 BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking
BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking
Nucleic Acids Research



- Altmetric
An increasing prevalence of hospital acquired infections and foodborne illnesses caused by pathogenic and multidrug-resistant bacteria has stimulated a pressing need for benchtop computational techniques to rapidly and accurately classify bacteria from genomic sequence data, and based on that, to trace the source of infection. BacWGSTdb (http://bacdb.org/BacWGSTdb) is a free publicly accessible database we have developed for bacterial whole-genome sequence typing and source tracking. This database incorporates extensive resources for bacterial genome sequencing data and the corresponding metadata, combined with specialized bioinformatics tools that enable the systematic characterization of the bacterial isolates recovered from infections. Here, we present BacWGSTdb 2.0, which encompasses several major updates, including (i) the integration of the core genome multi-locus sequence typing (cgMLST) approach, which is highly scalable and appropriate for typing isolates belonging to different lineages; (ii) the addition of a multiple genome analysis module that can process dozens of user uploaded sequences in a batch mode; (iii) a new source tracking module for comparing user uploaded plasmid sequences to those deposited in the public databases; (iv) the number of species encompassed in BacWGSTdb 2.0 has increased from 9 to 20, which represents bacterial pathogens of medical importance; (v) a newly designed, user-friendly interface and a set of visualization tools for providing a convenient platform for users are also included. Overall, the updated BacWGSTdb 2.0 bears great utility in continuing to provide users, including epidemiologists, clinicians and bench scientists, with a one-stop solution to bacterial genome sequence analysis.
INTRODUCTION
The history of the world is intertwined with the impact that bacterial infectious diseases have had on humans. The advent of antimicrobials has fostered the belief that, as Sir McFarland Burnett stated in 1962, ‘Almost all of the major practical problems of dealing with infectious disease had been solved’ (1). However, this was soon led to disillusion due to the later emergence and rapid dissemination of antimicrobial resistance (AMR). In particular, in the current era of globalization, increasing international travel and food transportation have led to cross-board bacterial transmission events and even pandemics (2). A well-known example in the recent decade is the large outbreak caused by Shiga-toxin-producing Escherichia coli O104:H4, which started in Germany in the summer of 2011 with the consumption of sprouts and later spread in only two months to at least 16 countries (3). Although relatively rare, such mode of transmission also take place with hospital-acquired infections. For example, Acinetobacter baumannii, a notorious multi-drug resistant nosocomial bacteria, was believed to have initially infected American soldiers in Iraq and from them to have been brought back to military hospitals in the United States, after which it disseminated rapidly throughout the entire nation (4). Given the possible cross-border transmission nature of pathogenic bacteria and the consequent threat to global public health, there is a pressing need for global surveillance and the early detection of infectious disease outbreaks.
Traditional epidemiological studies have usually focused initially on patients with certain epidemiological links, recovered bacterial isolates from these subjects’ clinical samples and finally determined whether their isolates had clonal relationships through bacterial typing techniques (5). However, such suspected links are often missing due to untimely or incomplete epidemiological surveys or asymptomatic carriers (6). Therefore, it is imperative to establish a reverse strategy, i.e. when bacterial isolates are found to be sufficiently similar by high-resolution typing techniques, they are deemed to be a consequence of infection from a common source (7). Due to its ultimate single base pair resolution, whole-genome sequencing (WGS) has fulfilled this demand and gradually replaced pulsed-field gel electrophoresis (PFGE) and conventional seven-locus multi-locus sequence typing (MLST) as the new ‘gold standard’ typing technique (8,9). Concomitantly, there is a pressing need to organize, standardize and share bacterial genome sequences in a worldwide accessible database to fully exert the power of this reverse strategy on international surveillance and the early outbreak detection of bacterial infections.
In the year 2015, we initially introduced BacWGSTdb, a bacterial whole genome sequence typing and source tracking database designed for nine bacterial organisms of medical importance (10). This database incorporates extensive resources from bacterial genome sequence data as well as the corresponding metadata retrieved from the NCBI GenBank and BioSample database (11,12). By implementing a reference genome-based single-nucleotide polymorphism (SNP) approach, BacWGSTdb provides instant comparisons between user-uploaded genomes and an unprecedentedly large global set of genomes. Thus, clinicians, microbiologists and epidemiologists who work in medical facilities or public health institutions with no specialist knowledge of bioinformatics can use the database for the determination of clonal relationships and source tracking. Bench scientists can also use BacWGSTdb as a convenient tool for preliminary evolutionary and comparative genomic analyses.
Since the first version of BacWGSTdb, a vast number of additional genomic sequences was determined experimentally and, more importantly, a variety of newly discovered phenotypic traits (e.g. AMR and virulence) could be associated with the WGS data in the public domain (13,14). The demand for database updates and a uniform platform for real-time in silico prediction of these phenotypic traits based on genomic sequence data has become urgent. Here, we updated BacWGSTdb to version 2.0, which reflects not only a large increase in the curated dataset of bacterial genome sequencing data for the existing bacterial organisms, but also the new addition of eleven common pathogenic species. In addition to its updated content, a novel module for tracking the source of newly sequenced plasmids has been incorporated. AMR has challenged the treatment of infectious diseases which pose a serious threat to public health. As the major vector carrying AMR genes, plasmids are prone to horizontal transfer and offer AMR to originally antimicrobial susceptible bacteria, making treatment even more difficult. In this sense, tracing the transmission of AMR-carrying plasmids is of equal importance to tracing that of bacteria. Furthermore, the phylogenetic analysis in BacWGSTdb 2.0 is enhanced by adding the core genome MLST (cgMLST) approach, thereby meeting different genotyping demands. Taken together, we expect that BacWGSTdb 2.0 will continue to benefit users by providing a one-stop repository for bacterial whole-genome sequence typing and source tracking.
DATABASE UPDATE AND ENHANCEMENTS
BacWGSTdb 1.0 included nine bacterial species, while in this update, eleven more species have been newly added. The detailed species and the number of isolates included in BacWGSTdb are listed in Table 1. Thereby, BacWGSTdb has covered almost all common nosocomial, community and foodborne bacterial pathogens.

| Species | Version 1 | Version 2 | Number of isolates (Version 1) | Number of isolates (Version 2) |
|---|---|---|---|---|
| Acinetobacter baumannii | √ | √ | 1026 | 4260 |
| Bacillus anthracis | √ | √ | 158 | 227 |
| Bacillus cereus | √ | 1017 | ||
| Campylobacter coli | √ | 820 | ||
| Campylobacter jejuni | √ | 1621 | ||
| Clostridioides difficile | √ | 2322 | ||
| Enterococcus faecalis | √ | 1523 | ||
| Enterococcus faecium | √ | 1885 | ||
| Escherichia coli | √ | √ | 6328 | 21 733 |
| Klebsiella pneumoniae | √ | √ | 3279 | 9020 |
| Listeria monocytogenes | √ | 3243 | ||
| Mycobacterium abscessus | √ | 1611 | ||
| Mycobacterium tuberculosis | √ | √ | 2532 | 6512 |
| Salmonella enterica | √ | √ | 5276 | 14 658 |
| Staphylococcus aureus | √ | √ | 4890 | 11 505 |
| Streptococcus agalactiae | √ | 1398 | ||
| Streptococcus pneumoniae | √ | √ | 3018 | 8347 |
| Streptococcus suis | √ | 1252 | ||
| Vibrio cholerae | √ | 818 | ||
| Yersinia pestis | √ | √ | 257 | 369 |
The usage of BacWGSTdb includes two function modules, ‘Tools’ and ‘Browse’. The former allows users to upload their own FASTA-formatted genome sequence(s) to find closely related records in the database. The latter allows users to browse the isolates in the database, compare their clinical and microbiological traits, and investigate their phylogenetic relationships. With the release of BacWGSTdb 2.0, we are introducing multiple major changes in both the backend and the web interface that provide users with more effective and efficient ways to browse and query the WGS data and to quickly cluster and identify related sequences for uncovering potential transmission sources. This helps clinicians or public health scientists investigate hospital-acquired or foodborne infection outbreaks. In addition, BacWGSTdb 2.0 also provides the online analyses of conventional seven-locus MLST, predictions of AMR and virulence genes, and source tracking of plasmid sequences. An overview of the database schematic is shown in Figure 1.

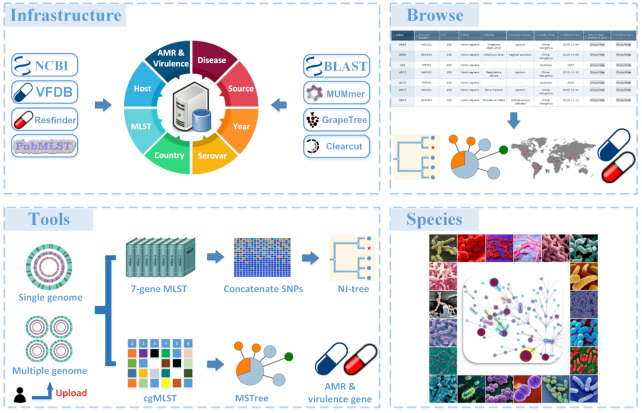
Overview of the content and function modules of BacWGSTdb 2.0. ‘Infrastructure’ lists the public database and tools integrated in BacWGSTdb 2.0. ‘Browse’ functions to visualize and compare the genetic relationships among isolates deposited in BacWGSTdb 2.0. ‘Tools’ functions for whole genome sequence typing and source tracking based on user uploaded sequence(s). ‘Species’ represents 20 bacterial species currently supported by BacWGSTdb 2.0.
Single genome analysis
In BacWGSTdb 1.0, the module Single Genome Analysis performs conventional seven-locus MLST analysis and searches of the genetically closest relatives in the database following the SNP approach upon the user's uploaded preassembled single genome. In detail, MUMmer 3.22 is used for the alignment with the reference genome and the subsequent SNP identification; and the phylogenetic tree is generated by Clearcut 1.0 (15,16). The most important update to this module in BacWGSTdb 2.0 is that it now offers both SNP and cgMLST analysis to compare the user uploaded genome sequence against those deposited in the database. The SNP approach compares single nucleotide differences between isolates to a designated reference genome, which can be used to investigate the clonal relationship among isolates sharing a high genetic relatedness (e.g. collected from outbreaks of hospital or foodborne infections). By comparison, the cgMLST approach, another widely used sequence typing approach for bacterial genomes, is an extension of conventional seven-locus MLST scheme that expands the range of target genes to whole genome level and is often used as a solution to provide highly detailed phylogenetic relatedness of a species and is suitable for investigating the middle/long-term evolutionary history of bacterial pathogens. Thus, the two approaches are complementary and meet different genotyping demands (17).
To perform a cgMLST analysis, a pre-defined reference database (cgMLST scheme) for each species, which contains all known allelic variants in the coding regions for all genomes deposited in BacWGSTdb, is prepared at the backend of BacWGSTdb 2.0 by LOCUST 1.0 (18). The user uploaded genomic sequence is compared to the species-specific cgMLST scheme using BLASTn (19). The identified allelic profile continues to make comparisons with that of each isolate deposited in BacWGSTdb 2.0 for determination of the closely related isolates according to their number of pairwise allelic differences. GrapeTree is used to construct and visualize the minimal spanning tree generated based on the cgMLST allelic profiles, which supports manipulations of both tree layout and the user specified metadata attributes (20) (Figure 2).
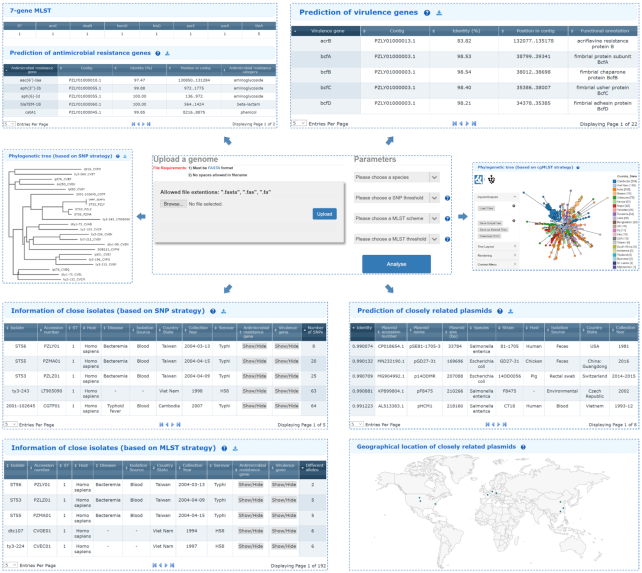
Screenshots of the updated web interface of the Single Genome Analysis module and the detailed outputs using a Salmonella enterica isolate as an example. After uploading the preassembled genomic sequence and setting the appropriate parameters, the analytical results return and can subsequently be classified into three sections: conventional seven-locus MLST, identification of AMR and virulence genes, and source tracking of plasmids and bacteria. In particular, the phylogenetic analysis revealed that the query isolate shows high degree of genetic relatedness in the database, suggesting that these isolates might have originated from the same source. The entire analysis process takes 3–5 min.
Another important update in this module is the integration of a novel function for typing and source tracking of plasmids. BacWGSTdb 2.0 deposits all complete plasmid sequences from the NCBI GenBank database as well as their metadata. When a draft bacterial genome, which usually contains the plasmid sequence of the sequenced isolate, is uploaded, its plasmid replicon types will be determined by searching the genomic sequence against the database of plasmid replicon genes with BLASTn (21). Moreover, pairwise distances between the users’ sequence and those of each of the plasmids in BacWGSTdb are computed using Mash 2.2 with maximal P-value set to 0.1 and minimal identity set to 0.9 (22). The analytical results include a table listing the records of similar plasmids based on Mash distance, together with a world map displaying the records with available geographical location data (Figure 2). This update makes it possible to trace the transmission routes of plasmids, which we believe are of at least equal importance with that of chromosomes.
Furthermore, the in silico identification of acquired AMR and virulence genes are also incorporated into BacWGSTdb 2.0. The user uploaded sequence is searched against the ResFinder 3.2 and VFDB 2019 database by BLASTn, with minimal identity and a threshold coverage of 90% (23–25).
Multiple genome analysis
The newly designed multiple genome analysis module can process up to 30 user uploaded genome sequences in a batch mode. In silico identifications of conventional seven-locus MLST, AMR and virulence genes are performed for each of the uploaded genomes. A more important purpose for this module is that when users collect multiple isolates which they suspect belong to the same outbreak event, the module can help determine whether there is a clonal relationship among these isolates according to the pairwise comparison of the cgMLST alleles or SNP differences. To this end, the phylogenetic relatedness among the user uploaded multiple genomic sequences will be determined following both the SNP and cgMLST approaches (Figure 3).
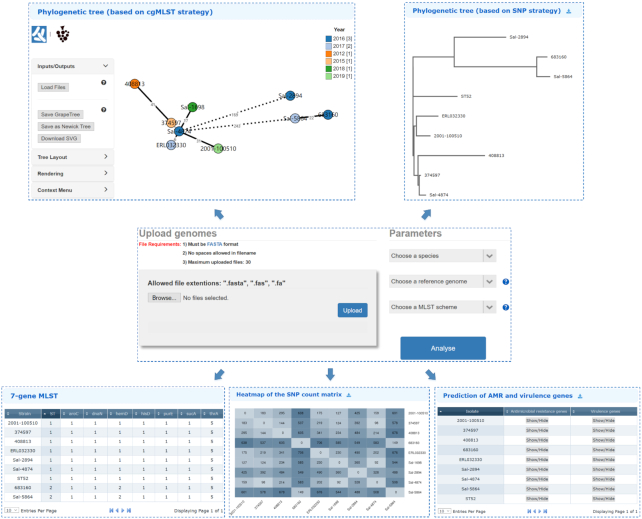
Screenshots of the updated web interface of the Multiple Genome Analysis module and the detailed outputs using nine S. enterica isolates as an example. The Results page lists for each of the uploaded genomes the conventional seven-locus MLST results and the predictions of AMR and virulence genes. In addition, the phylogenetic trees based on the SNP and cgMLST approaches both reveal that the query isolates did not involve an outbreak event, since they differ from one another by over 100 SNPs or cgMLST loci. The entire analysis process takes 5–8 min.
Browse and Search
The Browse and Search module is designed to visualize and compare the genetic relationships among isolates deposited in BacWGSTdb. Each ‘Browse’ page lists the clinical and microbiological metadata of each of the isolates deposited in the database, including their seven-locus MLST sequence type, host, clinical outcome, collection date and geographical location. Data can be sorted by clicking on a specific column header and downloaded as a tab-delimited file. The updated annotations on the AMR and virulence genes for each isolate are also displayed. In BacWGSTdb 1.0, users can select multiple isolates belonging to the same sequence type, and build phylogenetic trees following the SNP approach. This limit has been broken in BacWGSTdb 2.0: users can select and compare any isolate of interest, even from among those belonging to different sequence types. Both the SNP and cgMLST approaches will be applied for establishment of phylogenetic trees. In addition, a newly designed search function enables users to look up keywords (e.g. sequence types) of interest based on varied categories (Figure 4). We therefore believe that the updated Browse and Search module will allow users to retrieve information from the database in a convenient and time-saving manner.
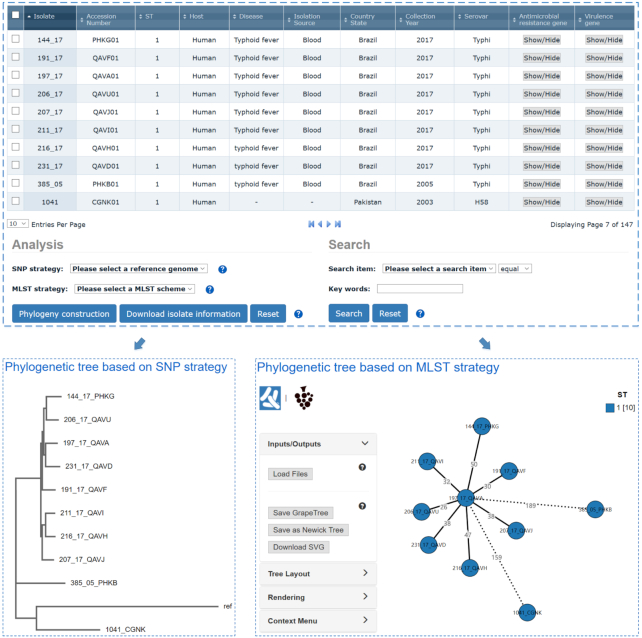
Screenshots of the updated Browse interface. Users can sort and select isolates based on their various attributes. For the selected isolates, phylogenetic trees following both the SNP and cgMLST approaches are provided.
CONCLUSIONS AND FUTURE PERSPECTIVES
Next-generation sequencing and bioinformatics are expediting pathogen characterization, transforming the response to infectious disease outbreaks, and providing new insights into disease emergence and transmission. Standardized and user-friendly online databases make WGS analysis more accessible, even to those lacking bioinformatics expertise. Here, we reported a major update of BacWGSTdb, including the significantly expanded content of the database, additional analytical and visualization tools, and a newly designed, user-friendly interface, all of which greatly facilitate the genomic epidemiological surveillance of bacterial pathogens. We believe these additions significantly enrich our database, which is expected to provide a one-stop solution to bacterial genome analysis and, more importantly, translate whole genome sequencing from proof-of-concept to routine use in clinical practice.
FUNDING
National Natural Science Foundation of China [31670132, 81401698]; Zhejiang Province Public Welfare Technology Application Research Project [LGF18H190001]. Funding for open charge: National Natural Science Foundation of China [31670132].
Conflict of interest statement. None declared.
REFERENCES
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.