 mMGE: a database for human metagenomic extrachromosomal mobile genetic elements
mMGE: a database for human metagenomic extrachromosomal mobile genetic elements
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

- Altmetric
Extrachromosomal mobile genetic elements (eMGEs), including phages and plasmids, that can move across different microbes, play important roles in genome evolution and shaping the structure of microbial communities. However, we still know very little about eMGEs, especially their abundances, distributions and putative functions in microbiomes. Thus, a comprehensive description of eMGEs is of great utility. Here we present mMGE, a comprehensive catalog of 517 251 non-redundant eMGEs, including 92 492 plasmids and 424 759 phages, derived from diverse body sites of 66 425 human metagenomic samples. About half the eMGEs could be further grouped into 70 074 clusters using relaxed criteria (referred as to eMGE clusters below). We provide extensive annotations of the identified eMGEs including sequence characteristics, taxonomy affiliation, gene contents and their prokaryotic hosts. We also calculate the prevalence, both within and across samples for each eMGE and eMGE cluster, enabling users to see putative associations of eMGEs with human phenotypes or their distribution preferences. All eMGE records can be browsed or queried in multiple ways, such as eMGE clusters, metagenomic samples and associated hosts. The mMGE is equipped with a user-friendly interface and a BLAST server, facilitating easy access/queries to all its contents easily. mMGE is freely available for academic use at: https://mgedb.comp-sysbio.org.
INTRODUCTION
Extrachromosomal mobile genetic elements (eMGEs), such as plasmids and bacteriophages, play critical roles in horizontal gene transfer (HGT) and microbial evolution within the microbial community by mediating intra- or intercellular DNA trafficking (1–4). Due to their high mobility and accessory genes related to antibiotic resistance (5–7), virulence factors (8,9) and auxiliary metabolic pathways (10–12), the eMGEs are essential for host fitness and the dissemination of drug resistance, which in turn shape microbial community structures. Furthermore, given that eMGEs frequently carry genes that encode toxins or other virulence factors, the prokaryotic hosts acquiring these genes have the potential to become deadly pathogens (13,14). Recently, disease-specific alterations of eMGEs have also been observed in several diseases (15–18), but the roles the eMGEs play in pathophysiology is still unclear, especially in a metagenomic setting.
With advances in sequencing technology, the accumulation of metagenomic data provides an unprecedented opportunity for detecting novel eMGEs (19). Recently, great progress has been made in identifying phages from metagenomic samples. For example, the human Gut Virome Database (GVD) (19) and the Integrated Microbial Genome/Virus (IMG/VR) database (20) detected phage genomes (and fragments) from assembled metagenomes. The Microbe Versus Phage (MVP) database established interactions between phages and prokaryotes based on a literature collection and a re-analysis of genomic and metagenomic sequences (21). Although those valuable resources significantly extend our knowledge of eMGEs, they focus only on phages from certain body sites, e.g. the gut, whereas plasmids were generally ignored (especially those derived from metagenomes). The existing plasmid databases, including PlasmID, for plasmid clone information and distribution (22), Plasmid ATLAS, for plasmid visual analytics and identification (23), and PLSDB, for complete bacterial plasmids (24), collected information of existing plasmids without exploring the emerging large number of metagenomes. While the ACLAME database (25) provides mobile genetic elements, including phages, plasmids and prophages, it has not been updated in the ten years prior to this publication. Thus, a comprehensive eMGE database, including both phages and plasmids, as well as detailed sample metadata and their host information, will be of great use in understanding the diversity and putative functions of eMGEs in humans.
We have thus constructed mMGE, a database of human metagenomic extrachromosomal mobile genetic elements. Currently mMGE contains a total of 517 251 non-redundant eMGEs, including 92 492 plasmids and 424 759 phages, that we identified from 66 425 human metagenomic samples. In addition to basic information (including sequence characteristics, interactions with prokaryotic hosts, gene contents and taxonomic annotations), the extensive metadata of the samples, the abundances and distributions of the eMGEs across samples, phenotypes and populations are also available, allowing users to explore their biological functions, biogeographical patterns and habitat preferences. In addition, users can browse or query eMGE records in multiple ways, including eMGE clusters, metagenomic samples and putative hosts. mMGE is equipped with a user-friendly interface and a BLAST server, facilitating users to access and query all its contents easily.
DATABASE CONSTRUCTION
Figure 1 illustrates the overall workflow of mMGE. In brief, 66 425 metagenomic human samples were collected, followed by data preprocessing, eMGE identification, abundance calculation and eMGE annotation. Below we provide more details of materials and methods used in this study.

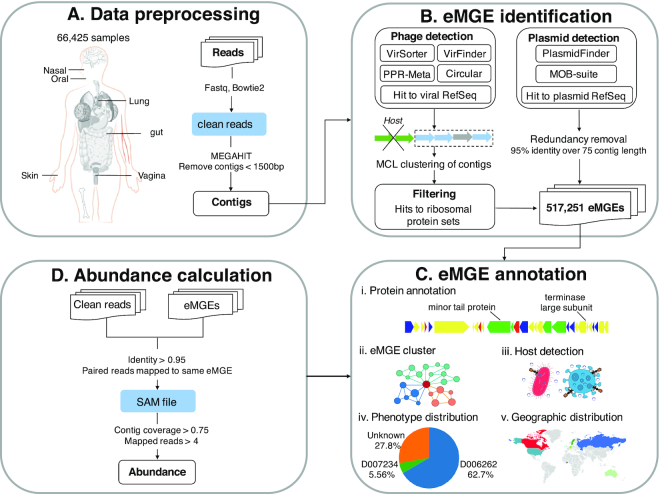
The overall workflow of mMGE. (A) Data pre-processing. A total of 66 425 human metagenomic samples and associated meta-data were collected, followed by pre-processing and assembly of raw sequencing reads. (B) eMGE identification. State-of-art toolsets were used to identify eMGEs. (C) eMGE annotation. Comprehensive annotations were provided for the eMGEs, including putative protein function and host information, etc; (D) Abundance calculation. Abundances and prevalence of the eMGEs across samples were also determined. See ‘Materials and Methods’ section for more details.
Data collection and processing of metagenomic sequencing reads
Raw sequencing reads of 80 889 human metagenomic samples, from 370 datasets, were downloaded from the NCBI SRA (Sequencing Read Archive, https://www.ncbi.nlm.nih.gov/sra; Supplementary Table S1) database. Meta-data, including experimental conditions, dates of sampling and human host information, were also retrieved from corresponding publications and/or the NCBI SRA database. Phenotypes associated with samples were organized according to MeSH (Medical Subject Headings) (26), a hierarchically organized controlled vocabulary for biomedical information, while the metagenomic samples were organized according to the Genome Online Database classification system (27).
The FastQC (v0.11.8, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to check the overall quality of the downloaded sequences, followed by the use of Bowtie2 (28) to remove host-derived reads through mapping to the human reference genome (hg38). To trim sequence and remove low-quality bases, fastp (29) was utilized with the following parameters:’-l 50 -x -q 20 -u 5 -M 20 -W 4′. The samples containing less than 10 000 reads were removed from the subsequent analysis to ensure quality of the data. Then MEGAHIT (v1.2.8) (30) was used to assemble the high-quality clean reads per sample. After assembling, contigs that were less than 1.5 kb were discarded and the redundancy was removed with a threshold of 95% identity over 75% of their length.
In total, after quality control, we collected 66 425 human metagenomic samples associated with 110 phenotypes across 18 body sites from 49 countries. Table 1 summarizes the statistics of the samples we have collected.

| Body site | #samples | #projects | #associated phenotypes | #associated countries |
|---|---|---|---|---|
| Gut | 41 841 | 233 | 63 | 42 |
| Oral cavity | 11 313 | 41 | 9 | 9 |
| Skin | 5384 | 30 | 7 | 7 |
| Blood | 2976 | 20 | 26 | 8 |
| Nasopharyngeal | 1930 | 16 | 9 | 6 |
| Vagina | 1028 | 6 | 1 | 3 |
| Sputum | 379 | 4 | 1 | 6 |
| Eye | 229 | 10 | 1 | 2 |
| Urethra | 123 | 5 | 3 | 3 |
| Tooth | 106 | 3 | 4 | 3 |
| Reproductive system | 76 | 1 | 0 | 1 |
| Milk | 60 | 2 | 0 | 2 |
| Trachea | 33 | 1 | 2 | 1 |
| Lung | 25 | 2 | 2 | 1 |
| Liver | 20 | 2 | 2 | 2 |
| Circulatory system | 12 | 1 | 1 | 0 |
| Lymphatic system | 11 | 3 | 3 | 2 |
| Excretory system | 1 | 1 | 1 | 1 |
eMGE identification, dereplication and clustering
After assembling and removing redundancy, all contigs were then piped through VirSorter (31), VirFinder (32) and PPR-Meta (33) for phage identification while went through PlasmidFinder (34) and MOB-suite (35) for plasmid identification. The phage sequences were identified by following the procedures described in (36) but with more stringent criteria. Firstly, the assemblies that met at least two of the following criteria were kept as putative phage contigs: (i) VirSorter positive (categories 1–2); (ii) PPR-Meta phage score > 0.7; (iii) VirFinder score > 0.6 and P-value < 0.05; (iv) Be circular; (v) Hit a phage genome from RefSeq with >50% identity and >90% coverage of contig length according to BLASTn (37). Subsequently, the candidate phage contigs obtained above were decontaminated using CheckV (38); those met the following criteria were discarded as described previously (36,39): (1) having more than three hits against ribosomal protein sets in COG (40) database or (2) having at least one ribosomal protein, VirSorter negative and non-circular and having less than three Hidden Markov Model (HMM) hits to the prokaryotic viral orthologous groups (pVOGs, E-value < 1e-5) (41) per 10 kb. For the detection of plasmid sequences, the contigs satisfying at least one of the following criteria were selected as putative plasmid contigs: (i) Predicted by PlasmidFinder as positive; (ii) Predicted by MOB-suite as positive; (iii) Hit a plasmid genome from RefSeq with >50% identity and >90% coverage of contig length according to BLASTn.
All the above identified eMGE contigs were dereplicated at the population level if they shared >95% nucleotide identity across >70% coverage according to Lincluster (42), resulting in 517 251 non-redundant eMGE populations (92 492 plasmids and 424 759 phages). Then, a sequence-based classification framework was adopted to group closely related eMGE genomes into clusters (43). As a result, a total of 70 074 eMGE clusters containing 316 926 eMGE fragments (ranging from 2 to 384 members per cluster) were obtained, with most clusters (46.96%) having only two members. In addition, the quality and completeness of each phage contig were evaluated with CheckV (38) and the ‘Minimum Information about an Uncultivated Virus Genome’(MIUViG) framework (44) was utilized to classify phage contigs as ‘Genome fragment’ or ‘High-quality draft genome’. Consequently, 23 738 high-quality genomes were obtained and 14 990 of them were complete.
Annotation of eMGE contigs
The open reading frames (ORFs) for each eMGE contig were predicted using prodigal (v2.6.3) (45). With the predicted proteins were subjected to all-vs-all Blastp with thresholds of E-value < 1e-5 and bit score > 50, the proteins were clustered into families by using a Markov Clustering Algorithm (MCL) with log-transformed E-value as similarity score and two for MCL inflation (46). The HMM profile for each protein family (protein family is also the protein cluster) was built with MAFFT (47) and hmmbuild (48). The functional annotations of all proteins were achieved by querying against PFAM (49), VOGdb (http://vogdb.org/) and eggNOG (50) databases. Consequently, about 40.86% of all predicted proteins had hits to at least one of the public databases, leaving the majority of the eMGE proteins as having unknown functions. For each protein family, its functional annotation was the one where >75% of its members were annotated with the functions (51), while those that could not be annotated in the previous way were queried against proteins from RefSeq with hmmsearch (52) (threshold of 1e-5 for E-value and 50 for bit score).
For each eMGE contig, taxonomy annotations were achieved through the following three steps. Firstly, 21 hallmark POGs recognized as taxon-specific signatures were used to assign a taxonomic lineage to eMGEs at different levels (order, family, subfamily and genus). Secondly, the eMGEs that clustered with genomes from RefSeq were able to be assigned to known taxonomic genera (53). Finally, for those contigs that could not be assigned to a specific taxonomy with above two steps, we tried to annotate them using a majority-rule approach. The proteins predicted in the contig were aligned against the proteins deposited in the UniProt database (54), and the taxonomy level was determined to a taxon if more than 75% of the proteins hit the same taxon (51).
eMGE-host identification
For each eMGE contig, its microbial hosts were determined by following the approaches described previously (19). Firstly, the eMGE fragments were aligned against NCBI RefSeq with BLASTn, and RefSeq genomes with >95% identity across more than 2500 bp were considered as putative hosts of eMGEs. Secondly, the bacterial genomes from NCBI RefSeq and metagenomic assemblies >1500 bp were used to build CRISPR-Cas spacer database. The CRISPR spacers in microbial genomes and assemblies were predicted using MinCED (55) with default parameters (4 110 100 spacers were obtained). The detected spacers were then aligned against phage fragments using BLASTn with the following options: -task blastn-short -word_size 5, E-value < 1e-5, bit score > 45, identity > 95% of full length, and a maximum of two mismatches was allowed. Finally, tRNAscan-SE (56) was separately used to identify tRNA genes from phage sequences and bacterial genomes, and bacterial genomes with tRNA genes matching phage tRNA genes at 95% identity across 100% of the length were considered as the corresponding host. The host of eMGE contigs can be determined in either of the three ways.
In total, we identified 2 032 843 eMGE-host associations, with the hosts spanning across 20 bacterial and archaeal phyla (Figure 2A). For each eMGE sequence and/or eMGE cluster in mMGE, its host range was determined by following the way described previously (21). Briefly, for eMGEs with only one host, the host range was assigned as the taxonomic rank of the host in the NCBI taxonomy database, while the host range was defined as the taxonomic rank of the Last Common Ancestor of all its hosts if an eMGE fragment infects multiple hosts. mMGE provides host information for 115 072 eMGE fragments (22.24% of all the eMGE fragments or genomes), and approximately 50% of eMGE clusters have host range at ‘species’ or ‘genus’ level (Figure 2B).
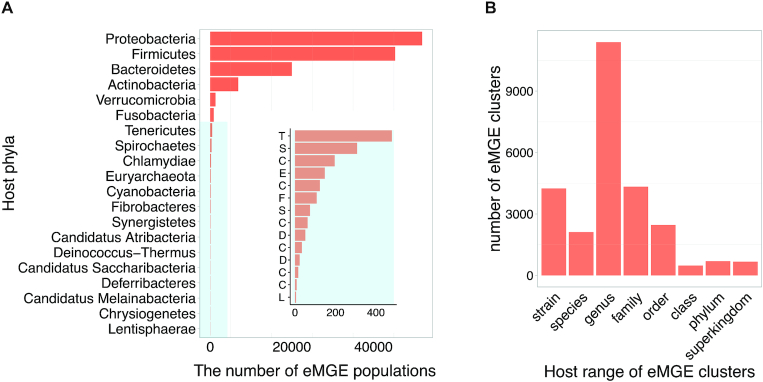
The distribution of identified eMGE-hosts. (A) The number of eMGE populations associated with their corresponding bacterial and archaeal host phyla; The inset with blue background provides resolution for the low frequency bacteria host phyla and each letter on the y-axis corresponds to the first letter of host phyla's name. (B) The number of eMGE clusters distributed across different host range levels.
Abundance and prevalence of eMGEs across samples
To calculate the relative abundance of different eMGE fragments, the quality-filtered reads were mapped to eMGE contigs using BWA mem (57), where the reads mapped with <95% identity or paired-reads mapped to different locations were removed. The Bedtools (58) genomecov was then used to calculate the coverage over contigs and only the eMGE contigs with >75% of length covered by reads were considered to be present in that sample (59). Then the number of mapped reads to a contig were normalized by the total number of clean reads per metagenomic sample, which was used as the approximate relative abundance for that eMGE fragment. The relative abundance of each eMGE cluster was calculated by summing the percentage of reads mapped to members belonging to that cluster.
DATABASE OVERVIEW AND FUNCTIONALITY
Overview of mMGE
The current version of the mMGE database contains 517 251 unique/non-redundant eMGEs (92 492 plasmids and 424 759 phages) identified from 66 425 metagenomic samples. Figure 3 shows the comparison of mMGE against multiple public databases, including IMG/VR (20), GVD (19) and plasmid RefSeq. Figure 3A shows the completeness of phage contigs from IMG/VR, GVD and mMGE estimated with CheckV, from which we can see that mMGE (Complete: 7.46%, high-quality: 4.36%, medium-quality: 20.32%, low-quality: 61.40%) has the highest percentage of complete genomes while has comparable quality compared with IMG/VR (Complete: 5.16%, high-quality: 2.45%, medium-quality: 23.11%, low-quality: 64.41%) and GVD (Complete: 7.10%, high-quality: 6.02%, medium-quality: 15.85%, low-quality: 65.97%). Compared with RefSeq, GVD and IMG/VR, mMGE significantly extends the number of phages and plasmids as shown in Figure 3B and C, and mMGE can successfully recover half of the phages detected by GVD or IMG/VR (56.67% in GVD and 47.01% in IMG/VR recovered by mMGE separately) which is much better compared with the overlap between GVD and IMG/VR (6.70 and 3.25% shared by GVD and IMG/VR separately).
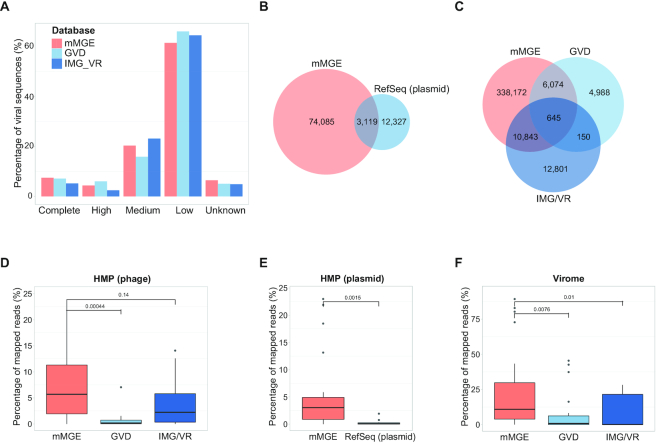
Contents of mMGE and comparisons with public databases. (A) The completeness and quality of phage contigs estimated by CheckV for mMGE, IMG/VR and GVD, where high denotes high quality and the same for medium and low; (B) and (C) The venn diagram of plasmids and phages from different sources, where all contigs were dereplicated at population level and decontaminated with CheckV and only phage populations from human samples were considered; (D–F) The percentage of mapped reads for phages or plasmids from HMP dataset (D and E) and Virome dataset (F), where the HMP dataset includes 20 samples from PRJNA48479 and the Virome dataset contains viral enriched samples that came from PRJNA588313.
Moreover, we evaluated the identification sensitivity of plasmids and phages from different databases by comparing their percentage of mapped reads. For this purpose, three datasets were used for a fair comparison, including 20 human gut metagenomic samples from PRJNA48419 (60) (HMP dataset), the viral enriched samples from PRJNA588313 (Virome dataset) and a plasmid dataset containing 131 plasmid contigs assembled from human metagenomes with metaplasmidSPAdes (61) (Metaplasmid data). Figure 3D–F separately shows the percentages of mapped reads for HMP and Virome datasets, from which we can clearly see that mMGE has the best identification sensitivity. For the Metaplasmid dataset, 63 of 131 plasmid contigs can be successfully recovered by mMGE while RefSeq plasmid covers only 29 of them. From the results shown in Figure 3, we can see that mMGE significantly extends the number of eMGEs with comparable quality but much higher identification sensitivity compared with existing databases.
Web interface
mMGE provides a user-friendly and interactive portal for browsing and querying all eMGEs and their associated information as shown in Figure 4. In the ‘eMGE’ page, the information of all eMGE fragments or genomes as well as their associated information can be easily browsed (Figure 4A). The detailed information page for a specific eMGE can be available by clicking on the eMGE ID of interest. With the relative abundance of eMGEs across samples, mMGE enables the users to investigate the distribution patterns of eMGEs across countries and human phenotypes. The detailed information of its hosts and the meta-data of metagenomic samples in which this eMGE can be detected is also available. All eMGE records can be browsed via eMGE clusters (Figure 4B), eMGE-host interactions (Figure 4C) and metagenomic samples (Figure 4D). In the ‘eMGE cluster’ page, mMGE enables users to browse the most relevant information of eMGE clusters, including the number of members in the cluster (‘eMGE members’), the number of samples in which they are present (‘Sample count’), the number of identified putative hosts (‘Total hosts’), the number of protein families they contain (‘Protein members’) and the predicted host-range (Figure 4B). All identified eMGE-host interactions are listed in the ‘Interactions’ page, where the interactive visualization of the eMGE-host interaction network is also provided. The ‘Data’ page provides manually curated meta-data of the metagenomic samples used whenever possible (Figure 4D). The metagenomic samples can be viewed according to the collection sites, body sites or phenotypes. To facilitate researchers to download the raw sequencing data, additional links to samples and projects from NCBI were also provided. For each metagenomic sample or project, we also summarized the total number of associated eMGEs and the associated eMGE sequences of each metagenomic sample can be obtained. In addition, mMGE provides comprehensive annotations for protein families associated with eMGEs in the ‘Proteins’ page (Figure 4E).
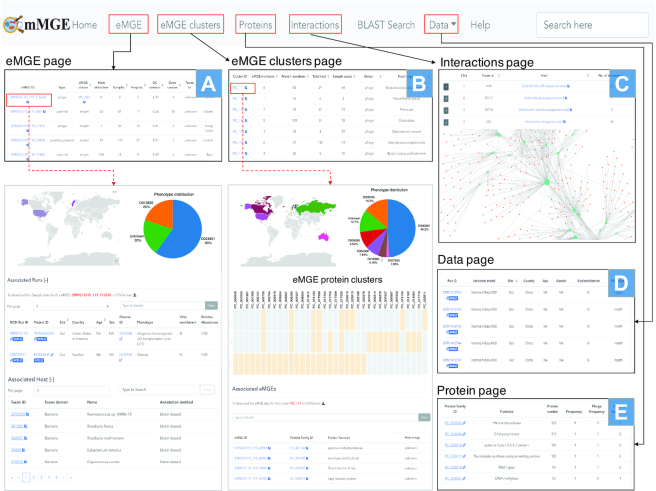
The user-friendly web interface of mMGE. (A) The ‘eMGE’ page shows the basic information of eMGEs; (B) The ‘eMGE cluster’ page shows the information about eMGE clusters; (C) The ‘Interaction’ page presents the interactions between eMGEs and their hosts; (D) The ‘Data’ page shows the information about each sample and project; (E) The ‘Proteins’ page presents the protein content of each eMGE. Those pages can be cross searched to provide more detailed information of eMGEs or eMGE clusters.
Protein clusters evolutionarily conserved within eMGE clusters
Despite the diverse gene contents across different plasmids and phages, it has been found that conserved genetic modules maybe shared between related eMGE genomes (62). To facilitate users to explore the evolutionary relationships between eMGEs within eMGE clusters, the visualization of the matrix of protein clusters across eMGEs was also provided (Figure 4B). For example, Figure 5 shows an example of a matrix view of protein clusters within the eMGE cluster ‘MC_62′, where the functions associated with each protein cluster are provided. We can see that the protein clusters shared by all members in ‘MC_62′ are involved in assembly (‘head’, ’capsid’ and ’neck’), DNA packaging (‘terminase’), infection (‘tail’, ’baseplate’ and ’virion’, ’portal’) and lysis (‘lysozyme’ and ’lysin’), indicating the essential functional roles of these proteins.
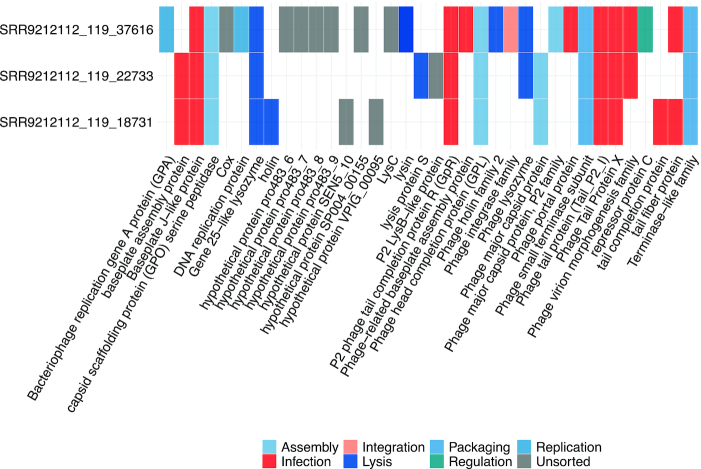
The example matrix view of protein clusters within the eMGE cluster ‘MC_62’. The columns correspond to the protein clusters while the rows represent eMGE members within this cluster. The protein clusters were colored according to their functional annotations.
Querying eMGE with BLAST
To facilitate users to query mMGE with their own sequences, we provided a BLAST server for the users to align their sequences against the eMGEs deposited in the mMGE database (63). In this way, users may easily check what phages or plasmids their sequences are based on hits to the eMGEs from mMGE. The BLAST search could be accessed at: https://mgedb.comp-sysbio.org/#/submitBlast.
FUTURE DIRECTIONS
Since eMGEs are a major source of antimicrobial resistance genes, virulence and pathogenicity related genes, insertion sequences and other transposable elements, an annotation and discovery pipeline will be provided in a future version. In addition to collecting eMGEs detected in human metagenomes, the future version will complement the data with eMGEs derived from other source such as animals or ocean. We note that there is much room to improve mMGE in the following directions: (i) Including a binning method, such as MetaBat (64), which can be used to merge contigs derived from the same population and extend assembly completeness; (ii) Improving the discovery pipeline so it can detect more eMGEs with higher quality; (iii) Incorporating Long-read metagenomic datasets, which can then be used to improve the assembly and identify more eMGEs (4).
CONCLUSION
Due to the high mobility of eMGEs and their complex interactions with microbial hosts, the analysis of eMGEs is essential for characterization of microbial communities and exploring their potential roles in regulating microbial communities. Here we have introduced mMGE, an integrative resource for environmental uncultivated eMGEs derived from diverse human body sites coupled with extensive annotations. With 66 425 samples collected from 18 body sites, 110 human phenotypes and 49 countries, we manually curated meta-data of all samples and applied stringent criteria to keep only high-confidence eMGE sequences. In total, 517 251 unique eMGEs were obtained, including 424 759 phages and 92 492 plasmids. Extensive comparisons with existing database indicated that mMGE contains more eMGEs with higher quality. To facilitate users to perform downstream analysis, mMGE provides precomputed relative abundance of eMGEs, their prevalence within and across samples as well as putative associations with phenotypes. Comprehensive annotations of each eMGE record including sequence characteristics, protein content, taxonomy affiliation and host-eMGE interactions are also available at the website. The web server allows users to browse the included eMGE records in multiple ways and uploaded nucleotide sequences can be searched in the database. mMGE provides a modern, interactive and user-friendly interface, enabling users to easily access and query all its contents. As metagenomic datasets continue to expand, we will continue developing mMGE in the near future by including eMGEs detected from more samples and more comprehensive annotations.
DATA AVAILABILITY
All data are freely available to all academic users. This work is licensed under a Creative Commons Attribution 3.0 Unported Licence (CC BY 3.0). In addition to downloading the data provided on certain web pages, the users can download all data from the ‘Data download’ section of the ‘Help’ page.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [61932008, 61772368]; Shanghai Municipal Science and Technology Major Project [2018SHZDZX01]; Shanghai Science and Technology Innovation Fund [19511101404]; National Key Research and Development Program of China [2018YFC0910503 to X.M.Z., 2018YFC0910502, 2019YFA0905601 to W.H.C]. Funding for open access charge: National Key Research and Development Program of China; National Natural Science Foundation of China; Shanghai Municipal Science and Technology Major Project.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
49.
50.
51.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.