 INTEDE: interactome of drug-metabolizing enzymes
INTEDE: interactome of drug-metabolizing enzymes
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first four authors should be regarded as Joint First Authors.

- Altmetric
Drug-metabolizing enzymes (DMEs) are critical determinant of drug safety and efficacy, and the interactome of DMEs has attracted extensive attention. There are 3 major interaction types in an interactome: microbiome–DME interaction (MICBIO), xenobiotics–DME interaction (XEOTIC) and host protein–DME interaction (HOSPPI). The interaction data of each type are essential for drug metabolism, and the collective consideration of multiple types has implication for the future practice of precision medicine. However, no database was designed to systematically provide the data of all types of DME interactions. Here, a database of the Interactome of Drug-Metabolizing Enzymes (INTEDE) was therefore constructed to offer these interaction data. First, 1047 unique DMEs (448 host and 599 microbial) were confirmed, for the first time, using their metabolizing drugs. Second, for these newly confirmed DMEs, all types of their interactions (3359 MICBIOs between 225 microbial species and 185 DMEs; 47 778 XEOTICs between 4150 xenobiotics and 501 DMEs; 7849 HOSPPIs between 565 human proteins and 566 DMEs) were comprehensively collected and then provided, which enabled the crosstalk analysis among multiple types. Because of the huge amount of accumulated data, the INTEDE made it possible to generalize key features for revealing disease etiology and optimizing clinical treatment. INTEDE is freely accessible at: https://idrblab.org/intede/

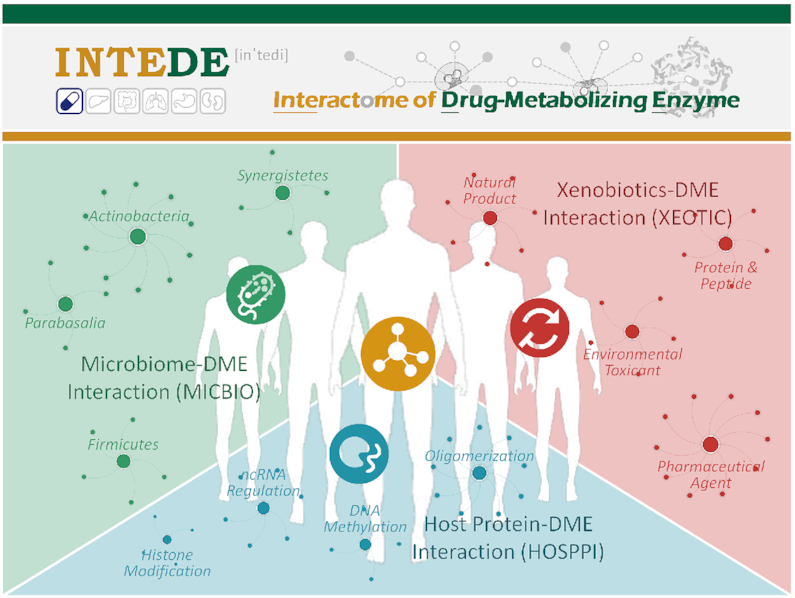
INTRODUCTION
Drug-metabolizing enzymes (DMEs) transform the parent drug to a metabolite with substantially different physicochemical and pharmacological properties, and are critical determinants of drug safety and efficacy (1,2). The interactome of DMEs has therefore attracted considerable attention (3–12). There are three major types of interactions: (i) microbiome–DME interactions (MICBIOs) determining the dynamic nature (5) and interpersonal variability (6) of drug metabolism; (ii) host protein–DME interactions (HOSPPIs) essential for predicting in vivo efficacy or clearance based on in vitro result (7) and revealing the mechanism underlying drug resistance (8) or adverse drug reaction (9); (iii) xenobiotics–DME interactions (XEOTICs) that are the key factor of metabolism-based drug–drug interaction (5) and a constant inspiration of clinical treatment optimization (10). Because the metabolism of drugs is collectively determined by multiple types of interactions, the accumulation of DME interactome data may give vital insight into the prediction of clinical consequences (7) and will have implication for the future practice of precision medicine (5).
Moreover, because of its dynamic evolvements (5), wide distribution (13), and rich repository of enzymes (14), the microbiome and its DMEs are found to be not only as critical as human DMEs in drug metabolism (10), but also key in the study of individual or tissue-specific metabolism of drugs (13,14) and the discovery of novel therapeutics targeting microbial protein (10). As a result, there was an explosive growth of interest in studying the interactome of microbial DME (15–19). Particularly, the MICBIO between actinobacteria Eggerthella lenta and microbial DME tyrosine decarboxylases was found to collectively regulate the metabolism of levodopa (10); the HOSPPI between host UDP-glucuronosyltransferases and microbial DME beta-glucuronidases could lead to an adverse drug reaction of irinotecan (20); the XEOTIC between antibiotic ciprofloxacin and bacterial DME cytidine deaminase was discovered to abrogate the resistance of gemcitabine (21). Thus, it is important to collect these interactome data, and collectively analyze the multiple types of interactions for not only human but also microbial DMEs (7,10,15).
Nowadays, various databases have been developed to provide DME-related data, the majority of which are freely accessible and remain active. Some of these databases (such as DrugBank (22), PDB (23), PharmGKB (24), Protein Atlas (25), TTD (26) and UniProt (27)) provide the enzyme or DME information as part of a broader collection of biological/pharmacological data, and some others (including BRENDA (28), KEGG (29), MetaCyc (30), Reactome (31), SMPDB (32) and VMH (33)) contain the general enzymatic classifications or metabolic reaction/pathway data for a comprehensive set of enzymes. Since all these databases do not include DME interactome data, some databases have been designed to provide the interactions between ∼2800 xenobiotics and ∼190 DMEs (including CTD (34), T3DB (35) and Transformer (36)). However, no database has been designed to describe the key role of microbial DME in drug metabolism, and the interaction data of neither microbiome (MICBIO) nor host protein (HOSPPI), that alter drug metabolism by affecting human/microbial DME, are provided in any available knowledge base. As the crosstalk among interaction types is crucial for drug metabolism (7,10,15), it is critical to construct a database that comprehensively describes all three types of DME interactions.
Here, a newly developed database, Interactome of Drug-Metabolizing Enzyme (INTEDE) was therefore introduced. First, a comprehensive literature review of all (>1900) drugs approved by U.S. FDA and >3000 drugs in clinical trial or preclinical research was performed. Different from the small amount of DMEs (∼30) that were officially described in the U.S. FDA guidance (37,38), 553 DMEs (241 and 312 from human and microbiome, respectively) were confirmed in INTEDE to metabolize approved drugs, 421 DMEs (188 from human and 233 from microbiome) were to metabolize the drug in clinical or preclinical test, and 494 DMEs (208 from human and 286 from microbiome) were to metabolize the pharmaceutically investigative agents. Second, for all these newly confirmed DMEs, INTEDE comprehensively provided all three types of their interactions (MICBIOs, HOSPPIs and XEOTICs, as shown in Figure 1), which allowed the crosstalk among multiple interaction types (7,10,15) and could thus facilitate the study of disease etiology and the optimization of clinical treatment. All in all, since such crosstalk is key for drug metabolism, the INTEDE is expected to have implications for the future practice of precision medicine.
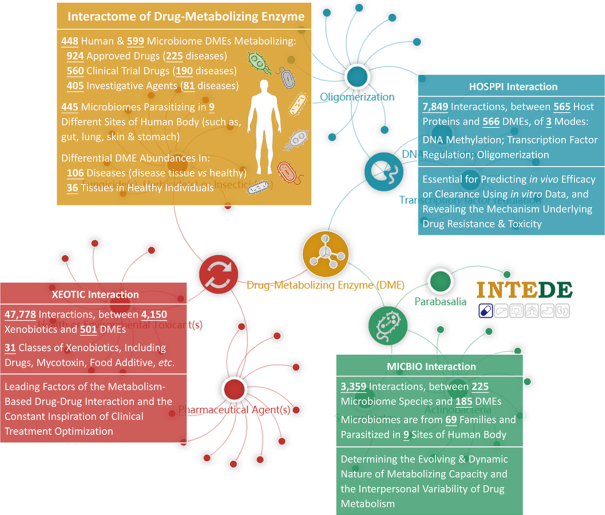
INTEDE statistics of (i) the DMEs, and their corresponding drug(s) and tissue/disease specific protein abundances (orange) and (ii) three types of DME interactions: MICBIOs (green), HOSPPIs (blue) and XEOTICs (red). MICBIO determines the evolving and dynamic nature of metabolizing capacity and the interpersonal variability of drug metabolism; HOSPPI is essential for predicting in vivo efficacy/clearance based on the in vitro data, and revealing the mechanisms underlying drug resistance and toxicity; XEOTIC is one of the leading factors of the metabolism-based drug-drug interaction and the constant inspiration of clinical treatment optimization.
FACTUAL CONTENT AND DATA RETRIEVAL
Confirmation and collection of human/microbial DMEs
The DMEs collected in INTEDE were first confirmed by the drugs of clinical importance (either approved or clinical/preclinical/investigative). Particularly, a comprehensive literature review of all drugs approved by FDA (1921 in total, collected from the U.S. FDA official site), 2958 drugs in clinical trial (data from the ClinicalTrial.gov (39) and TTD (40)) and 10 213 pharmaceutically investigative agents (obtained from the TTD (40)), was performed to confirm their corresponding DMEs by searching the PubMed (41) using such keyword combinations as ‘‘Drug Name’ + drug metabolizing enzyme’, ‘‘Drug Name’ + metabolism’, ‘‘Drug Name’ + enzyme’, ‘‘Drug Name’ + drug metabolism’ and ‘‘Drug Name’ + metabolized’. Moreover, to facilitate the discovery of microbial DMEs, >2000 microbial species (from bacteria, fungi, metamonada, amoebozoa and archaea (3,42,43)) colonizing throughout the human body (such as bladder, blood, eye, gut, lung, oral cavity, skin, stomach, urethra and vagina (44–46)) were collected, and the microbial DMEs were then discovered by the literature review of PubMed (41) using such keyword combinations as ‘‘Species Name’ + ‘Drug Name’’, ‘‘Species Name’ + drug metabolism’, ‘‘Species Name’ + drug metabolized’ and ‘‘Species Name’ + metabolizing enzyme’.
As a result, 553 DMEs (241 host and 312 microbial) were identified to metabolize 924 approved drugs, 421 DMEs (188 host and 233 microbial) were to metabolize 560 drugs in preclinical and clinical, and 494 DMEs (208 host and 286 microbial) were to metabolize 405 pharmaceutically investigative agents. In total, there were 1047 unique DMEs in INTEDE (448 from human and 599 from microbes). On one hand, 448 human DMEs were grouped to diverse enzymatic families (43 families in total defined by the second level of the Enzyme Commission (EC) nomenclature (28)), and the Top-5 popular DME families were: Paired Donor Oxidoreductase (EC: 1.14, 67 DMEs), CH-OH Donor Oxidoreductase (EC: 1.1, 47 DMEs), Ester Bond Hydrolase (EC: 3.1, 41 DMEs), Glycosyltransferase (EC: 2.4, 34 DMEs) and Kinase (EC:2.7, 32 DMEs). On the other hand, 599 microbial DMEs also belonged to diverse enzymatic families (29 families in total defined by the second level of EC nomenclature), and the Top-5 popular DME families were: Carbon-Nitrogen Hydrolase (EC: 3.5, 99 DMEs), Glycosylase (EC: 3.2, 91 DMEs), Paired Donor Oxidoreductase (EC: 1.14, 33 DMEs), CH-NH Donor Oxidoreductase (EC: 1.5, 31 DMEs), and Acyltransferase (EC: 2.3, 20 DMEs). Experimentally assessed kinetic parameters (Km) and catalytic efficiencies (Kcat/Km) between 235 drugs and 190 human/microbial DMEs were also offered. As shown in Figure 2, a full list of drugs metabolized by certain DME were displayed in INTEDE.
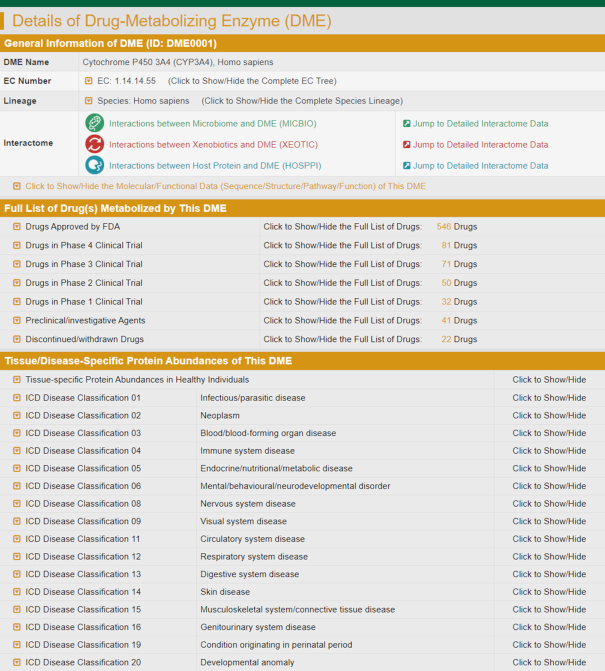
A typical INTEDE page providing the details of DME, which included: (i) the general information (DME name, EC nomenclature, taxonomy lineage, hyperlink to detailed interactome and molecular or functional data); (ii) the full list of drugs metabolized by this DME (categorized based on clinical status: FDA approved, clinical, preclinical, investigative and discontinued. By clicking on each category, the detail information of each drug can be viewed); (iii) the tissue and disease specific protein abundance of the DME (the differential abundance profiles of 394 DMEs in 60 tissues and 106 diseases, and the expression profiles of 348 DMEs across 36 human tissues).
Tissue and disease specific protein sbundances of DMEs
Tissue and disease specific DME abundances are critical for the mitigation of drug toxic reaction (47) and alteration of drug pharmacokinetics (48). In particular, tissue specific DME abundances can not only maintain the functional homeostasis of drug, but also mitigate their tissue-dependent adverse reactions, such as cardiotoxicity, nephrotoxicity and neurotoxicity (47); the variations in DME abundances among different disease indications (such as infection, immunodeficiency and inflammation) can significantly affect hepatic/renal clearance, and therefore reduce drug efficacy or aggravate adverse drug reaction (48). Since these tissue/disease-dependent variations in DME abundances were expected to provide new therapeutic strategies (49–52), the relevant data should be accumulated and further analyzed to promote modern drug discovery.
Disease specific DME abundance data were collected as follows, the detailed procedure of which was illustrated in Supplementary Method S1. First, 5304 series records from the same microarray platform (Affymetrix HGU133 Plus 2.0) were gathered from the Gene Expression Omnibus (53). Second, the collected records were sequentially processed by data normalization, transformation, integration, perfect match correction, quantile, robust multiarray average and median polish (54). Third, the differential expression analysis (55) was conducted by comparing the DME abundance among sample groups (defined in Supplementary Method S1). Fourth, the violin-plot describing the differential expression pattern of the studied DMEs among sample groups was drawn (shown in Supplementary Figure S1). Furthermore, tissue specific DME abundances data were collected using the following procedure, and the detailed process was elaborated in Supplementary Method S1. First, a benchmark dataset that contained the protein expression data across 36 human tissues (56) was collected. Second, the intensities of each DME were processed via data integration and scaling. Third, the expression bar plot of studied DME across 36 tissues was drawn (Supplementary Figure S2). Overall, the differential abundance profile of 394 DMEs in 20,663 patients and healthy individuals of 60 tissues and 106 diseases, and the expression profile of 348 DMEs across 36 human tissues were provided in and downloadable from the website. As shown in Figure 2, the tissue and disease specific DME abundances were described.
The interactome of human and microbial DMEs
INTEDE comprehensively provided all three types of DME interaction data (as shown in Figure 1) for those 1047 unique DMEs (448 from human and 599 from microbe) confirmed in previous section. These interaction data included: (i) microbiome–DME interactions (MICBIOs); (ii) host protein–DME interactions (HOSPPIs) and (iii) xenobiotics–DME interactions (XEOTICs).
Interactions between microbiome and DMEs
The microbial manipulation of drug metabolism by interacting with DME is characterized by (i) the vast diversity of involved microbial species (including various bacteria or fungi from dozens of taxonomic phyla (6,10,20,21,57–59)) and (ii) the wide distribution of these species throughout the human body (not only in gut, but also in skin, vagina, and many other sites (17,21,44,60–62)). Since the microbiome-DME interactions (MICBIOs) are key in determining the dynamic feature (5) and interpersonal variability (6) of drug metabolism, it is very essential to have the MICBIOs data for understanding the controls/effects of the circadian timing system (63) and individualized microbiota compositions (21,61) on the metabolism of certain medication (Figure 1).
Because the MICBIOs data were largely dispersed in literatures, PubMed database (41) was first searched to find the interactions between microbiome and human/microbial DMEs. In particular, the keyword combinations of ‘‘Microbial Species Name’ + ‘DME Name’’, ‘‘Species Name’ + ‘Drug Name’’ and ‘‘Species Name’ + drug metabolism’ were used in literature reviews, and the discovered publications were manually assessed for retrieving any MICBIOs-related information. Moreover, due to the collective determination by multiple factors present in a drug delivery route (64), the metabolism of a drug should be assessed by simultaneously considering its DME(s) and the colonizing microbes, if both DME and microbe are involved in the same route (64). Therefore, additional literature reviews were then conducted using the keyword combination of ‘‘Microbial Species Name’ + ‘DME Name’ + ‘Drug Name’’. The studies that describe the DME and microbe that metabolized the same drug and present in the same delivery route were identified, and these publications were also manually evaluated for retrieving the MICBIOs-related data.
As a result, the collected data included the taxonomic lineage of each microbe (kingdom, phylum, class, order, family, genus and species), the human body site distributed by each microbe (bladder, blood, eye, gut, lung, oral cavity, skin, stomach, urethra and vagina), the list of drugs affected by the interaction between microbial species and DME, and the mechanism of how drug metabolism is influenced by the corresponding interactions. All in all, the latest version of INTEDE contained 3359 MICBIOs between 225 microbes and 185 human/microbial DMEs. These microbes came from 3 kingdoms, 19 phyla, 31 classes, 40 orders, 69 families and 107 genera as defined by the NCBI Taxonomy database (65), and colonized in 10 different human body sites (such as: bladder, blood, eye, gut, lung, oral cavity, skin, stomach, urethra and vagina). The MICBIOs data can be assessed and retrieved by various search strategies in both the Home page and the subpage entitled ‘Microbiome-DME interaction’ of INTEDE (as shown in Figure 3).
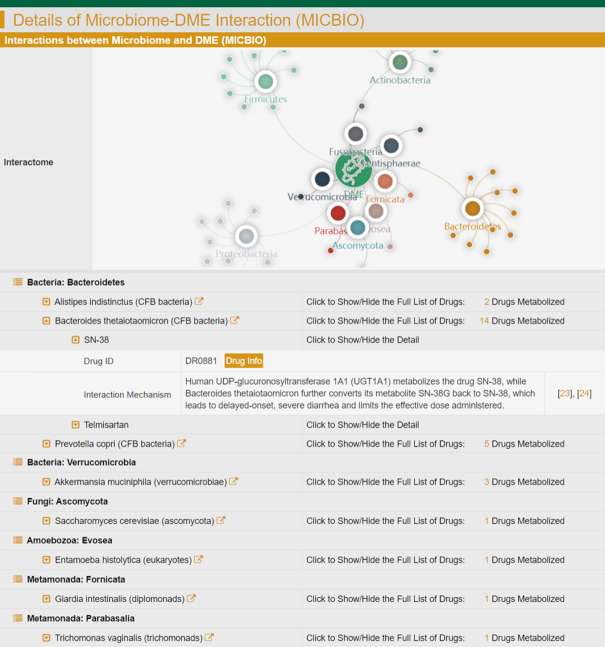
A typical INTEDE page offering the data of microbiome-DME interaction (MICBIO). The INTEDE contained 3359 MICBIOs between 225 microbes and 185 DMEs. These microbes came from 3 kingdoms, 19 phyla, 69 families and 107 genera, and colonized in 10 different host body sites (bladder, blood, eye, gut, lung, oral cavity, skin, stomach, urethra and vagina).
Interactions between host proteins and DMEs
Host protein–DME interactions (HOSPPIs) were frequently encountered in drug metabolism, and widely acknowledged to be critical determinant of drug safety and efficacy (7–9,66–69). A variety of HOSPPIs were of great interest in recent metabolic study, which included (i) oligomerizations that are essential for predicting in vivo drug efficacy or clearance based on the in vitro results (7), (ii) transcription factor regulations that are critical for revealing the mechanisms underlying drug resistance (8) and metabolic variation (70,71), (iii) epigenetic regulations (like DNA methylations, histone modifications and non-coding RNA regulations) that lead to inter-individual variability in drug responses and adverse reactions, and highlight the implications for personalized medicine (9,72,73). Furthermore, the effects of HOSPPIs on altering the drug metabolisms varied between healthy and pathological condition, and differed among disease indications (74). It was therefore essential to collect HOSPPIs to facilitate the prediction of clinical consequence (Figure 1).
The HOSPPIs data was identified by literature search using different keyword combinations. For example, when searching for the oligomerization of a DME, ‘‘DME Name’ + oligomer’, ‘‘DME Name’ + oligomerization’, ‘‘DME Name’ + dimer’, ‘‘DME Name’ + trimer’, ‘‘DME Name’ + dimerization’, ‘‘DME Name’ + trimerization’ and ‘‘DME Name’ + protein-protein interaction’ were used; when discovering the transcription factor regulations on a DME, ‘transcription factor + ‘DME Name’’ and ‘transcriptional regulation + ‘DME Name’’ were reviewed; when finding the DNA methylations of a DME, ‘‘DME Name’ + methylation’, ‘‘DME Name’ + epigenetics’ and ‘‘DME Name’ + methylate’ were searched. Moreover, the additional DNA methylation data for all DMEs in INTEDE were further collected using the following processes. First, 1377 series records of a very popular microarray platform (Illumina HumanMethylation450 BeadChip) were collected from the Gene Expression Omnibus (53), and 136 series records with both patients and healthy individuals data were selected, which contained 16 256 samples of 86 disease indications. Second, all these collected records were sequentially processed by quality control, normalization and batch effect correction (75–78), and differential methylations were discovered by calculating both P-values and delta-beta values (79,80). According to the definition in previous reports, the threshold of P-values was set to 0.05 (P-value < 0.05 was considered to be statistically significant (79)), and the cutoff of delta-beta values was set to 0.2 and 0.3 (the absolute value of delta-beta >0.3 was considered as ‘significant methylation’ (79), and 0.3 ≥ |delta-beta| >0.2 was reported as ‘moderate methylation’ (80)). Third, the above differential methylations were discovered among four different sample groups, which included: (i) DME methylation in the normal tissue adjacent to the disease tissue of patients (blue), (ii) DME methylation in the disease tissue of patients (red), (iii) DME methylation in the normal tissue of healthy individual (green) and (iv) DME methylation in the tissue other than the disease tissue of patients (yellow). Fourth, the methylation plot across samples was drawn using ggplot2 in R environment, and the violin plots of methylation variation between two studied groups was generated using ggplot2 and ggbeeswarm in R. All plots can be viewed online (Supplementary Figure S3) and downloaded from the website.
Consequently, the HOSPPI information included the interaction types (oligomerization, transcription factor regulation, DNA methylation and drug co-metabolism), the studied diseases (95 diseases standardized in a latest version of International Classification of Diseases (81), ICD-11), the targeted cell lines (289 cell lines, such as A549, Caco-2, HEK293, HepG2 and MCF-7), the levels of DNA methylation (significant or moderate) together with the corresponding P-value and delta-beta value, the drugs which metabolism was affected by studied HOSPPIs, the detailed mechanism of a HOSPPI on drug metabolism, and the violin/bar plots illustrating the differential methylation between different sample groups. All in all, INTEDE contained 7849 HOSPPIs between 565 host proteins and 566 DMEs: oligomerization (homo-/hetero-), transcription factor regulation (activation/repression), DNA methylation (hypo-/hyper-), drug co-metabolism (direct/indirect). HOSPPIs data can be assessed in INTEDE (Figure 4).
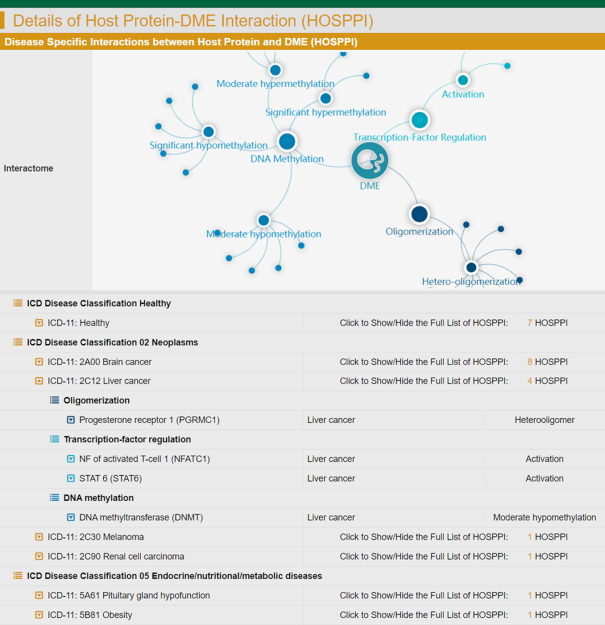
A typical INTEDE page offering the data of host protein-DME interactions (HOSPPIs). The INTEDE contained 7849 HOSPPIs between 565 host proteins and 566 DMEs. Various types of HOSPPI were in the INTEDE, which included: oligomerization (homo-/hetero-), transcription factor regulation (activation/repression), and DNA methylation (hypo-/hyper-).
Interactions between xenobiotics and DMEs
The activity or expression of DMEs could be inhibited or induced by xenobiotics, which, in turn, affected the hepatic clearance of drug and led to the reduced efficacy and therapeutic failure (82–85). These xenobiotics included: (i) pharmaceutical agents (FDA approved, clinical, preclinical, patented and investigative), (ii) health toxicants (biotoxin, carcinogen, environmental pollutant, mycotoxin, and neurotoxin), (iii) natural substances (biological extract, natural product/mixture and traditional medicine), (iv) agricultural chemicals (fungicides, herbicides and insecticide), (v) additive agents (food additives and cosmetic additives) and (vi) proteins/peptides. Since all these xenobiotics are one of the leading factors of the metabolism-based drug–drug interactions (5) and the constant inspiration of clinical treatment optimizations (10), it was essential to collect those xenobiotics-DME interactions (XEOTICs), and clarify the effects of an XEOTIC on modulating the activity or expression of human/microbial DMEs.
Therefore, the XEOTIC data were systematically searched by literature review in PubMed based on the keyword combinations between ‘DME Name’ and ‘biotoxin’, ‘environmental pollutant’, ‘extract’, ‘fungicides’, ‘inducer’, ‘inhibitor’, ‘insecticides’, ‘nature product’ and ‘pesticides’. Those discovered literatures were evaluated manually to identify the xenobiotics affecting DMEs. The collected data included the name of xenobiotics, the modulation types (inducer and inhibitor), the xenobiotic classifications (additive, agricultural chemical, health toxicant, natural substance, and pharmaceutical agent), the modulation activity (measured by MIC/IC50/Ki values), and the affected cell system (choriocarcinoma, hamster ovary, and high five). All in all, the INTEDE provided 47,778 XEOTICs between 4150 xenobiotics and 501 human or microbial DMEs. These miscellaneous xenobiotics cover all the xenobiotic types described in the previous paragraph (from (i) pharmaceutical agents to (vi) proteins/peptides). All XEOTICs can be assessed in the subpage of INTEDE entitled ‘Xenobiotics-DME Interaction’ (Figure 5).

A typical INTEDE page offering the data of xenobiotics-DME interaction (XEOTIC). The INTEDE provided 47 778 XEOTICs between 4150 xenobiotics and 501 DMEs, which were diverse. Particularly, the types of xenobiotics included (i) pharmaceutical agents (FDA approved, clinical, preclinical and investigative), (ii) health toxicants (biotoxin, carcinogen, environmental pollutant, mycotoxin and neurotoxin), (iii) natural substances (biological extract, natural product and traditional medicine), (iv) agricultural chemicals (fungicides, herbicides and insecticide), (v) additive agents (food additive and cosmetic additive) and (vi) proteins /peptides.
Data standardization, access and retrieval
To make the access and analysis of INTEDE data convenient for all users, the collected raw data were carefully cleaned up and then were systematically standardized, which included the disease standardization, EC classification, unit unification, structure availability and crosslink to various reference databases. Detailed description on the way to clean and standardize data was provided in Supplementary Method S2. Furthermore, INTEDE has been smoothly running for months, and tested from different sites around the world. All data could be viewed, accessed, and downloaded (Supplementary Figure S4). Currently, the INTEDE is freely assessed without login requirement by all users at: https://idrblab.org/intede/.
INTERACTION CROSSTALK AND PERSPECTIVES
Although the data of each type of DME interaction in INTEDE were essential for current clinical studies, the crosstalk among different interaction types had emerged to be increasingly important due to the extremely complex mechanisms underlying drug metabolism (10,20,57,86,87). Taking the primary medication for treating Parkinson's disease—levodopa—as an example, it must enter the brain and be converted there by the human DME aromatic amino acid decarboxylase (AADC) to its active form—dopamine. However, the delivery of levodopa to blood–brain barrier is greatly restricted by its intestinal metabolisms of both human and gut microbiome (10). Particularly, the host intestinal AADC and gut microbes Enterococcus faecalis and Eggerthella lenta can convert levodopa into a different chemical before it reaches the brain, which causes unwanted side effects (10). Therefore, levodopa should be co-administrated with other drug by collectively considering multiple DME interactions: (i) the MICBIO between the above microbes and brain DME AADC; (ii) the XEOTIC between xenobiotics carbidopa (inhibitor of intestinal AADC) and AADC (10). In other words, the crosstalk between the MICBIO and XEOTIC of human DME AADC inspired a new strategy that combines AFMT (a microbe inhibitor) and carbidopa (a DME inhibitor) with levodopa, for effectively overcoming the serious side effects of anti-Parkinson drug (10).
Similarly, the HOSPPI between host protein carboxylesterase and DME CYP3A4 was found as the leading cause of drug resistance, which could therefore be reversed by the XEOTIC between xenobiotics vitamin D and DME CYP3A4 (87); the HOSPPI between host protein UGT1A1 and microbial DME beta-glucuronidase was reported to cause diarrhea, which needed be relieved by the XEOTIC between xenobiotic amoxapine and the microbial DME beta-glucuronidase (20,57). Thus, the multiple types of DME interaction in INTEDE might keep inspiring new strategies for dealing with drug resistances and side effects.
To make the crosstalk analysis of each DME possible, the data of multiple interaction types were systematically collected. As a result, 453 (43.3%) out of 1047 DMEs were described in INTEDE with multiple interaction types. Moreover, the number of interaction data for each type was from thousands to tens of thousands, which made it highly possible to identify the differential features or generalize the common characteristics from these DME-related big data. Meanwhile, because of the lack of multiple interaction types for the remaining 56.7% DMEs, it is essential to continue drug metabolism studies for a further extension of our knowledge on DME interactome.
In summary, INTEDE is unique in: (i) providing the largest number of DMEs that are manually curated and systematically confirmed based on their metabolizing drug(s), (ii) covering the novel DME data of diverse microbes (parasitizing in different sites of human body) together with their metabolizing drug(s), (iii) describing the comprehensive interactome data from three perspectives for both human and microbial DMEs and (iv) enabling the crosstalk analysis among various types of DME interaction. With the extensive efforts made on describing DME interactome (3–10) and revealing the crosstalk among interaction types (10,20,57,86,87), those immense, connected and structuralized data provided in INTEDE are expected to have implications for the future practice of clinical treatment optimization (88–92) and personalized medicine (93–96).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2017YFC0908600, 2018YFC0910500]; National Natural Science Foundation of China [U1909208, 81872798]; Leading Talent of the ‘Ten Thousand Plan’ – National High-Level Talents Special Support Plan, Key R&D Program of Zhejiang Province [2020C03010]; Fundamental Research Funds for the Central Universities [2018QNA7023, 10611CDJXZ238826, 2018CDQYSG0007, CDJZR14468801]; Information Technology Center, Zhejiang University; China Knowledge Centre for Engineering Sciences and Technology [No. CKCEST-2019-1-12]. Funding for open access charge: National Natural Science Foundation of China [81872798].
Conflict of interest statement. None declared.
REFERENCES
1.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
20.
21.
22.
23.
24.
25.
26.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.