 dbGuide: a database of functionally validated guide RNAs for genome editing in human and mouse cells
dbGuide: a database of functionally validated guide RNAs for genome editing in human and mouse cells
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

- Altmetric
With the technology's accessibility and ease of use, CRISPR has been employed widely in many different organisms and experimental settings. As a result, thousands of publications have used CRISPR to make specific genetic perturbations, establishing in itself a resource of validated guide RNA sequences. While numerous computational tools to assist in the design and identification of candidate guide RNAs exist, these are still just at best predictions and generally, researchers inevitably will test multiple sequences for functional activity. Here, we present dbGuide (https://sgrnascorer.cancer.gov/dbguide), a database of functionally validated guide RNA sequences for CRISPR/Cas9-based knockout in human and mouse. Our database not only contains computationally determined candidate guide RNA sequences, but of even greater value, over 4000 sequences which have been functionally validated either through direct amplicon sequencing or manual curation of literature from over 1000 publications. Finally, our established framework will allow for continual addition of newly published and experimentally validated guide RNA sequences for CRISPR/Cas9-based knockout as well as incorporation of sequences from different gene editing systems, additional species and other types of site-specific functionalities such as base editing, gene activation, repression and epigenetic modification.
INTRODUCTION
CRISPR/Cas9-based genome editing has been an indispensable technology for understanding the biology of living organisms (1,2). As a result, a tremendous effort has been invested in aiding researchers in designing and executing CRISPR-based experiments by providing critical resources such as reagents and protocols as well as software tools that allow a user to rapidly identify guide RNA sequences with specific predicted on-target and off-target characteristics (3–18) (Table 1). However, these sequences are essentially predictions, often requiring assessment of multiple candidate guide RNAs.

Over the last 7 years, there has been an explosion of literature utilizing CRISPR/Cas9 to knockout specific genes or genomic regions, of which the majority is in human or mouse model systems. The culmination of all of this work has provided an opportunity to mine this information for guide RNA sequences shown to be functional at either a genotypic and/or phenotypic level. Variation in reporting standards, typographical errors and relegation of sequences into supplementary materials are among a few reasons why curation of this information has been non-trivial.
To this end, we present dbGuide, a database of functionally validated guide RNA sequences for CRISPR/Cas9 mediated knockout experiments in human or mouse cells (http://sgrnascorer.cancer.gov/dbguide). We have manually curated guide RNA sequences from over 1000 peer-reviewed articles with each sequence having direct reference to the original publication. We are also making results of targeted amplicon sequencing for ∼2000 unique sgRNA sequences tested individually in human (293T) or mouse (NIH-3T3 or P19) cultured cells publicly searchable in our database. In total, these efforts encompass nearly 6000 unique guide RNA sequences for which some level of validated activity exists. To our knowledge, this represents the largest database of functionally validated guide RNA sequences. We expect this to be a continually growing resource through multiple mechanisms. We have provided a downloadable template to encourage researchers to submit their newly published/validated sequences and our computational framework will also allow for the inclusion of sequences for CRISPR systems other than for Streptococcus pyogenes, modalities such as base editing, gene activation/repression and epigenetic modifications, and sequences used in other species.
MATERIALS AND METHODS
Master list of human and mouse sgRNA sequences
In addition to functionally validated sgRNA sequences, in order to provide further utility to the database, computationally designed sequences were also obtained from a variety of sources, primarily focused on protein-coding genes (17,19–26). These sources are listed in Supplementary Table S1. CSV-delimited files containing all sgRNA sequences present in our database for human (https://sgrnascorer.cancer.gov/downloads/hg_guide_info.csv.gz) and mouse (https://sgrnascorer.cancer.gov/downloads/mm_guide_info.csv.gz) can be downloaded directly from the dbGuide home page.
Published sgRNA sequences
A broad search of the PubMed database for ‘CRISPR OR Cas9’ was performed and yielded over 15 000 indexed citations. Subsequently, review articles, publications not using human or mouse cells, publications not using S. pyogenes Cas9 and publications not performing knock-out experiments were excluded. In total, guide RNA sequences were sourced from a total of 1322 peer-reviewed articles (Supplementary Table S2) from which the guide RNA sequences used could be ascertained. Although it is likely some published sequences have been missed, a data template is provided for which these missed sequences can be submitted and incorporated into subsequent database updates.
Targeted amplicon sequencing data
Quantitative editing data for nearly 2000 sgRNA sequences were generated internally from either transfection of Cas9/sgRNA plasmids (1 µg) or Cas9 protein (4 µg)/in vitro transcribed (IVT) (2.25 µg) RNA into mouse (NIH-3T3 or P19) or human (HEK293T) cells in a 24-well format. Cas9 protein was produced using plasmid Addgene-62731, a gift from Niels Geijsen (Addgene plasmid # 62731; http://n2t.net/addgene:62731; RRID:Addgene_62731) (27). IVT RNA was produced using a similar protocol as previously published (28). Cas9/RNP complexing and transfection using Lipofectamine 2000 were also performed similarly to as previously described (29). Polymerase chain reaction from genomic DNA was performed and amplicons were sequenced using the Illumina MiSeq V2 300 cycle kit using the PE (2 × 150) format. List of all sgRNAs sequences tested are listed in Supplementary Table S3.
Mapping information and on/off-target scoring metrics
For all sgRNA sequences obtained, genomic locations of the corresponding target sites were obtained/verified using UCSC BLAT against either the hg38 or mm10 reference genome sequences downloaded from the UCSC Genome Browser (30,31). Subsequently, sgRNA locations were cross-referenced with the Gencode V32 (human) or Gencode VM23 (mouse) gene/transcript annotations to determine which transcript(s) each sgRNA could target.
For on-target metrics, sgRNA Scorer 2.0 (32), Rule Set 2 (17) and FORECasT (33) scores were downloaded/calculated for each sgRNA and if a score could not be obtained, a value of ‘NV’ was denoted. For off-target analysis, Guidescan 1.0 (34) values were generated for each guide and similarly, for those guides for which a score could not be obtained, a value of ‘NV’ was given.
Analysis of targeted amplicon sequencing data
Paired end raw FASTQ files were merged using FLASH (35), filtered for low quality bases, subsequently mapped to the designated genomic locations in hg38/mm10 using bwa mem, and then sorted and indexed bam files were generated using samtools (36). A custom python snakemake (37) pipeline was made to calculate non-homologous end joining (NHEJ) mutation frequencies. This analysis pipeline is publicly available at https://github.com/rajchari2/ngs_amplicon_analysis.
RESULTS AND DISCUSSION
dbGuide uses a simple HTML interface which utilizes the datatables and highcharts javascript libraries for displaying data in tabular and graphical formats, respectively (Figure 1A). The application is built in python using django with a MySQL database used for data storage and retrieval. The database can be accessed without e-mail registration or login.

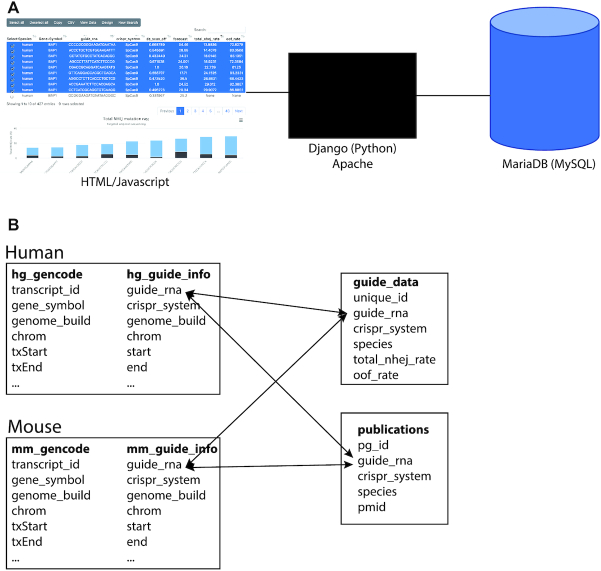
Structure of the dbGuide database. (A) Components of the dbGuide database. The user interface was developed using html and javascript and the application is managed on an Apache web server using django. All underlying data is stored in a MariaDB (MySQL) database. (B) Schema of the MySQL database. Each species, currently limited to human and mouse, has a table of sgRNA sequences with pre-computed metrics and a table with gene annotation information. sgRNA sequences obtained from publications are stored in a single table and the ‘species’ field is used to determine which species the guide RNA was used. Similarly, sequences from targeted amplicon sequencing data also have the ‘species’ field for this purpose.
Within the MySQL relational database, for both human and mouse, there was central sgRNA table with genome target position and all metrics pre-calculated and a gene annotation table which has the location of all protein coding genes based on Gencode annotation. Finally, there are two separate tables for summarizing the amplicon sequencing data and the publication-validated sequences. A depiction of the schema is shown in Figure 1B.
Opening user interface
The introductory user interface is very simple (Figure 2A). The user first specifies whether to search in the human or mouse genome and then can provide either a chromosomal position (in BED format), gene symbol, Ensembl gene or transcript ID, or an sgRNA spacer sequence (without the PAM). In addition, a link to an excel spreadsheet template is provided for researchers wishing to contribute to the database.
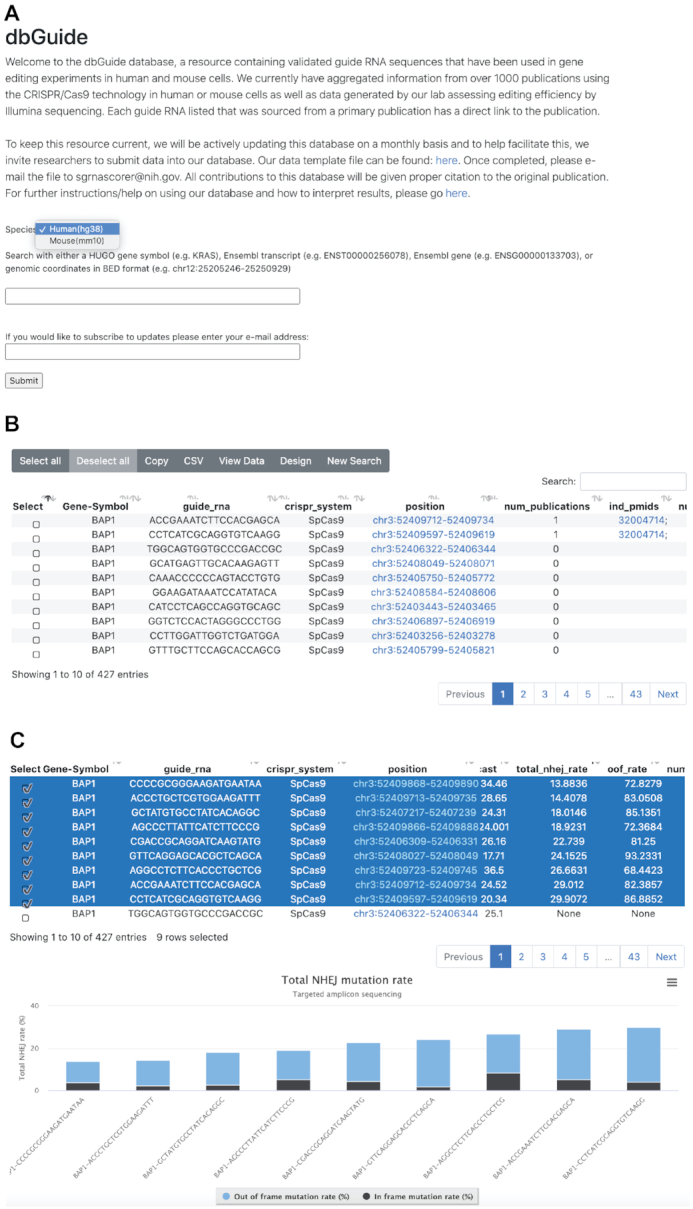
Screen shots of the key user interfaces. (A) Opening window of the dbGuide database. The user must first select whether to search within the human/mouse genome and then can specify a HGNC gene symbol, chromosomal coordinate in BED format, or ENSEMBL gene/transcript ID. (B) Window depicting results of a search by gene symbol. Selectable rows of sgRNA sequences are returned with various pre-computed metrics and PubMed identifiers, if the sequence had been used in a peer-reviewed publication. (C) Graphical representation of sgRNA sequences for which targeted amplicon sequencing data were generated. Stacked bar plots display the percentage of NHEJ mutation events that results in an ‘in-frame’ (black) or ‘out of frame’ (blue) amino acid change.
Search results when searching by gene/coordinate/accession
Upon entering a valid search term, the user will be shown a table of results detailing all of the guide RNA sequences identified based on the search criteria (Figure 2B). The table displays the following columns:
Gene-Symbol: most recent official gene symbol for a gene,
guide_rna: sgRNA spacer sequence (without PAM),
crispr_system: Cas9 ortholog, currently restricted to SpCas9,
position: genomic location of the sgRNA target site in either human (hg38) or mouse (mm10) assembly
in_protein_coding_exon: whether the guide RNA targets a protein coding exon,
num_transcripts: the number of GENCODE transcripts targeted,
transcript_id_list: list of GENCODE transcripts (by ENSEMBL ID),
sgrnascorer: predicted activity using the sgRNA Scorer 2.0 algorithm (−3 to 3). Higher the value, greater the predicted activity,
rule_set_2: predicted activity using the Rule Set 2 algorithm (0–1). Higher the value, greater the predicted activity
guide_scan_off: guidescan off-target score (0–1). Higher the value, higher the specificity
forecast: Favored Outcomes of Repair Events at Cas9 targets (FORECasT) score for predicting mutational outcomes (in frame indel %, lower values preferred for knockouts),
total_nhej_rate: if amplicon sequencing data exists, value between 0 and 100, or ‘None’ otherwise
oof_rate: out of frame mutation rate which is the proportion of mutated reads which would lead to an out of frame mutation
num_publications: number of published articles using the selected guide RNA sequence
ind_pmids: list of articles by PubMed ID which used selected guide RNA sequence
num_screens: number of pooled CRISPR screens in which the guide RNA sequences was enriched or depleted
screen_pmids: list of articles by PubMed ID of CRISPR-based pooled screens in which this guide was enriched or depleted
sources: list of sources from which the guide RNA sequence has been curated from
status
status: if the sgRNA sequence was functionally validated in a publication or targeted amplicon sequencing data exists, it is termed ‘validated’. Otherwise, it is termed ‘design’. This field allows the user to rapidly filter the list of returned sequences by typing ‘validate’ in the upper-right search box.
Viewing sequence editing data
In addition to sorting by total NHEJ rate, one can view the data in a graphical format. By selecting sgRNA sequences for which data exists, the user can click the ‘View Data’ which will show a stacked bar plot breaking down the total NHEJ rate between ‘in frame’ and ‘out of frame’ percentage (Figure 2C). This can be specifically important for knockout experiments where a higher out of frame (OOF) mutation percentage gives higher probability of protein loss.
Downloading sequences for use in experiments
Upon identifying guide RNA sequences of interest, the user can export the sequences in a ‘ready to order’ format by clicking on the ‘Design’. A tab-delimited text file is generated listing oligonucleotides needed for either ligation into a plasmid or in vitro transcription as well as a PDF protocol describing what compatible vectors can be used and how they can be obtained. It is highly recommended to select and download multiple guide RNA sequences when performing knockout experiments to ensure any functional consequences observed may not be due to spurious off-target activity from a single guide RNA.
DATA AVAILABILITY
Sorted BAM files for amplicon sequencing data are available from NCBI under Project Identifier PRJNA664634.
ACKNOWLEDGEMENTS
We would like to acknowledge Troy Taylor, William Gillette, Jane Jones and Dominic Esposito for kindly providing Cas9 protein. We would also like to acknowledge the Biomedical Informatics and Data Science (BIDS) and Frederick Research Computing Environment (FRCE) for providing assistance in web hosting and computational support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Frederick National Laboratory for Cancer Research's Technology and Training Fellowship (to T.P.S.). Funding for open access charge: Frederick National Laboratory for Cancer Research.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
31.
32.
33.
34.
35.
36.