 GIMICA: host genetic and immune factors shaping human microbiota
GIMICA: host genetic and immune factors shaping human microbiota
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

- Altmetric
Besides the environmental factors having tremendous impacts on the composition of microbial community, the host factors have recently gained extensive attentions on their roles in shaping human microbiota. There are two major types of host factors: host genetic factors (HGFs) and host immune factors (HIFs). These factors of each type are essential for defining the chemical and physical landscapes inhabited by microbiota, and the collective consideration of both types have great implication to serve comprehensive health management. However, no database was available to provide the comprehensive factors of both types. Herein, a database entitled ‘Host Genetic and Immune Factors Shaping Human Microbiota (GIMICA)’ was constructed. Based on the 4257 microbes confirmed to inhabit nine sites of human body, 2851 HGFs (1368 single nucleotide polymorphisms (SNPs), 186 copy number variations (CNVs), and 1297 non-coding ribonucleic acids (RNAs)) modulating the expression of 370 microbes were collected, and 549 HIFs (126 lymphocytes and phagocytes, 387 immune proteins, and 36 immune pathways) regulating the abundance of 455 microbes were also provided. All in all, GIMICA enabled the collective consideration not only between different types of host factor but also between the host and environmental ones, which is freely accessible without login requirement at: https://idrblab.org/gimica/


INTRODUCTION
The microbial communities (MCs) in different sites of human body are known to be instrumental in host physiology and health (1–4). The composition of these MCs has been generally considered as susceptible to a variety of environmental factors (5–7) (including: diet, lifestyle, disease, etc.), which thus mediates changes in host metabolic status (5) and regulates the development of chronic disease (6–9). Besides these environmental ones, the host factor has recently emerged as another crucial determinant that shapes human microbiota (10–12). Particularly, different from environmental ones, host factors are able to define the chemical and physical landscape (including nutrient availability and activity threshold for immune system) inhabited by host microbiota (13–18), which therefore determine the heritable nature of microbiome (12) and immune system activity (13,19).
There are two popular host factors that are frequently considered in current MC studies (19–23): (i) host genetic factors (HGFs, including: single nucleotide polymorphism (SNP), gene copy number variation (CNV) and non-coding ribonucleic acid (RNA) regulation) are crucial in exploring the heritability of microbiota compositions (24), revealing the mechanisms underlying the adaptation to environmental fluctuations (11), and discovering promising drug target (25) and targeted therapeutics (26,27); (ii) host immune factors (HIFs, including: checkpoint molecule, cytokine, antibody and lymphocyte) are critical for maintaining the symbiotic balance between host and microbiota (28), promoting the activities of immune system (13,20), and boosting the efficacy of immunotherapy (29). Since the composition of human MCs is greatly affected by their inhabited chemical/physical landscapes (13), the accumulation of human factors that define such landscapes can give significant insights into current MC studies.
So far, a variety of human MCs-related databases have been constructed and currently active, the majority of which focus on providing sequencing/metagenomic data (dbCAN-seq (30), eHOMD (31), gcMeta (32), GMrepo (33), HPMCD (34), MAHMI (35), MBGD (36), MetaRef (37), MDB (38)), while some others are specialized in containing the data of either metabolites/metabolic reactions (HMDB (39), mVOC (40), and VMH (41)) or taxonomic profiling (MMDB (42)). Only three databases are available now to describe the environmental factors affecting the compositions of MCs: Disbiome (43) linking the microbiome to disease indication, gutMDisorder (44) collecting the immense data of diet, drug and lifestyle that lead to microbial dysbiosis and aBiofilm (45) providing some environmental substances that inhibit bacteria. However, no database has yet been designed to provide the host factors shaping human microbiota.
Herein, a new database of host genetic and immune factors shaping human microbiota (GIMICA) was therefore introduced. First, a comprehensive literature review on human MC was conducted. Thousands of microbe species were discovered as inhabiting human blood, gut, lung, mucosa, nose, ocular surface, oral cavity, skin, and vagina. These microbe species were very diverse and belonged to >50 taxonomic phyla (such as: Firmicutes, Actinobacteria and Ascomycota). Second, based on these microbes, the HGFs and HIFs for each species were collected via literature review, and the host data of both genetic and immune factors were then provided in the GIMICA database. Third, the crosslinks of each microbe to other MCs-related knowledge bases, especially the ones describing the environmental factors affecting the composition of MCs, were systematically discovered. These crosslinks were essential because they allowed, for the first time, the collective considerations not only among different host factors but also between the host and environmental ones. All in all, the GIMICA comprehensively provided the host genetic and immune factors that shape human microbiota (as illustrated in Figure 1), which allowed the collective considerations among the different types of factor (between HGF and HIF; between host and environmental factors). Since the composition of MCs should be considered as a complex shaped by the combination of both host and environmental factors (46–48), the GIMICA is expected to have implication for the future practice of MCs-related studies on human physiology and health (1–3,49–53).

GIMICA comprehensively described host genetic and immune factors shaping human microbiota, which allowed the collective consideration among different types of factors (between host genetic factors and host immune factors). By providing the crosslinks to available databases (Disbiome (43), gutMDisorder (44) and aBiofilm (45)) with environmental factors information, GIMICA further enabled the collective consideration between the host and environmental factors.
FACTUAL CONTENT AND DATA RETRIEVAL
Collecting and confirming microbes inhabiting human
The data of microbe species inhabiting human body were first collected by literature reviews in PubMed (54) using the keyword combinations of: ‘microbial community + human’, ‘microbe + human’, ‘microbiota + human’, etc. Second, several MCs-related databases (such as: eHOMD (31), GMrepo (33), VMH (41) and gutMDisorder (44)) were reviewed, and hundreds of microbes in these databases were collected for further assessing their suitability to be included in GIMICA. In total, 6438 microbes were identified based on the researches in these steps above. Third, the names of all these identified microbes were standardized by searching NCBI Taxonomy database (55), which resulted in 5386 microbe species with well-defined Taxonomy ID. A repository that contained the diverse synonyms of each species were further constructed using NCBI Taxonomy (55) data. Fourth, for those microbes collected from MCs-related databases, literature search in PubMed (54) was performed based on the keyword combinations of ‘'Microbe Name’ + human’, ‘‘Microbe Synonym’ + human’, etc. During this literature review and the review in the first step, the body sites in human, where each microbe species inhabited, were carefully identified and systematically recorded. Consequently, 4257 microbes were confirmed to inhabit nine sites of human body. Particularly, 281, 2436, 513, 225, 197, 164, 468, 1012 and 276 microbe species (bacteria, fungi, metamonada, amoebozoa and archaea (56–58)) were found to colonize in human blood, gut, lung, mucosa, nose, ocular surface, oral cavity, skin and vagina, respectively (59–63). These microbes are diverse and belong to 58 taxonomic phyla (such as Firmicutes, Actinobacteria, Ascomycota, Euryarchaeota, Bacteroidete, Proteobacteria and Tenericutes). Finally, the affiliated information for each human microbe were systematically collected using literature review, which contained taxonomic lineage (kingdom, phylum, class, order, family, genus, and species), oxygen dependency/tolerance (aerobe, anaerobe, aerotolerant and aerophilic), mechanisms of microbial metabolism (such as catalytic, fermentative, oxidative, proteolytic, respiratory and saccharolytic), sites of human body inhabited (such as gut, oral cavity and skin), Gram-staining classifications (negative, positive and variable), impact on/relation to host (probiotic, commensal, opportunistic and pathogen) and other general descriptions (disease-relevance, metabolic product, mechanism of symbiosis, the number of genes/transcripts and genome size).
Host genetic factors shaping human microbiota
The host genetic factors that shape human microbiota are characterized by a variety of regulatory types, which contained single nucleotide polymorphisms (upstream or downstream gene variants, untranslated region variants, intron variants, etc.), copy number variations (duplication, deletion, etc.), and non-coding RNA (lncRNA, rRNA, miRNA, miscRNA, sRNA, pseudogene, etc.). Such factors were essential in exploring the heritability of microbiota compositions (24), revealing the mechanism underlying the adaptation to environmental fluctuations (11), and discovering the drug target and targeted therapeutics (26). Because the HGFs data were largely dispersed in literatures, the PubMed (54) was systematically searched to discover the regulation of human microbiota by HGFs. Particularly, keyword combinations of ‘genetic variant + ‘Microbe Name’’, ‘host genetic factor + ‘Microbe Name’’, ‘genetic variation + ‘Microbe Name’’, ‘gene copy number variations + ‘Microbe Name’’, ‘CNV + ‘Microbe Name’’, ‘nucleotide polymorphism + ‘Microbe Name’’, and ‘SNP + ‘Microbe Name’’ were adopted for literature reviews, and the resulting publications were manually assessed for retrieving any HGFs-related information.
As a result, the collected data included the various regulatory types of host genetic factors (single nucleotide polymorphisms, copy number variations and non-coding RNAs), the detail sub-types of each HGF type (such as upstream variant, gene duplication and lncRNA), the genetic sequence, genome location and strand type for each HGF, and the impacts of each HGF on studied microbe species (up/down-regulation of microbe abundance, promotion/inhibition of the gene expression in microbes). As a result, current version of GIMICA had 1,368 single nucleotide polymorphisms (590 intron, 74 missense, 54 synonymous variants, 43 upstream gene, 26 non coding transcript variant, 17 UTR, 7 downstream gene, etc.) regulating the relative abundances of 267 microbes that primarily inhabit host gut, mucosa, and vagina; 186 copy number variations (37 duplications and 149 deletions) modulating the expressions of 156 microbes that live mainly in human oral cavity, gut, lung, skin, blood, and mucosa; 1297 non-coding RNA regulations (514 miRNAs, 683 lncRNAs, 14 sRNAs, 9 pseudogenes, etc.) altering the gene expression of 251 microbe species that reside mostly in host lung, oral cavity, gut and skin. All these HGFs data can be assessed and retrieved using various search strategies in both the Home page and the subpage (which is entitled as ‘Host Genetic Factors’) of the GIMICA database. The example host genetic factors in GIMICA were illustrated in the Figure 2.

Example host genetic factors in GIMICA. 1368 single nucleotide polymorphisms regulating the relative abundance of 267 microbes that primarily inhabit human gut, mucosa and vagina; 186 gene copy number variations modulating the expression of 156 microbes that live mainly in human oral cavity, gut, lung, skin, blood and mucosa; 1297 non-coding RNA regulations altering the expression of 251 microbes that reside mostly in host lung, oral cavity, gut and skin.
Host immune factors shaping human microbiota
The host immune factors that shape human microbiota contained a number of immune molecules and cells regulating microbiota compositions (such as checkpoint molecule, cytokine, complement component, eosinophils, immunoglobulin, innate lymphoid cell, macrophage, monocyte, natural killer cell, retinoic acid receptor, toll-like receptor, etc.). These factors are critical for maintaining symbiotic balance between host and microbiota (28), promoting the activities of immune system (13), and boosting the efficacy of immunotherapy (29). Similar to the collection of the HGF data, the HIF data were identified by literature search based on different keyword combinations, which included ‘host immune factors + ‘Microbe Name’’, ‘immune cell + ‘Microbe Name’’, ‘immune system + ‘Microbe Name’’, ‘immunity + ‘Microbe Name’’, ‘checkpoint molecules + ‘Microbe Name’’, ‘complement component + ‘Microbe Name’’, etc. The resulting publications were also manually assessed for retrieving any HIF-related information.
Consequently, the collected data included various regulatory types of host immune factors (HIF), such as checkpoint molecules, cytokines, complement component, eosinophils, immunoglobulin, etc. Moreover, other critical data were also collected and provided, which included the names of immune-related molecules/cells, the sequence, protein family, signaling pathway, and molecular function for each HIF, and the impacts of each HIF on a studied microbe (up/down-regulation of microbe abundance, promotion/inhibition of the gene expression in microbe). All in all, GIMICA contained 126 lymphocytes (including helper/cytotoxic/regulatory T-cells, plasma/regulatory B-cells, natural killer cells, etc.) and phagocytes (including macrophages, neutrophils, dendritic cells, monocytes, eosinophils, mast cells, etc.) affecting the expression of 258 microbes that are mainly located in human blood, gut, skin, oral cavity and ocular surface; 387 immune proteins (cytokines, immune receptors, checkpoint proteins, complement components, etc.) altering the compositions of 235 microbes that primarily reside in human gut, oral cavity, blood, ocular surface, and vagina; 36 immune-related signaling pathways (toll-like receptor, interleukin, etc.) modulating the gene expressions of 65 microbe species that live in human blood, gut and skin. All these HIF data can be assessed and retrieved using various search strategies in both the Home page and the subpage entitled ‘Host Immune Factors’ of GIMICA. The example host immune factors in GIMICA were illustrated in the Figure 3. Figure 4 shows the body sites distribution of microbes inhabiting human (Figure 4A) and the statistics of host genetic factors and immune factors shaping human microbiota in GIMICA (Figure 4B).
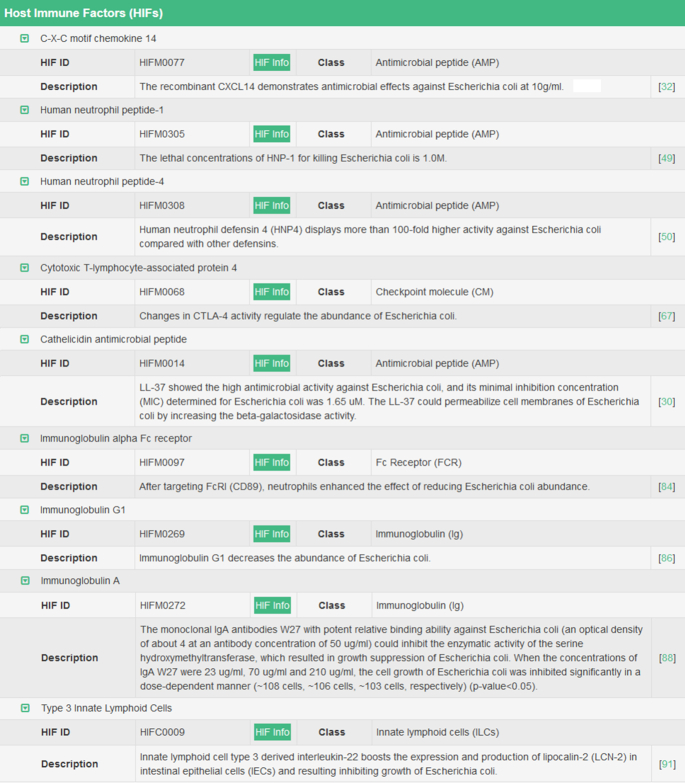
Example host immune factors in GIMICA. 126 lymphocytes/phagocytes affecting the expressions of 258 microbes that are mainly located in human blood, gut, oral cavity and ocular surface; 387 immune proteins altering the compositions of 235 microbes that primarily reside in human gut, oral cavity, blood and vagina; 36 immune-relevant signaling pathways modulating the expressions of 65 microbes that live in human blood, gut and skin.
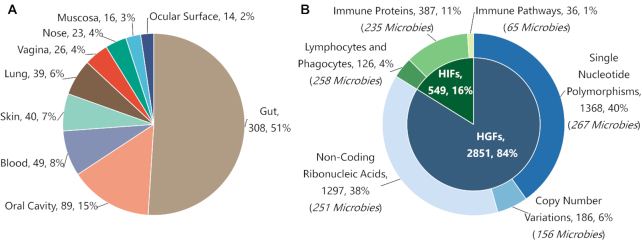
The statistic of microbes and host factors in GIMICA. (A) The body sites distribution of microbes inhabiting human; (B) The statistics of host genetic factors (HGFs) and host immune factors (HIFs) shaping human microbiota in GIMICA.
Collective consideration among different factors
Human microbiota composition was frequently discovered to be collectively shaped by multiple host factors (46,47). For example, genetic variation in host protein NOD2 (HGF) and the immune response stimulated by the NF-kappa B pathway (HIF) could collectively increase the abundance of microbes in the Enterobacteriaceae family, and this microbiota dysbiosis had been discovered as a leading cause of neonatal meningitis (13). In other words, when associating the alteration of microbiome composition with a studied disease, both host genetic and immune factors should be assessed. To make this collective consideration possible, both types of host factor (HGF and HIF) were systematically reviewed and collected in GIMICA for each microbe. As a result, 36.4% of all microbes in GIMICA database were identified with both HGF and HIF reported.
Moreover, human microbiota composition was also found susceptible to both environmental and host factors (46,64). For example, a genetic polymorphism in host interlukin-5 gene (host factor) and the environmental factor (such as high-fat diets) could collectively elevate the concentration of Prevotella melaninogenica in vaginal microbiota, which in turn led to a chronic inflammation (65). Since the environmental factors had been long studied, the collective considerations of both host and environmental factors were of great interests and significant importance. To enable this collective consideration, each microbe in GIMICA was cross-referenced and linked to available databases (Disbiome (43), gutMDisorder (44) and aBiofilm (45)) that provided environmental factor. As a result, 88.4% of all microbes in GIMICA were identified with both host and environmental factors.
Data standardization, access and retrieval
To make the access and analysis of GIMICA data convenient for all users, the collected raw data were carefully cleaned up and then systematically standardized. These standardizations included: (i) all diseases in GIMICA were standardized by the latest version of International Classification of Diseases (ICD-11, officially released by World Health Organization (66)), which was expected to serve comprehensive health management (67); (ii) all microbe species were standardized using NCBI Taxonomy database (55) and (iii) the extend data of each microbe could be accessed using crosslinks to ICD-11 (66), UniProt (68), KEGG (69), NCBI Taxonomy (55), gutMDisorder (44), Disbiome (43), etc. GIMICA has been smoothly running for months and tested from various sites around the world. All data can be viewed, assessed and downloaded. GIMICA is freely assessable without login requirement by all users at: https://idrblab.org/gimica/
CONCLUSION
In sum, GIMICA is unique in: (i) providing both host genetic and immune factors shaping human microbiota and (ii) enabling collective consideration not only among various host factors but also between the host and environmental ones. With the extensive efforts made on describing the host factors that are capable of shaping human microbiota (10–12) and discussing the collective determination by both host and environmental factors (46,47,64), those immense, connected and structuralized data provided in GIMICA are expected to have implication for the future practice of MCs-related study on human physiology and health (1–3,70–73).
FUNDING
National Key R&D Program of China [2018YFC0910500]; National Natural Science Foundation of China [U1909208, 81872798]; Project supported by the China Knowledge Centre for Engineering Sciences and Technology [No. CKCEST-2019-1-12]; Leading Talent of the ‘Ten Thousand Plan’ – National High-Level Talents Special Support Plan, Key R&D Program of Zhejiang Province [2020C03010]; Technology Innovation and Application Demonstration Project of Chongqing [cstc2018jscx-msybX0287]; Fundamental Research Funds for the Central Universities [2018QNA7023, 10611CDJXZ238826, 2018CDQYSG0007, CDJZR14468801, 2019CDYGYB005]; Information Technology Center, Zhejiang University. Funding for open access charge: National Natural Science Foundation of China [U1909208, 81872798].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
69.
70.
71.
72.