebauer group and (1) for a transcriptome-wide, computational study by us that establishes a causal link between local RNA structure features, RNA editing, alternative splicing and Alu repeats (see also (3)).
Here, we present a new computational method, called CoBold (in German, a ‘Kobold’ denotes a mischievous, but mostly well-meaning little devil that play tricks on people) that is capable of identifying candidate transient helices of all functional classes shown in Figure 1 and Table 1 and their relationship to a known reference RNA structure. Depending on the input evidence supplied by the user (i.e. either a single input sequence or a corresponding multiple-sequence alignment (MSA)), our method can readily identify new RNA structure features that can impact the co-transcriptional formation of the reference RNA structure, either in a positive or negative way. When used in a comparative way, i.e. with an input MSA, CoBold is capable of assigning estimated reliability values (i.e. P-values) that allow for the filtering and ranking of the predicted transient features. We investigate the merits of CoBold for five biological cases where functional RNA structures and transient RNA structure features are known and show that our method can be used to swiftly gain valuable insight into RNA structure formation in vivo that could otherwise be only obtained via dedicated experiments. Our hope for the future is that CoBold will help identifying more experimentally confirmed cases of functional transient RNA structure features which can then in turn be used to improve computational prediction methods such as CoBold. We make CoBold accessible via two dedicated web-servers at www.e-rna.org where users can run CoBold analyses and also visualize the CoBold predictions in a number of ways.
CoBold is the first method of its kind and will hopefully aid in identifying transient features that are difficult to investigate experimentally while also playing key functional roles in diverse biological systems in vivo ranging from a viral, bacterial to eukaryotic.
MATERIALS AND METHODS
We here describe our new computational method CoBold which can either be used in a non-comparative or comparative way depending on the input evidence that the user can supply for the RNA transcript of interest.
Non comparative part of CoBold
Without any evidence on orthologous transcripts from evolutionary related species, CoBold takes the sequence of a single RNA transcript as input and calls a highly modified version of CoFold (18). In its original implementation, the RNA structure predicted by CoFold is derived from a 2D matrix which stores the entries of the underlying dynamic-programming algorithm via a traceback-procedure which starts at the left top corner of this matrix depicted in Figure 4. In order to identify relevant transient RNA structure features that could clash with the reference RNA structure, we modified the original traceback procedure of CoFold so that it is now capable of collecting information on all relevant transient RNA structures of the different functional classes shown in Figure 1 in a single traversal of the optimisation matrix shown in Figure 4.
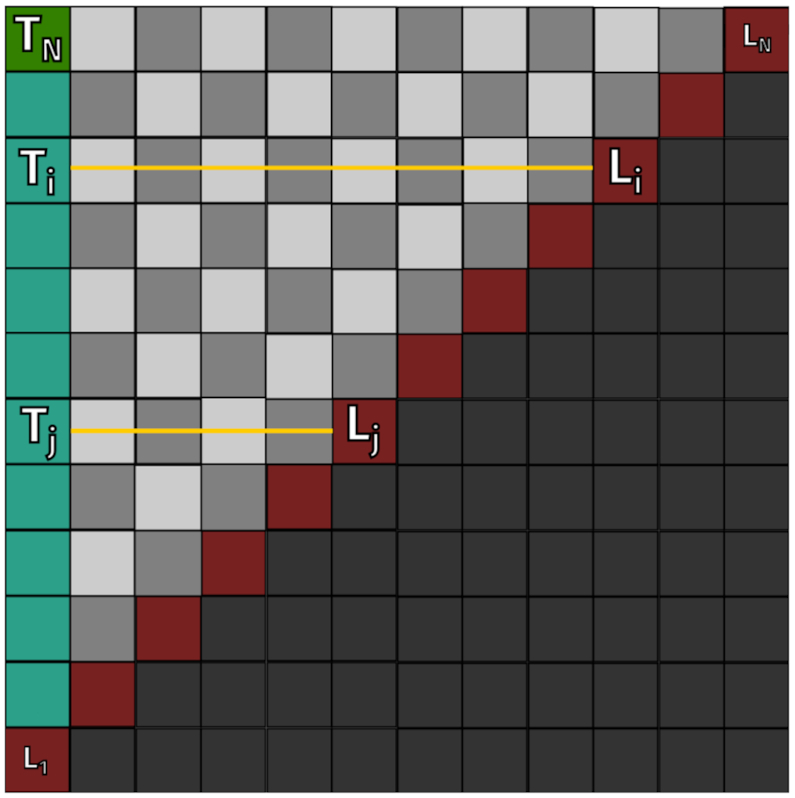
Two-dimensional dynamic programming matrix underlying the algorithm of the non-comparative part of CoBold. Both axes correspond to an input sequence of length L. Each Ti = (1, i) entry in this matrix corresponds to the optimal RNA structure Si that is co-transcriptional formed for a sub-sequence Li, Li ≤ L, of the original input sequence. By starting the traceback procedure at field Ti = (1, i) of this matrix, we can retrieve Si by subsequently following the structural information stored in each subsequent node, which also points to the following one in the path. At the end, the RNA secondary structure for sub-sequence Li is obtained.
The key idea behind the new trace-back procedure is to realise that this matrix not only contains information on the optimal co-transcriptionally folded RNA structure for the fully extended input sequence of length L, but also for all sub-sequences Li ≔ (x1, x2, …, xi) of length Li ≤ L, see Figure 4. The new trace-back procedure traverses all optimal co-transcriptionally folded RNA structures Si for all sub-sequences Li and thereby gathers information on all different classes of transient helices within these Si that could impact the formation of the known reference RNA structure for length L. The minimum length required for a transient helix is a global parameter in CoBold that can be adjusted by the user (default value is 2 bp).
Overall, the non-comparative part of CoBold thus has the same time-and-memory requirements as the original CoFold—namely time and memory for calculating the matrix shown in Figure 4—and requires only time for the modified traceback procedure for an input sequence of length L. The non-comparative part of CoBold thus corresponds to a computationally efficient tool for identifying different functional classes of potentially relevant transient RNA structure features that could impact a given reference RNA structure.
Comparative part of CoBold
If the user supplies a multiple-sequence alignment (MSA) containing the transcript of interest as well as a corresponding reference RNA structure as input to CoBold, it becomes conceptually possible to computationally detect and rank transient helices based on their degree of evolutionary conservation.
In order to detect potential transient helices, CoBold utilises Transat (33), a fully probabilistic method contributed by us earlier that is capable of detecting conserved helices of functional RNA structures, including transient, pseudo-knotted and alternative structures. Transat takes as input a MSA of homologous sequences and a corresponding evolutionary tree (if the tree is not known, it can be readily inferred by Transat via a maximum-likelihood method). Briefly, Transat first calculates all possible candidate helices for each un-gapped sequence in the input alignment. These features are then mapped onto the input MSA and the corresponding base-paired alignment columns considered as candidate helices. For each candidate helix within the MSA, Transat then calculates a so-called log-likelihood score which employs two dedicated probabilistic models of evolution in order to quantitatively compare the likelihood of two competing hypothesis: one, that the pair of alignment columns evolved as base-pairs, two, that the same set of alignment columns evolved independently. Finally (and optionally), Transat estimates P-values for the the predicted helices in order to take into account that different input MSAs have different overall propensities to form spurious helices. This propensity depends on a number of complex features of the input alignment such as the dinucleotide composition within the sequences and the gap-pattern. For this, it generates a sufficiently large number of (carefully) randomised versions of the original input alignment in order to determine a corresponding null-distribution of log-likelihood scores. This distribution is then used to assign P-values to the log-likelihood scores of the helices detected in the original input MSA.
For CoBold, two features of Transat are particularly noteworthy. First, the fact that Transat is a fully probabilistic method that does not employ the MFE strategy and that is capable of assigning reliability values to the detected helices. Second, that its performance has been shown to be independent of the length of the input alignment. Moreover, it is computationally efficient, requiring only time for an input alignment of N sequences and length L. Given a known reference RNA structure and an input alignment of decent quality, it is thus possible to assess the presence and potential impact of conserved transient helices irrespective of sequence length.
As CoBold takes a given reference RNA structure as input, the conserved helices identified by Transat can be readily assessed with respect to the given reference structure and classified into the categories of clashing helices specified in Figure 1. In the current implementation of CoBold, users can choose to report only the statistically most significant transient helix that clashes with any helix of the reference RNA structure. It is also possible to impose an overall threshold value for the statistical significant of any predicted helices, via a P-value threshold of 10−2. CoBold can report the detected helices sorted by functional class, see Figure 1. It is thus for example easy to identify the most significant transient helix of type 5′-Cis that could prevent the formation of a key helix of the reference structure.
The comparative mode of CoBold allows users to readily explore different evolutionary time-scales on which transient helices have been conserved by utilising different input MSAs capturing different species and evolutionary time scales. This type of analysis can provide valuable additional insight into the potential functional roles of select transient helices.
Finally, it is possible to use the predictions of the comparative part of CoBold as filter to post-process the predictions of the non-comparative part of CoBold, e.g. to focus on well-conserved transient helices or transient helices with a significant statistical significance.
Web-server of CoBold for visualisation
The predictions by CoBold can offer unique insight into the complex interplay of transient helices and distinct structural features of the reference RNA structure. In order to explore these relationships in a visual and more intuitive manner while also highlighting the underlying quantitative information, we have built two dedicated CoBold web-servers at www.e-rna.org for running CoBold analyses and for visualising and manipulating CoBold predictions. The visualization server is technically based on R shiny(http://www.rstudio.com/shiny/) (34) and utilises our own R4RNA R-package for RNA structure visualisation (35).
The visualisation tool allows to directly compare the predicted transient helices to the given reference structure. This is achieved via a so-called double-arc plot. Any base-pair is shown as arc (or semi-circle) linking the two base-paired sequence position along the horizontal line which can represent the input sequence or the input alignment. The double-arc plot shows the features of the reference RNA structure on top of the horizontal line and the relevant predicted features below, see e.g. Figure 5. The user can then click on key structural features of the reference RNA structure to highlight the relevant predicted transient features. When visualising predictions of the comparative mode of CoBold, the user can impose a P-value threshold.
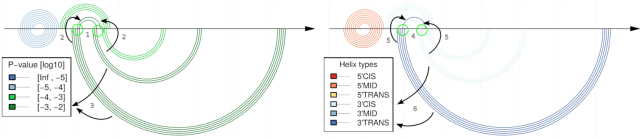
Example figure of an arch plot: The horizontal line represents the input MSA. Each arc or semi-circle corresponds to a single base-pair connecting the corresponding two sequence positions. Arcs on top represent the reference RNA structure, in this case the permissive conformation of the Levivirus. Arcs below the horizontal line depict the transient helices predicted by the comparative mode of CoBold. For each shown transient helix, it is easy to identify the corresponding helix or helices it is clashing with (in this case indicated by the arrows) as these share base-paired positions along the horizontal line, see the feature highlighted by the circles and arrows (1–2, 4–5). The left part of the figure shows the predicted helices and the reference helix/helices they are clashing with in the known structure colored by statistical significance (i.e. P-value) (5) whereas the right part of the figure shows them colored by functional class (6). In this case, the most prominent feature is thus the predicted transient helix comprising four base-pairs of high statistical significance (see dark green color and legend, as indicated by the arrow on the bottom left) and of beneficial type (i.e. 3′-Trans) (see dark blue color and legend on the bottom right) which may aid in the formation of the 3′ helix of the reference RNA structure conformation.
When considering predictions of both modes in CoBold, both sets of predictions can be combined, for example in order to focus on helices with a high degree of evolutionary conservation. Moreover, users can focus on transient helices of a distinct functional class, e.g. those that are potentially disruptive (e.g. 5′-Cis) or particularly beneficial (e.g. 3′-Trans). And finally, the web-server allows for the download of the resulting high-quality figures in pdf and png format that can be readily integrated into publications.
RESULTS
We focus the evaluation of CoBold on five select data sets whose transient features and known RNA structures have been experimentally confirmed and whose functional roles have been well established (25,36). These examples showcase how functional features and regulatory feedback loops can be encoded into sequences via structural features such as helices that are mutually exclusive. The overview below includes information on the respective accession numbers of the reference sequences for which the reference RNA structures are known as well as the IDs of the multiple sequence alignments employed.
HDV ribozyme (Figure 6):
The Hepatitis Delta Virus (HDV) is a single-stranded (ss) RNA virus that aggravates symptoms of human hepatitis. This particular ribozyme regulates the self-cleaving process required for viral replication. This is achieved via two different structural configurations that either allow or prevent replication depending on the environmental conditions. In the active state, the region on the sequence where the self-cleaving happens is exposed, so HDV can undergo the process and replicate. During poor conditions for replication, however, the self-cleaving region is sequestered by a hairpin, thereby preventing replication. Notwithstanding, if optimal replication conditions are achieved once more, the 5′ region of the sequestering hairpin is itself sequestered, so the self-cleaving region is exposed again and the replication can occur. The HDV ribozyme sequence has a length of 152 nt. In this particular case, we employed the alignment RF00094 from Rfam (37), with M28267.1 635-775 as reference sequence.
5′-UTR of Leviviridae Levivirus (Figure 7):
The Levivirus is a ssRNA bacteriophage, i.e. a virus that kills certain bacteria. Its 5′ UTR controls the translation of its proteins by regulating the binding of the ribosome and the replicase, which compete for the same region. If the Levivirus is not in need of protein translation, the ribosomal binding site is sequestered by an upstream complementary region and trapped in a helix. However, if protein translation is needed, this complementary region is paired with a different region further upstream in the sequence, thereby releasing the originally mentioned region and enabling ribosome binding. This particular sequence has a length of 158 nt. For this analysis we used a custom alignment, choosing GQ153927.1 1-132 as reference sequence.
SAM riboswitch (Figure 8):
This particular riboswitch is a structure widely shared among bacteria which allows for a switch in gene expression depending on the presence of a certain co-enzyme (in this case, S-adenosylmethionine (SAM)). With SAM binding, the riboswitch adopts an RNA structure that stops transcription. This structure comprises two helices, one called anti-anti-terminator and one called terminator (in 5′ to 3′ order). Without SAM binding, this RNA structure is partly re-arranged. In this rearrangement, the two anti–anti-terminator and terminator helices are destroyed and converted into a single new helix that pairs the 3′ arm of the former anti–anti-terminator helix and the 5′ arm of the former terminator helix into a new anti-terminator helix. This anti-terminator helix allows for transcription to proceed. The SAM riboswitch has a length of 215 nt. In this particular study we employed a custom alignment, with AL009126.3 1258276-1258464 as reference sequence.
Tryphtophan (Trp) operon leader (Figure 9):
This structure regulates the transcription of the trp operon in E. coli. Similarly to the above examples, it senses if the optimal conditions for transcription are given or not. In this case, however, the region is responsive to the transcription speed itself. While transcription is being conducted by the ribosome as its nominal speed, a terminator hairpin forms which stops transcription. If conditions are not optimal for transcription and the ribosome stalls, an anti-terminator structure emerges that sequesters the terminator which in turns allows the ribosome to transcribe the whole sequence, instead of just a partial one. The sequence of the trp operon leader has a length of 152 nt. For the purpose of this analysis we used Rfam alignment RF00513 with AE005174.2 2263095-2263188 as reference.
ZTP riboswitch (Figure 10):
This structure switch regulates transcription in bacteria via the presence or absence of the purine bio-synthetic intermediate 5-aminoimidazole-4-carbozamide riboside 5′-triphosphate (ZTP) molecule. Here, we focus on the riboswitch appearing in the bacterium Clostridium beijerinckii whose structure is commonly designated as Clostridium beijerinckii pfl. This riboswitch’s structure was recently studied in great detail experimentally (25) and its cofolding path was extracted by analysing the SHAPE values of its different sub-sequences. We wanted to check if our proposed method is able to recover that particular structure that was extracted experimentally with a significant amount of effort and time. The ZTP riboswitch we use has a length of 172 nt. In this study, we employed an improved version of the RF01750 alignment from (25) with CP000721.1 1211931-1212029 as reference sequence. Our improvement consists of removing all-gap columns from the alignment, as well as sequences that do not contribute to the overall evolutionary information contained in the alignment.
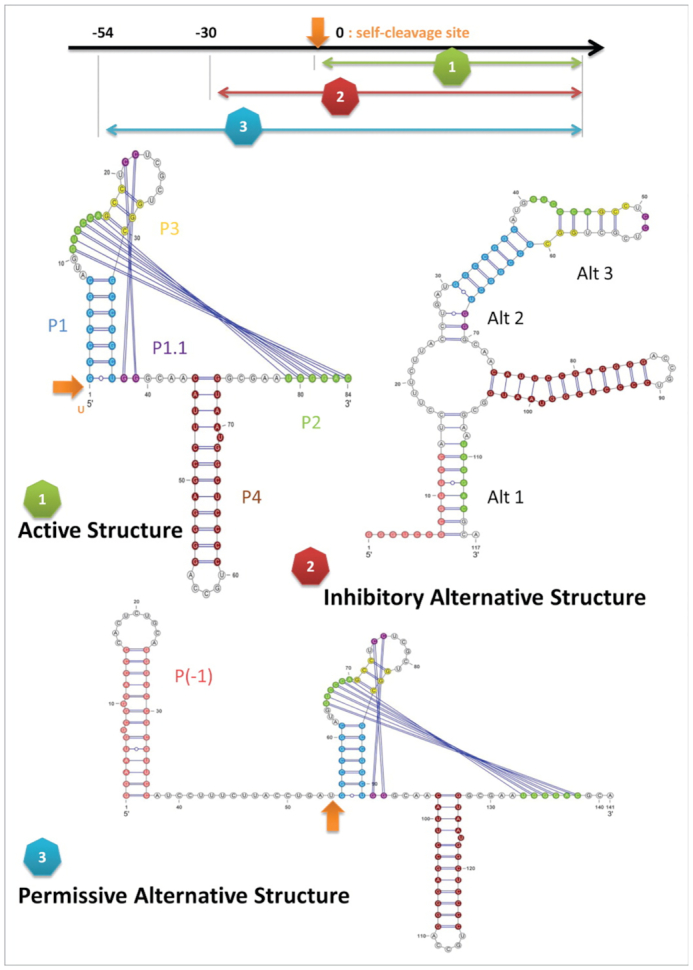
Three different functional conformations the HDV ribozyme: the active structure, the inhibitory structure and the permissive alternative structure. The self-cleaving site is indicated by the orange arrow, being exposed in the active and permissive structure and sequestered in the inhibitory one. Picture from (36).
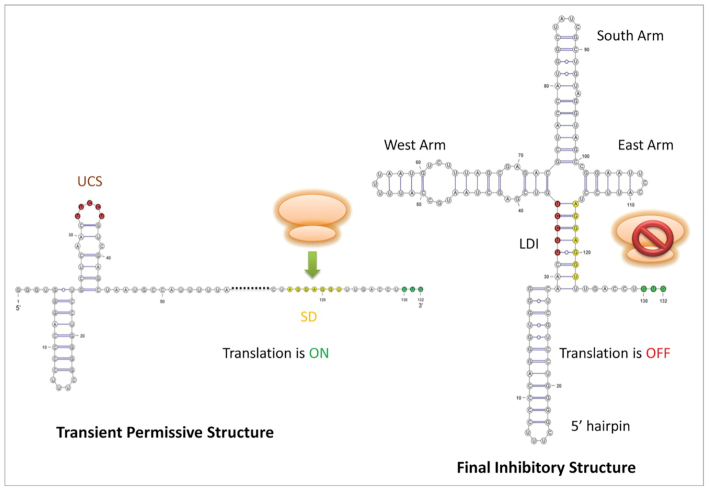
Two functional conformations of the 5′ UTR of the Leviviridae Levivirus: the inhibitory conformation and the permissive alternative conformation. The ribosome binding site (SD) site is indicated by the yellow nucleotides, being exposed in the permissive structure and sequestered in the inhibitory one. Picture from (36).
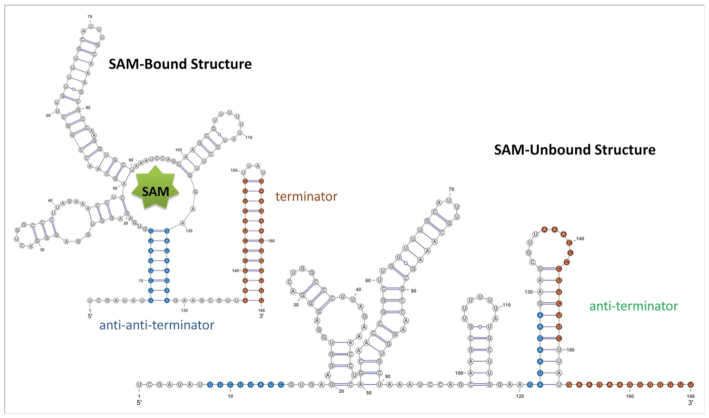
Two functional conformations of SAM-responsive riboswitch: the bound structure and the unbound structure. The terminator, anti-terminator and anti-anti-terminator hairpins are indicated in brown, green and blue, respectively. It can be seen how the terminator is sequestered in the unbound state and free at the bound one. Picture from (36).
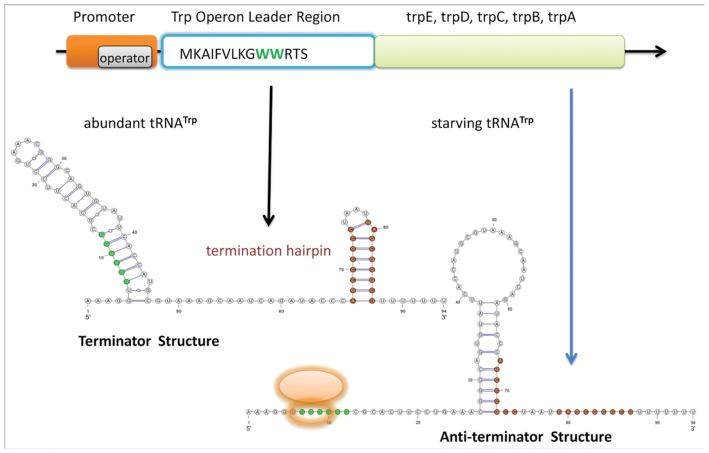
Two functional conformations of the tryphtophan (TRP) operon leader: the terminator structure and the anti-terminator structure. The ribosome binding site and anti-terminator hairpin are indicated in green and brown respectively. It can be seen how the binding site is sequestered in the terminator state and free and accessible at the anti-terminator one, where the terminator is sequestered by the anti-terminator. Picture from (36).
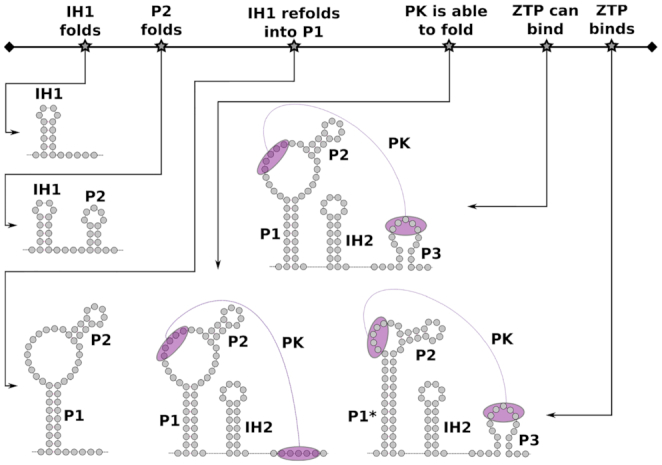
Co-transcriptional folding pathway of the ZTP riboswitch: Picture showing the cofolding path as well as the different conformations for the ZTP riboswitch for the ZTP-bound and the ZTP-unbound state. The time-line at the top outlines the relevant structural steps happening during transcription with the corresponding RNA structures shown below. Figure adapted from (25).
In the following, we focus on two of the above examples to highlight that CoBold can not only help in identifying these structural features for a given reference RNA structure, but also aid in their functional interpretation. Please refer to the supplementary information (SI) for a detailed discussion of the other systems (Supplementary Figures S3–S5), where their corresponding results (Supplementary Figures S6–S25) will be discussed in a similar manner as we will do in the following text with the two selected ones (Supplementary Figures S1 and S2). For each case, we proceed as follows:
As input to CoBold, we use the reference RNA sequence, a corresponding MSA and evolutionary tree as well as a reference RNA structure.
Using the comparative mode of CoBold, we detect all evolutionarily conserved helices that are clashing with the reference RNA structure, rank them in terms of significance (i.e. by P-value) and sort them by helix type, see Figure 1.
Using the non-comparative mode of CoBold, we detect all co-transcriptionally emerging transient helices that are clashing with the reference RNA structure, rank them in terms of stability and sort them by helix type. The stability of a helix is measured in terms of its minimum-free energy.
Depending of the desired analysis and its stringency (e.g. in terms of P-value threshold imposed by the user), we either consider the predictions of the non-comparative and comparative mode separately or, alternatively, compute the intersection of helices predicted by both modes in CoBold.
When the different clashing helices have been predicted, we interpret their relevance and potential functional role in terms of their type (see Table 1), as they could either prevent or perturb the formation of the reference structure or, on the contrary, aid its formation. In addition, we can quantitatively assess the extent of their evolutionary conservation and, thus, likely functional relevance via their estimated P-values, for cases when the user can supply an input alignment for the sequence of interest.
Detecting evolutionary-conserved clashing helices
The 5′ UTR of the Leviviridae Levivirus has two distinct functional conformations, the inhibitory and permissive conformation, see Figures 11 and 14. They share some structural features, but contain contain mutually exclusive helices. Using the comparative mode of CoBold, we can identify a conserved transient helix that is clashing with the region located near the UCS, which is a requirement for the formation of the cruciform structure of the inhibitory conformation. This particular helix is threatening both functional conformations of the Levivirus. When we consider only the most significant clashing helices (see Figures 13 and 15), we find that these, well-conserved helices are typically of the beneficial type (3′-Cis) or neutral (5′-Trans or 3′-Trans) and may thus explicitly aid in the formation of the known functional configuration. The helix clashing with the 5′ hairpin is the only one that clashes with a structure shared by both conformations. It is officially classified as having a neutral effect, but—due to its significance in terms of P-value—may play a beneficial role by protecting the formation of that specific helix. There are only some neutral helices competing with some nucleotides in the cruciform, mostly at the west and south arms.
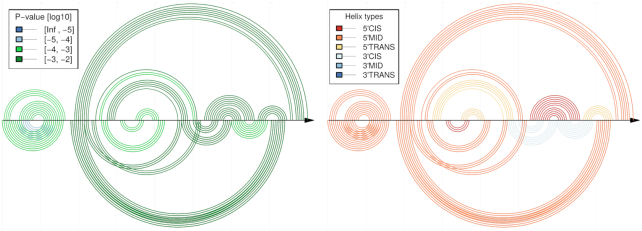
Comparative CoBold predictions for the inhibitory conformation of the Levivirus with the inhibitory RNA conformation shown on top and the predicted transient helices shown below the horizontal line, once colored in terms of statistical significance (left) and once in terms of type of transient helix, see also the previous figure for more general information on this type of plot.
For the other four functional families, the observations are mostly similar to the ones for the Levivirus. In particular, when focusing on the most significant helices, most of the predicted transient helices are of the beneficial or neutral type.
Detecting features of the cofolding pathway of the ZTP riboswitch
The co-transcriptionally folding pathway of the ZTP riboswitch was recently studied experimentally in great details (25). When analysing the ZTP riboswitch with the comparative mode of CoBold and zooming into significant helices with a maximum P-value of 10−3, we recover all the relevant regions of the functional structure with only a few false positives, see Figure 12. Only two structural features are missing: two small, independent helices which have not been evolutionary conserved. These two structural elements, even though they are involved in the formation of the final structure, are the least biologically relevant as they (1) do not fulfil an explicit known biological role and (2) are not directly involved in the formation of the pseudonot and the functioning of the riboswitch. CoBold thus allows us to recover the key features from the recent, detailed experimental investigation based on sequence-based information from a multiple-sequence alignment alone.
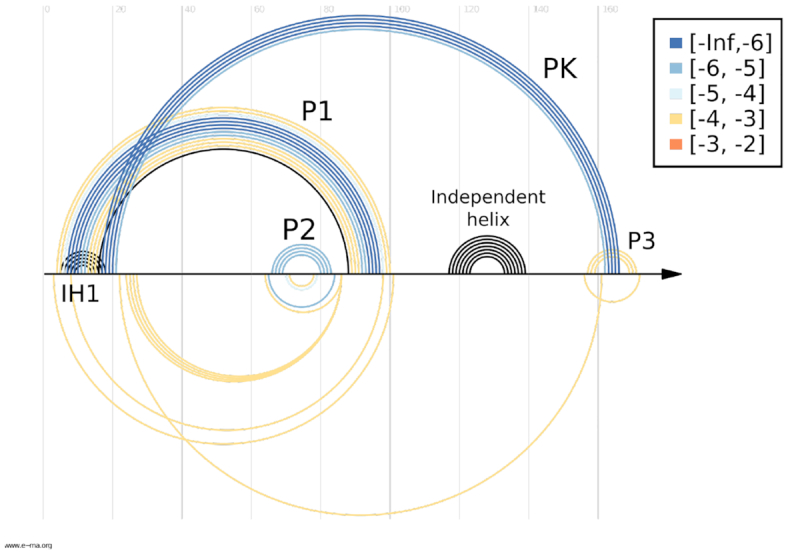
Sensitivity-specificity plot for the comparative CoBold predictions for the ZTP riboswitch for a P-value threshold of 10−3. The top of this figure shows the known RNA structure features. All non-black features correspond the features that have been correctly predicted by CoBold. These are colored depending on the statistical significance of the corresponding predicted helix. Only the independent helix is missing from the CoBold predictions. The bottom part of the figure shows only a few predicted base-pairs that are not part of the known RNA structure. The bottom part of the figure thus illustrates the high specificity of CoBold (which would be perfect if no features were shown below the line), whereas the top part of the figure thus conveniently summarises the high sensitivity of the CoBold predictions (hence the name sensitivity-specificity plot for this kind of figure). Overall, CoBold thus readily recovers the experimentally confirmed RNA structure features that matter for the co-transcriptional folding pathway of the ZTP-riboswitch with high sensitivity an specificity.
Comparative CoBold predictions of minimal P-value for the inhibitory conformation of the Levivirus. Compared to the similar, earlier Figure 11, this figure only shows the statistically most significant transient helix for each helix of the known reference RNA conformation which is shown on top, as usual. It thus represents a reduced version of the predictions in Figure 11 and allows to quickly assess the most relevant features viewed from the perspective of helix in the reference RNA structure.
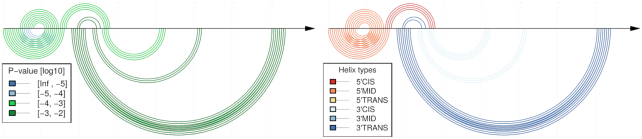
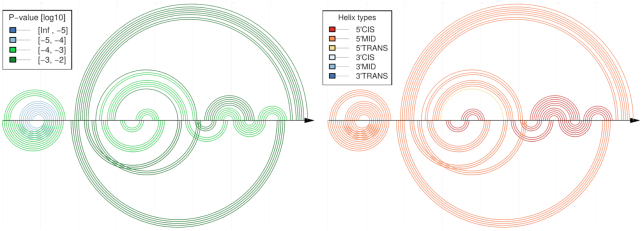
Comparative CoBold predictions for the permissive conformation of the Levivirus, see Figure 11 for the corresponding results for the inhibitory conformation of the Levivirus. As before, the top shows the reference RNA structure features and the bottom the predicted transient helices.
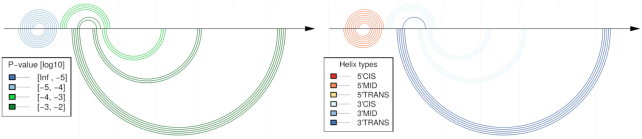
Comparative CoBold predictions of minimal P-value for the permissive conformation of the Levivirus. As before for Figures 11 and 13 for the inhibitory conformation, this figure highlights only the statistically most significant transient helix for each helix of the known RNA confirmation when compared to the predictions shown in previous Figure 14. As before, the top shows the reference RNA structure features and the bottom the predicted transient helices.
Detecting co-transcriptionally relevant transient helices
We now consider the insight that can be gained from considering the non-comparative predictions of CoBold. Figures 16 and 17 (left side) showcase the resulting predictions for both known functional conformations of the Levivirus. We can readily identify the known helices that appear during transcription and that interfere with the known reference structure, most of which are evolutionarily well conserved.
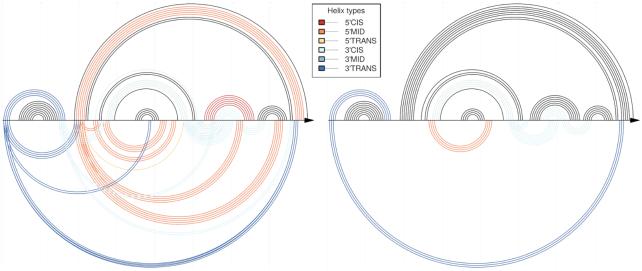
Non-comparative CoBold predictions for the inhibitory conformation of the Levivirus. Compared to Figure 11, this figure shows the predictions of CoBold that are based on the single RNA input sequence alone rather than an input multiple-sequence alignment. The right part of this figure shows the same predictions when also filtered by the comparative predictions and thus highlights only the most evolutionarily conserved transient features that are predicted by the non-comparative mode by CoBold. Note that coloring by P-value is not possible when using the non-comparative mode of CoBold, hence only the coloring by type of transient helix. As before, the top shows the reference RNA structure features and the bottom the predicted transient helices.
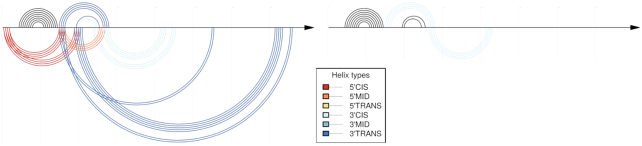
Non-comparative CoBold predictions for the permissive conformation of the Levivirus. Compared to Figure 14, this figure shows the predictions of CoBold that are based on the single RNA input sequence alone rather than the corresponding input multiple-sequence alignment. The right part of this figure shows the same predictions when also filtered by the comparative predictions which does not yield any evolutionarily conserved features. Note that coloring by P-value is not possible when using the non-comparative mode of CoBold, hence only the coloring by type of transient helix. As before, the top shows the reference RNA structure features and the bottom the predicted transient helices.
The majority of the identified transient helices belong to the neutral or beneficial kind and disruptive helices (5′-Cis) are almost absent. On closer inspection, i.e. when comparing these non-comparative with the comparative results of CoBold), these disruptive helices turn out to be not evolutionarily conserved and can thus be expected to not play a distinct functional role on the investigated evolutionary time-scale.
The most prevalent transient helices are of type 5′-Mid, 3′-Cis and 3′-Trans for the inhibitory Levivirus confirmation, which actively promote the co-transcriptional formation of the known helices and which are often found between the two known structural conformations.
For the other four families, we observe similar findings. The vast majority of the identified transient helices are beneficial or neutral with respect to the known structural conformations. A fair number of these is located between the mutually exclusive structures, thereby effectively serving as potential stepping-stones on a refolding pathway. One notable exception is the TRP operon leader, for which CoBold identifies an unusual number of disruptive (5′-Trans) transient helices that are very well conserved. These helices may potentially serve as highly-sensitive sensors that may prevent the formation of the nominal RNA confirmation unless the correct in vivo conditions are met. The TRP operon leader is also unusual in the sense that most of the transient features identified by the non-comparative mode are evolutionarily well conserved and in the sense that most of these features are predicted to affect both known reference RNA structures. CoBold is also capable of identifying the small transient helix that features during the co-transcriptional folding of the long E. coli 23S rRNA that was experimentally identified using SPET-seq by Incarnato et al. in 2017 (24).
DISCUSSION
Exciting recent research from diverse biological systems reveal that RNA structure features are responsible for regulating diverse and important steps in gene expression within the transcriptome. Yet, how these RNA structures form and change in a living biological system in vivo remains poorly understood and particularly challenging to study, both experimentally and computationally.
We here present a new computational method, called CoBold, that is capable of identifying different functional classes of transient features that may play distinct roles in guiding or preventing the formation of a known reference RNA structure in vivo. CoBold bases its predictions on sequence information only, which is either supplied by the user in terms of an single RNA structure (non-comparative mode of CoBold) or a corresponding multiple sequence alignment of evolutionarily related species (comparative mode of CoBold). CoBold employs two distinct computational algorithms to generate predictions in a comparative or non-comparative way. The non-comparative mode of CoBold explicitly takes the overall effects of co-transcriptional folding into account when generating its predictions and has been shown to generate high-quality RNA structure predictions, especially for long RNA transcripts for which the kinetic effects of co-transcriptional folding are expected to play a prominent role. The comparative mode of CoBold utilises a probabilistic method as well as dedicated, quantitative models of evolution to identify evolutionarily conserved transient helices and is also capable of estimating the reliability of the predicted helices. Provided that the a multiple-sequence alignment of decent quality can be supplied by the user, the comparative mode of CoBold should provide the most reliable prediction of transient helices with a potential functional role. Compared to the non-comparative mode of CoBold, it should also allow the identification of transient features that rely on trans interactions with ligand or other molecules which is something that the non-comparative mode of CoBold is conceptually incapable of modelling. Both modes of CoBold allow the user to generate predictions in a computationally efficient way, both in terms of memory and time, thereby readily enabling the analysis of transcripts that are significantly long than 200 nt. Predictions by CoBold can be easily generated and visualised via two dedicated web-servers at www.e-rna.org which allow for an intuitive as well as quantitative in-depth analysis of the predicted transient helices and their detailed impact on features the known reference RNA structure.
The CoBold analysis for five biological families with multiple known RNA structure configurations and known transient features shows that CoBold is able to identify known transient features and their likely impact on the formation of the known RNA structure configuration(s) for a varied arrangement of systems. Most families encode evolutionarily well-conserved transient helices that could explicitly aid the formation of the known RNA structure or serve as stepping-stones on the folding trajectory between two distinct RNA structure conformations. Our analysis of the TRP operon also discovered a number of well-conserved transient features that could easily hinder the formation of the known RNA structure confirmation(s). These features may serve as fine-tuning regulators in vivo that may prevent the formation of the known RNA structure unless the correct cellular conditions are met.
As CoBold provides very detailed insight into the transient helices, their level of evolutionary conservation and their likely impact on distinct features of a known RNA structure confirmation, we hope that the predictions of CoBold will not only aid the experimental identification of novel transient features, but will also significantly facilitate the investigation of their functional roles in vivo and help to improve computational prediction methods such as CoBold. Finally, with the exciting, recent advent of nucleotide-based therapeutics for a range of human diseases, we also hope that CoBold can significantly aid in the design of therapeutic agents to engineer co-transcriptional folding pathways in vivo or to prevent or encourage the formation of specific RNA features with distinct functional roles (39–41,41).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Helmholtz Association; M.M. gratefully acknowledges a PhD stipend from the Max Delbrück Centre for Molecular Medicine in Berlin, Germany. Funding for open access charge: Helmholtz Association.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.