 Guide-target mismatch effects on dCas9–sgRNA binding activity in living bacterial cells
Guide-target mismatch effects on dCas9–sgRNA binding activity in living bacterial cells
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

- Altmetric
As an effective programmable DNA targeting tool, CRISPR–Cas9 system has been adopted in varieties of biotechnological applications. However, the off-target effects, derived from the tolerance towards guide-target mismatches, are regarded as the major problems in engineering CRISPR systems. To understand this, we constructed two sgRNA libraries carrying saturated single- and double-nucleotide mismatches in living bacteria cells, and profiled the comprehensive landscape of in vivo binding affinity of dCas9 toward DNA target guided by each individual sgRNA with particular mismatches. We observed a synergistic effect in seed, where combinatorial double mutations caused more severe activity loss compared with the two corresponding single mutations. Moreover, we found that a particular mismatch type, dDrG (D = A, T, G), only showed moderate impairment on binding. To quantitatively understand the causal relationship between mismatch and binding behaviour of dCas9, we further established a biophysical model, and found that the thermodynamic properties of base-pairing coupled with strand invasion process, to a large extent, can account for the observed mismatch-activity landscape. Finally, we repurposed this model, together with a convolutional neural network constructed based on the same mechanism, as a predictive tool to guide the rational design of sgRNA in bacterial CRISPR interference.
INTRODUCTION
CRISPR–Cas9 system and its derivatives are recently adopted as versatile DNA targeting tools in a programmable manner and thus widely used in genome editing (1,2), target mutagenesis (3–5), genetic screening (6–8), chromosome painting (9,10), synthetic circuit construction (11) and in vitro nucleic acid detection (12,13), etc. For each application, the binding process is an essential step that starts with PAM(NGG) recognition and the melting of its nearby nucleotides (14), followed by strand invasion which finally results in the formation of an R-loop structure (15,16). However, binding to unexpected loci sometimes occur, leading to unpredictable results. This phenomenon, referred to as ‘off-targeting’, to a big extent, is due to the fact that the binding process can bear several mismatches (2,17) or bulges (18) between sgRNA and target DNA. The off-target effects are regarded as the major problems in engineering CRISPR systems.
Hitherto, various approaches have been designed to understand the target specificity of CRISPR–Cas9 system, where most of them are focusing on the location of imperfect base-pairs. These results (2,17,19–21) showed that mismatches within PAM-proximal 7–12 bp seed region have a severe impact on binding or cleavage. In contrast, the PAM-distal region can tolerate multiple mismatches. However, the lack of fine-grained quantitative profile in living cells may hinder our in-depth understanding of this issue, since binding efficiencies are influenced by intracellular conditions (22). In terms of research on mechanisms, although empirical conclusions or models (7,22–26) present a rough overview of the binding behaviour, the systematic interpretation of the whole process remains inadequate; as for analytical modeling, although many efforts have been made in explaining off-target cleavage (27–30), the progress in off-target binding lags behind (20,21). Considering the key differences between cleavage and binding such as different mismatch tolerance (31), we cannot take it for granted to use models or principles established from cleavage data directly in binding activity prediction. Thus, there is a critical need to get a comprehensive landscape of binding properties in vivo, and furthermore, to build a conceptual model based on real biophysics process that can not only explain the results, but also facilitate our understanding on the sequence-determinants of CRISPR–Cas9 systems.
To address these issues, exhaustive mappings between the sequence features and binding activities are required; and any potential bias needs to be bypassed when quantifying binding activities in a high-throughput manner to minimize noise. For this purpose, we applied a pooled screening approach to assay the binding affinity of different sequences. Endonuclease-deficient Streptococcus pyogenes Cas9 (dSpCas9) was used in experiments to focus on the binding property of Cas9 protein. In order to unbiasedly characterize the binding activity of dCas9 protein, we constructed a counter-selection system that can couple binding affinity with growth of bacterial cells. This system thus enables us to quantify the guide-target mismatch effects on dCas9 binding affinity through a massively parallel approach by measuring the abundance of each mutant with particular mismatches in the library using next generation sequencing (NGS). Based on the derived dataset, we took computational approaches to comprehensively interrogate the impact of mismatch positions, types and combinations on binding affinity. Moreover, the strand invasion process was adopted as a framework to simulate the binding behavior of dCas9, where the output of this dynamic system is largely determined by the thermodynamic properties of perfect or mismatched base pairing. This model, with a solid biophysical foundation, achieves the state-of-the-art performances via mapping the sequence features to binding activities and can help design sgRNAs with tailored activities.
MATERIALS AND METHODS
DNA manipulations and reagents
Plasmid extraction and DNA purification were performed using kits from Omega Bio-Tek. Restriction enzyme FastDigest Eco31I (namely BsaI) was purchased from Thermo Scientific. PCR reactions were carried out using KAPA HiFi HotStart polymerase from KAPA Biosystems (NGS library preparation) and Q5 High-Fidelity DNA polymerase from New England Biolabs (cloning). Plasmids were constructed through Gibson assembly. Antibiotic concentrations for kanamycin, ampicillin and chloramphenicol were 50, 100 and 7 mg/l, respectively. All strains and plasmids used in this work are listed in Supplementary Table S1. All oligonucleotides were ordered from Taihe Biotechnology and Genewiz (Supplementary Table S2).
Cell growth conditions and strain construction
In all experiments, bacteria were grown in LB medium or on LB agar plates. Selective agar plates were prepared by adding 250 g/l filter-sterilized sucrose stock solution to autoclaved sodium chloride-free LB broth (1.8% agar) to a final concentration of 5 g/l. Cells were normally grown at 37°C while the counter-selection against sacB-expressed strains was carried out at 30°C. Molecular cloning was performed with Escherichia coli DH10B (BioMed) as the host. E. coli K12 MG1655 was obtained from the ATCC (700926). The host E. coli strain MCm which used in the screening was constructed from previous work by integrating a chloramphenicol expression cassette cloned from pKM154 (Addgene plasmid #13036) into the smf locus of wild-type E. coli K12 MG1655 (8). Escherichia coli s17-1 sfGFP (superfolder GFP) was a kind gift of the George Guoqiang Chen laboratory at Tsinghua University (32).
Plasmid construction
The Cas9 expressing plasmid pCas9-J23109, dCas9 expressing plasmid pdCas9-J23109, pdCas9-J23111 and pdCas9-J23113 were constructed from previous work (8). The vector for sgRNA expression used in the transformation assay was derived from pTargetF (Addgene plasmid #62226). As for the counter-selection system, in order to flexibly reconstruct the system for different sgRNAs, we built two plasmids in advance, terms pTar-sacB and pTest-pMB1. First, pTar-sacB was digested with BsaI to get the backbone; while the insert fragment was amplified using pTest-pMB1 as the template with the sgRNA expressing region embedded in forward primer, and the target DNA region in reverse primer. Hence, the sgRNA and DNA target can be easily customized and the plasmid pN20test, which was finally used in screening, was assembled from these fragments (Supplementary Figure S1). To optimize the sensitivity of the counter-selection system, we modulated the expression strength of sacB by three different iGEM Anderson promoters (J23105, J23113, and J23114). We inserted the target of sgRNA r0 (TACACTTGAACTACCGCGAG) into the upstream of –35 region of these three promoters, respectively. As a result, three plasmids were constructed, pN20test-J23105/113/114-r0-exp. Similarly, we constructed three negative control plasmids, pN20test-J23105/113/114-r0-NC by replacing the r0 sgRNA cassette with one N20 sequence that cannot recognize the r0 target upstream of sacB promoter. We transformed them into MCm/pdCas9-J23111 and subsequently incubated them on a selective agar plate overnight to check the performance of these selection systems. Finally, J23114 was chosen as it exhibited robust difference in terms of colony growth between r0 and negative control plasmid, suggesting this particular level of sacB expression can efficiently discriminate sgRNAs with different binding activities (Supplementary Figure S2).
Verification of the system's ability to identify mismatched sgRNAs with altered binding activity
To further test whether the synthetic circuit can effectively distinguish sgRNAs that lead to different binding activities of CRISPR–dCas9 system, we replaced sacB open reading frame in pN20Test-114sacB with mcherry, and introduced mutations into sgRNA r0. Six plasmids pN20test-114mcherry-r0-m1/2/3/4/5/6 (Supplementary Figure S3A) were thus constructed. We assumed that if the system is robust, sgRNAs with six different substitutions, due to their different binding affinities to the target region upstream of mcherry promoter, should result in a variety of expression levels of mcherry. The results showed that different mismatched sgRNAs indeed led to altering expression levels of mCherry (Supplementary Figure S3B). Further validation of this mechanism was carried out by replacing the promoter of dCas9 with J23109 and J23113, which have lower strength. As expected, we observed higher fluorescence intensity from mCherry expression, derived from the decrease in dCas9 expression (Supplementary Figure S3B). Meanwhile, it could also be found that the decrease in dCas9 expression would lead to a reduced resolution to distinguish different mismatched sgRNAs. Thus J23111 was applied for dCas9 expression in subsequent work.
Quantification of mismatch effects on binding activity using transformation assay
To mimic the real screen experiment in prior, we further applied a transformation assay to quantify the mismatch effects based on the sacB counter-selection system. The single colony–derived overnight seed cultures of host strains Mcm/pdCas9-J23111 was grown in LB broth at 37°C until an OD600 of 0.6 was reached. Cells were collected by centrifugation at 8000 × g for 5 min at 4°C, washed five times in ice-cold sterile water with the same condition and finally resuspended in 15% (v/v) glycerol (one-sixteenth the volume of the original culture). We then transformed plasmids carrying wildtype, mismatched or negative control sgRNAs (pN20test-114sacB-r0/r349-exp/m3/NC, Supplementary Figure S4A) into the prepared competent cells (MCm/pdCas9-J23111) via Eppendorf 2510 Electroporator using the optimized parameter setting (1800 V, 50 ng plasmids/100 μl competent cells). The transformed cells were incubated in sodium chloride-free LB broth (four times the volume of the competent cells) for 1 h at 30°C for recovery. We then streaked each sample onto 90 mm sodium chloride-free LB agar plates (with kanamycin and ampicillin) with (selective) or without (control) 5 g/l of sucrose, three biological replicates were made for each sgRNA. As different expression levels of sacB would result in varying degrees of cytotoxicity, the survival rate defined as Equation (1) could measure the mismatch effects on binding.
As a result, different sgRNAs showed varying survival rates, which proved the system's ability of quantification of mismatch effects (Supplementary Figure S4B).
Construction of mismatched sgRNA libraries
We synthesized two mismatched oligo libraries based on sgRNA r0 (TACACTTGAACTACCGCGAG) and sgRNA r349 (AGTGTCATTGTTGAATTCTA). For each library, during each step of solid phase DNA synthesis for the 20 nucleotides belonging to the N20 region in sgRNA, a mixture consisted of 90% of the correct nucleotide and 10% of others (3.3% of each) were added, resulting in oligos with 10% single-mutation-rate at each position of N20. Hence the number of mismatches would follow a binomial distribution B(20, 0.90) (Supplementary Figure S5), and the library mainly consisted of single or double mismatched sgRNAs. Each of the oligomers was flanked by two BsaI sites used for further cloning via Golden Gate assembly. The oligo library was amplified by a PCR reaction and the products were inserted into the BsaI-digested pN20test-ran0/349–114sacB-preLib as the backbone vector (Supplementary Figure S6); the assembled plasmid libraries were then transformed in DH10B chemically competent cells, leading to 200 000 CFUs for each library, ∼100-fold coverage for all possible single- or double-mismatched sgRNAs. We further confirmed the quality of the library by Sanger sequencing of 10 colonies picked from the agar plate after transformation, all of them perfectly mapped back to candidate sequences of the libraries. The plasmids for each library was then extracted and used in subsequent screening experiments. We referred these two libraries as r0 and r349 mismatched plasmid libraries thereafter.
Screening experiments
The schematic illustration of the sgRNA activity screening experiments is shown as Figure 1A. The competent cells were prepared as described above (quantification of mismatch effects on binding activity). The library plasmids were then transformed by electroporation into the prepared competent cells (MCm/pdCas9-J23111), which was performed via Eppendorf 2510 Electroporator using the optimized parameter setting (1800 V, 50 ng plasmids/100 μl competent cells). Two biological replicates were made for each host strain by independent transformations. To achieve proper coverage for each sgRNA library, we prepared 19 transformations for each replicate, yielding totally of four working samples for the two libraries with two replicates each.

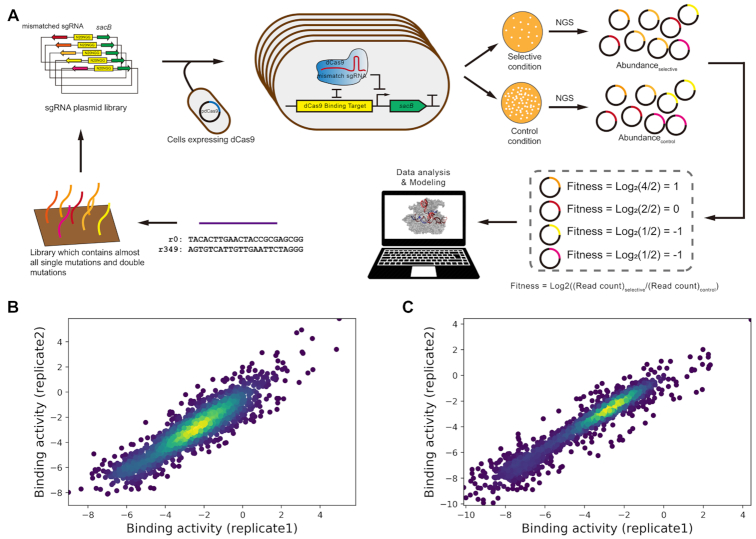
Pooled screening experiments produce high-quality data for binding activities. (A) Schematic diagram of high-throughput profiling of dCas9’s binding activities mediated by mismatched sgRNAs. Two comprehensive single- and double-mismatched libraries were constructed based on a sacB-based counter-selection system in E. coli. For each library, each individual mismatched sgRNA would guide dCas9 protein to block the expression of sacB by targeting to an N20NGG cassette upstream –35 box in promoter, leading to a decline in sacB expression; after growth on selective condition (sucrose), the cells harbouring the library would possess varying growth rates (colony number and size), which were quantified via NGS. The profile of read count number was used to decipher the binding behaviour of CRISPR-Cas9 system. The raw data revealed strong correlations between biological replicates for (B) library r0 (n = 1705, Pearson correlation coefficient = 0.906) and (C) library r349 (n = 1719, Pearson correlation coefficient = 0.956).
The transformed cells were incubated in sodium chloride-free LB broth (four times the volume of the competent cells) for 1 h at 30°C for recovery. We then streaked each sample onto twenty 150 mm sodium chloride-free LB agar plates (with kanamycin and ampicillin) with (selective) or without (control) 5 g/l of sucrose. Via measuring the transformation efficiency in prior, we made sure that no more than 5000 bacterial cells that were successfully transformed on each plate, with a density of around 25 colonies/cm2 and an average distance of ∼2.5 mm between two colonies. After overnight incubation at 30°C, we added 4 ml of sodium chloride-free LB broth on each plate and scraped cells off using spreaders. The cell suspension from the same replicate would be collected and mixed together. We then took 5 ml of each cell suspension to extract plasmids for NGS library preparation. We note that such well-controlled density of colonies on selective plate and growth at only 30°C prevent the overgrowth of colonies that inhibiting the resolution of screen.
Preparation of NGS library and sequencing
The purified plasmids were used as templates for PCR to amplify the N20 region of the mismatched sgRNA libraries (50 μl × 4 reactions per library; 50 ng template per reaction; pF/R_mismatch primers (Supplementary Table S2); KAPA HiFi HotStart polymerase (KAPA Biosystems); 95°C 3 min, 25 cycles [98°C, 20 s; 67.5°C, 15 s; 72°C, 30 s], 72°C for 1 min. The sequencing library was prepared following the manufacturer's protocol (TruSeq DNA Nano Library Prep Kit for Illumina). Sequencing for the mismatched sgRNA library was carried out using a 2 × 150 paired-end configuration and ∼30 million reads were collected for each library. Illumina HiSeq x10 by the PE150 technique was applied for sequencing (GeneWiz Inc.).
NGS data processing
There are totally 10 raw datasets (two replicates for each of the two libraries with two different conditions, r0-selective, r0-control, r349-selective, r349-control; and two plasmid libraries before selection, r0/349-pre) were collected via NGS. After production of clean data by de-multiplexing and removing adaptor regions, pairs of paired-end data were merged by FLASH script and those reads without detected pairs were removed. Python scripts generated in house were then used to search for the ‘TAGTN20GTTT’ 28-mer in the sequencing reads (and the reverse complementary sequence), and those carrying mutations within the upstream (TAGT) or downstream (GTTT) flanking regions (4 bp each) were removed. The read counts were then adjusted using (Equation 2) (n = number of sequencing libraries) to normalize the different sequencing depths of each library. Finally, sgRNAs with <20 read counts in the plasmid library (r0/r349-pre) were removed to increase statistical robustness.
The fitness of each sgRNA was calculated as Equation (3) by comparing its read count between selective condition and control condition (33), which was then normalized by wild type sequence (Equation 4). Here, log2 transformation was used to weaken the effect of extreme values (such as two mismatches in seed region) and thus enhance the resolution of subsequent modeling to elucidate the contribution of members with only moderate effect (such as only one mismatch in distal region from PAM).
Subsequently, the binding activities for each sgRNA in the two biological replicates were averaged as the arithmetic mean.
Regression analysis
After NGS data processing, we collected 1761 entries of binding activity data for library r0, and 1768 for library r349. Those data were first filtered to create high-quality datasets for regression analysis. To ensure the quality of data, we eliminated those data with low consistency between replicates. In detail, if the difference of binding activities between two biological replicates was >3, the related data would be removed from the dataset. After this filter step, 1705 sgRNAs remain for library r0 and 1719 sgRNAs remain for library r349, excluding 0.3% and 2.4% of data, respectively. We then followed a featurization protocol to encode the sgRNA sequences. One-hot encoding was applied here to transform the sequences into 32D vectors. Among these dimensions, 20 of them represent the possible positions of mismatch occurred (MT1∼20), while the other 12 represent all kinds of mismatch types (dArA, dArC, dArG, dTrU, dTrC, dTrG, dCrA, dCrU, dCrC, dGrA, dGrU, dGrG).
We used L1-regularized linear regression (Lasso) in our experiments to interpret the results. The training dataset (80% of all raw data) was used for hyperparameter tuning to optimize the performance of the model by five-fold cross-validation. After obtaining the optimized hyperparameter, we trained the model again using the whole training dataset, the remaining 20% held-out data was used to test the generalization ability of the trained model. All these approaches were performed using scikit-learn package (1.17.4) in Python.
Transformation assay of mismatch effects on Cas9 cleavage activities
The host strains E. coli K12 MG1655 carrying pdCas9-J23109 or pCas9-J23109 were used to prepare competent cells using the method described above (quantification of mismatch effects on binding activity). Plasmids carrying the sgRNA expression cassette (pTargetF) were transformed by electroporation into the prepared competent cells expressing Cas9 or dCas9. The electroporation was performed via a BTX Harvard apparatus ECM 630 High Throughput Electroporation System using an optimized parameter setting (2.1 kV, 1 kΩ, 25 μF). The transformed cells were incubated in LB medium (four times the volume of the competent cells) for 1 h at 37°C for recovery. We streaked the resulting culture onto the LB agar plates (with kanamycin and ampicillin) automated by EasySpiral Pro (Interscience). The colonies were counted after overnight cultivation. The survival rate for each sgRNA was calculated by comparing the CFU of Cas9-expressing cells with the CFU of dCas9-expressing cells. This ratio was further normalized by determining the colony number after transformation with a negative control sgRNA plasmid to minimize the impact of differences in electroporation efficiency that were due to competent cell preparation (Equation 5).
Characterization of mismatch effects through cytometry
The host strain E. coli s17–1 sfGFP carrying pdCas9-J23109 was used to prepare competent cells, after transformation of sgRNA expression cassette (pTargetF), cells were recovered and streaked on to LB agar plates (with kanamycin and ampicillin). We then picked up two single colonies from each sgRNA as two biological replicates and cultivated them into 5 ml LB broth overnight. Subsequently, Cultures were diluted to 1 ml pre-chilled PBS to make the final OD600 ∼0.01. The fluorescence intensity distribution was measured by S3e Cell Sorter (Bio-Rad). We took the arithmetic mean of median fluorescence intensity for each sgRNA in the two biological replicates to represent the expression level of sfGFP. The mismatch effect of each sgRNA is calculated as Equation (6).
Thermodynamic model
To account for the thermodynamic properties in R-loop formation process, we have to calculate energy change in the annealing process of sgRNA-complementary strand helix and the melting process of DNA–DNA duplex simultaneously. However, it should be noted that the parameters related to RNA/DNA (34,35) helix from literature are described for annealing process that starts at 5′ end of RNA (Supplementary Figure S8B). As for the R-loop formation process, the initial base-pair between sgRNA and the target forms in 3′ end of sgRNA. Fortunately, as different initiations have the same thermodynamic value for RNA/DNA helix, we can reparameterize the process with parameters that encode the opposite direction and then easily derive that (Supplementary Figure S8B, C)
Using the above formula, we can calculate the change in Gibbs free energy of each state transition (Figure 5B, except for the initial transition) via Equation (8).
Then consider the energy compensation carried out by dCas9 protein. The above energy change can be rewritten as Equation (9).
Suppose that the system is in equilibrium, the relationship between the probabilities of adjacent states can be denoted as the Equation (10).
Where
Furthermore, we defined the ground state as the dissociation state (), the free energy of each state relative to the ground state can be calculated as Equations (12 and 13). For clarity, we showed a comprehensive diagram of R-loop formation process for sgRNA r0 in Supplementary Figure S9.
From Equations (10 and 13), we can derive that
Finally, we can derive the probability of each state via Equations (11 and 14), which is shown as Equation (15).
There is no doubt that the binding activity is negatively related to the probability of dissociation (). However, due to the lack of thermodynamic parameters for the initial transition (), we took an approximation that ignored the initial state, which is shown as Equation (16).
where
The probability of dissociation would be proportional to the probability of the .
Equation (19) was applied in this work to calculate the probability of dissociation. We note that since the currently available parameters for mismatched base-pairs between DNA and RNA are incomplete, we can only calculate for mismatch types of dArA, dTrU, dCrC and dGrG (35), and positions of mismatches cannot be adjacent.
To determine the compensation term accounting for the interaction between dCas9 and nucleotides, we performed a grid-search approach using the Spearman correlation coefficient between binding activities and probabilities as the objective function. Finally, –3000 J/mol/base was selected as the energy compensation.
Construction of the convolutional neural network
The convolutional neural network (CNN) we designed is based on the thermodynamic model, and its structure is shown as Figure 6A. The first layer of the CNN is used to extract sequence features. As shown in Equation (9), the change in free energy of each state transition (except for the initial transition) is related to 7 bases. Among these bases, two Watson-Crick base-pairs must exist ( with , with ). Therefore, there are total 1024 (45) possible combinations of nearest neighbors. These nearest neighbors are transformed through one-hot encoding and served as convolutional kernels to extract sequence features. It should be noted that if the data is the same as the kernel, the product of them would be 6, otherwise would less than 6 (Supplementary Figure S12). The convolution results are subtracted by 5 and then activated via rectified linear units (ReLU, Equation 20), resulting in one-hot encoded combinations of nearest neighbors. As for the initial transition, there are 16 possible kernels, the convolution results were subtracted by 2 and activated through RuLU.
The rest layers of the CNN are used to interpret the sequence features into thermodynamic properties. The thermodynamic parameters are treated as trainable parameters in convolutional kernels, which are used to convolve with the results of the previous step, generating the change in Gibbs free energy of each transition. To realize Equations (13 and 15), the results are taken into a cumulative sum function (Equation 21) and then transformed into a probability distribution through a softmax function (Equation 22).
The output binding activity is defined as Equation (23).
Therefore, this approach can encode sequences into binding activities using the same calculation method as the thermodynamic model. Also, we randomly split the data into 80% of training dataset and 20% of test dataset to evaluate the performance of CNN. This network was built using Tensorflow (r2.0) in Python.
RESULTS
Profiling the landscape of mismatch effect via high-throughput screening
In order to unbiasedly interrogate the impact of mismatch on the binding activity of dCas9 protein, we constructed a counter-selection system that can couple binding affinity with growth of bacterial cells. With this system, we can quantify such effect via measuring the abundance of each mutant carrying particular mismatches in the library by NGS before and after selection. This genetic circuit includes (i) a constitutive promoter with an upstream N20NGG proximal to its –35 box, which serves as the target DNA and (ii) a sacB gene driven by this promoter whose expression is lethal to E. coli in the presence of sucrose (36). Hence, when the promoter is blocked by a dCas9–sgRNA complex with high activity, there would be a decline in the expression of sacB, leading to a robust growth under selective condition. In contrast, mismatches severely impairing the binding affinity results in normal expression of sacB and subsequent cell death. In this system, the binding sites for dCas9 can be flexibly switched and located at the same position from the target promoter (Supplementary Figure S1). This design bypasses the potential bias compared with targeting to a series of different sites in promoter or coding region, where context such as distance from transcriptional start site matters in determining CRISPRi activity (37). To more exhaustedly explore the sequence space, we prepared two mismatched libraries derived from two different sgRNAs, hereafter we named them as r0 (TACACTTGAACTACCGCGAG) and r349 (AGTGTCATTGTTGAATTCTA). These two sequences were randomly generated and have no off-target hit in E. coli genome via previous criteria (8). Each library contained saturated single and double nucleotide mismatches towards relevant DNA target. Besides, we also verified that the counter-selection systems could resolve the mismatch effects on dCas9 binding activities (see Methods). After construction of plasmid libraries, we transformed them by electroporation into an E. coli strain constitutively expressing dCas9 protein. The transformants were divided into aliquots and spread on LB agar plates with (selective condition) or without (control condition) 5 g/l sucrose. After incubation overnight at 30°C, we collected cells from plates and extracted plasmids. The sgRNA coding regions of each mutant were PCR amplified and then quantified through NGS. The abundance change relative to wild type sgRNA was applied to characterize the binding activity of each sequence (see Materials and Methods, Figure 1A).
After obtaining the sequencing results, we firstly performed pretreatment to filter low-quality entries (see Methods). As a result, there were 1705 sgRNAs left for library r0, covering all single mutants and 96.2% of all possible double mutants; as for library r349, 1719 remaining sequences included all single mutants and 97.0% of possible double mutants. The Pearson correlation coefficient between biological replicates (0.91 for r0, 0.96 for r349) indicated the reliability of these experiments. Therefore, we got two exhaustive maps of binding activities of single- and double- mutants with high-quality through this experimental framework (Figure 1B, C).
Mismatches in seed region have synergetic effects
The results suggest that (Figure 2B, C) mismatches in PAM-proximal region (seed region) would lead to manifest declines in binding activities, which was consistent with previous studies (2,17,19–21) as seed region serves as a nucleation site for R-loop formation (15). In contrast, the mismatches in PAM-distal region only had moderate impact on dCas9 binding. Additionally, we noticed that in seed region, double mutants exhibited apparently lower binding affinities than the sum of effects of two relevant single mutants (Figure 2D, E), suggesting the synergistic effects between mismatches in seed region. This phenomenon was also observed in an in vitro profiling of dCas9 binding behaviour at longer time scales (21). In contrast, we observed strictly additive effects out of seed region. These results indicated that the binding activity of CRISPR–dCas9 system could be quantitatively customized via inserting mutation(s) in seed to amplify differences; or adding mismatches out of seed region so as to linearly decrease the binding activity.
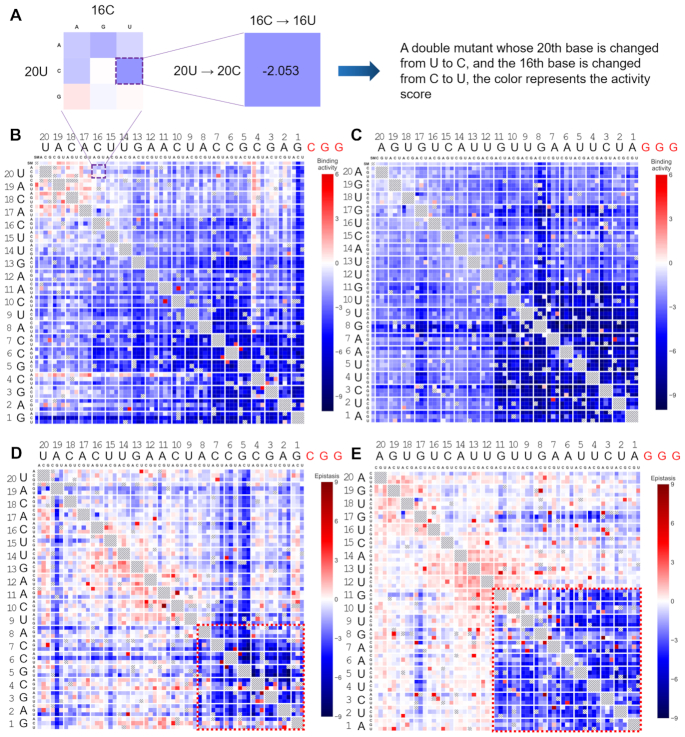
Comprehensive profiling of binding activities of CRISPR-dCas9 system mediated by mismatched sgRNAs. (A) Zoom in on one particular mutant with double mismatches in the heatmap, the outer character represents nucleotide in wild-type sgRNA which complementary to the target, the inner bases are possible mutations. (B) Binding activity score for single- (the first row and column, ‘SM’ stands for single mutant) and double- (other rows and columns) mutants across replicates (above and below the diagonal) for library r0 and (C) library r349. The bar on the right of each panel shows the color-encoding of the activity score. (D) Epistasis for double mutants for library r0 and (E) library r349, these values were calculated by subtracting activity scores of two corresponding single mutants from double mutants (Epistasis = Activityi,j – (Activityi + Activityj)). Positions exhibiting strong synergetic effect are highlighted by the red dashed frame. The bar on the right of each panel shows the color-encoding of the epistasis effect.
Mismatch type of dDrG (D = A, T, G) only has moderate effect on binding activity
Besides the positions of mismatches, what is still exclusive in this field is a comprehensive insight about how mismatch types affect binding activities. To discriminate the effects of mismatch positions and mismatch types, we adopted a regression approach, where the coefficient of each variable after fitting can be used to quantify the impact of the relevant terms on binding affinity, such as mismatch types (Figure 3A). We encoded each sgRNA sequence as a 32D vector (see Methods). L1-regularized linear regression (Lasso) was applied to fit the data. To avoid overfitting, we randomly split dataset into two subgroups with 80% of data were used as training dataset to optimize the hyperparameter through 5-fold validation as well as train the model parameters. The remaining 20% were used as test dataset to check the generalization capacity of the model. The performances in test dataset (r0: r2 = 0.63, r349: r2 = 0.65, Figure 3B, C) indicated that the model has reasonable generalization capacity and captures biological signals. We then analysed the features’ contributions to sgRNA activities (Figure 3D, E). Among the coefficients of 12 different mismatch types, we found dDrG (D = A, T, G) exhibited less impairment on binding than others. Importantly, this effect can be observed in both models trained from two different datasets (Figure 3D, E). To ensure that this discovery was not correlated to the sequence context, we further used the combinations of mismatch sites and mismatch types to extract sequence features, the same training scheme was applied and the results also showed that dDrG was more tolerable compared to other types of mismatch (Supplementary Figure S7). Furthermore, we found this observation was consistent with the thermodynamic stabilities of mismatched nucleotides between DNA and RNA (35), and was also partly discovered in previous study (17). Moreover, a structural study (15,38) also found a base-specific interaction between Arg71 and guanine in PAM-proximal region of sgRNA. Thus, it's reasonable to infer that dDrG was more tolerable than other types of mismatch.
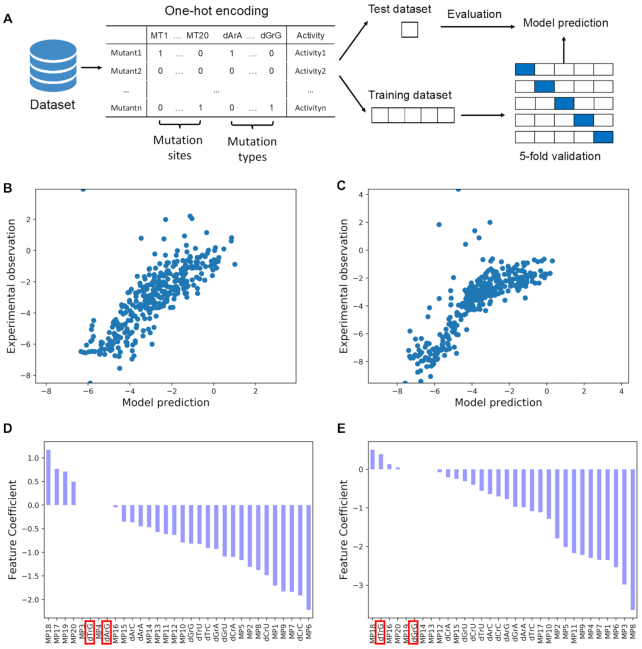
Regression approach demonstrates the effect of mismatch types on binding. (A) Schematic diagram of the computational process. 32 features including 20 for mutation sites and 12 for mutation types were extracted for each sgRNA. Data were random split into training dataset containing 80% of raw data and test data set containing the rest 20% of data. Lasso regression was used here to interpret the relationship between features and activities. The hyperparameter was optimized through 5-fold cross-validation, after that, the whole training dataset was used to train model parameters. Finally, the test dataset was used to evaluate the generalization capacity of the model. The model performance in test dataset showed a good generalization capacity for both (B) library r0 (n = 341, r2 = 0.63) and (C) library r349 (n = 344, r2 = 0.65). (D, E) Coefficients of features that contribute to the prediction power of the Lasso regression for (D) library r0 and (E) library r349.
To validate whether dDrG impairs binding activity to a less extent, we additionally measured mismatch effects via testing gene expression repressed by different dCas9–sgRNAs using cytometry (see Method). Using a chromosome-integrated sfGFP as the marker, we chose two sgRNAs targeting its non-template strand in ORF region (sgRNA1, sgRNA2) as well as 1 sgRNA targeting its promoter (sgRNA3); derived from these 3 sgRNAs, we constructed mutant sgRNAs covering saturated single substitutions in PAM-proximal 10 bp region of these sequences to minimize the effect of mismatch position (Figure 4A). As a result, dDrG showed stronger fluorescence signals than (or equal to) others, consistent with the hypothesis that dCas9 binding is less sensitive to dDrG mismatch (Figure 4B). Furthermore, we checked whether this phenomenon would extend to Cas9 cleavage activities. To this end, based on the fact that Cas9 induced DNA double strand break is lethal to bacteria, we carried out a transformation assay to measure the cleavage activities for mismatched sgRNAs via quantifying CFUs (see Methods). We chose 3 sgRNAs targeting E. coli genome (mocA-262, artP-627, araE-1205) which showed high activities in our previous study (39). Based on these sequences, we constructed several mutants with single mismatch by substituting some nucleotides in seed (Figure 4C). The results also revealed that dDrG had less impact on Cas9 cleavage activity compared to other mismatch types (Figure 4D). Overall, these results suggested that the mismatch type of dDrG generally has moderate effect on CRISPR-Cas9 systems, indicating that there may be a relationship between activities and thermodynamic properties of nucleic acids.
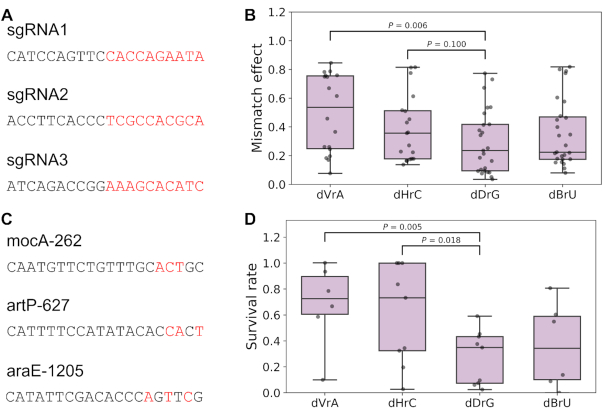
Mismatch type of dDrG exhibits less impairment on binding and cleavage. (A) Characterization of mismatch effects on dCas9 binding. Single mutants were constructed based on three targets belonging to a chromosome-integrated sfGFP expression cassette. Mutant sgRNAs were constructed carrying saturated single substitutions in PAM-proximal 10 bp region (shown in red). (B) Mismatch type of dDrG showed slightly lower impairment on binding activity (see Method) than dVrA (P = 0.006, one-tailed t-test) and dHrC (P = 0.100). (C) Transformation assay of mismatch effects on Cas9 cleavage activities. Mutants with single mismatch were constructed based on three targets in E. coli genome by replacing several nucleotides in seed (shown in red). (D) Mismatch type of dDrG showed slightly less impact on cleavage (see Method) than dVrA (P = 0.005) and dHrC (P = 0.018).
Binding activity is predicted by a model using independent thermodynamic data of nucleic acids
Although the abovementioned linear models can effectively predict the binding activities of protospacers in specific contexts, their generalization abilities were limited, which were specifically reflected in the slightly different patterns when training on different datasets (Figure 3D, E). Besides, the linear models could not account for the causal relationship between sequence and its binding behaviour either due to its ‘black box’ manner (40). To address these issues, we aimed to construct a conceptual model based on real biophysics, which is expected to be more general and basic. The thermodynamically more stable mismatch type of dDrG with less effect on binding identified by the linear model inspired us to focus on the thermodynamic features of nucleic acids. Here, our aim is to develop a model that uses nucleotide thermodynamics data from independent sources as much as possible. In principle, this model can quantify the free energy change during the strand invasion process in dCas9 binding for any sgRNA sequence of interest, the input of the model; and the calculated free energy landscape can be used to predict the binding activity of the given sgRNA, the output of model.
In nucleic acid thermodynamics, the nearest neighbor approach was developed to predict the thermodynamic stability of secondary structures, where each parameter represents the thermodynamic impact of adding a distinct base-pair to a double-stranded helix (41). We can use those parameters to calculate the thermodynamic stability of each step of either annealing or melting process. In the process of R-loop formation, these two procedures happen simultaneously (Figure 5B, Supplementary Figure S9). Thus, we can calculate the overall free energy landscape during R-loop formation using nearest neighbor approach. Suppose that the system during R-loop formation is in thermodynamic equilibrium, the total process can be treated as a Markov chain (the process is thus regarded as reversible). Hence, the probability of each state can be calculated de novo using the overall free energy landscape derived from the thermodynamic data of base pairing from independent sources. The probability of exit of R-loop formation (dissociation) can be regarded as reversely correlated with the binding activity (Figure 5A, see Materials and Methods). This is the basic framework of our model.
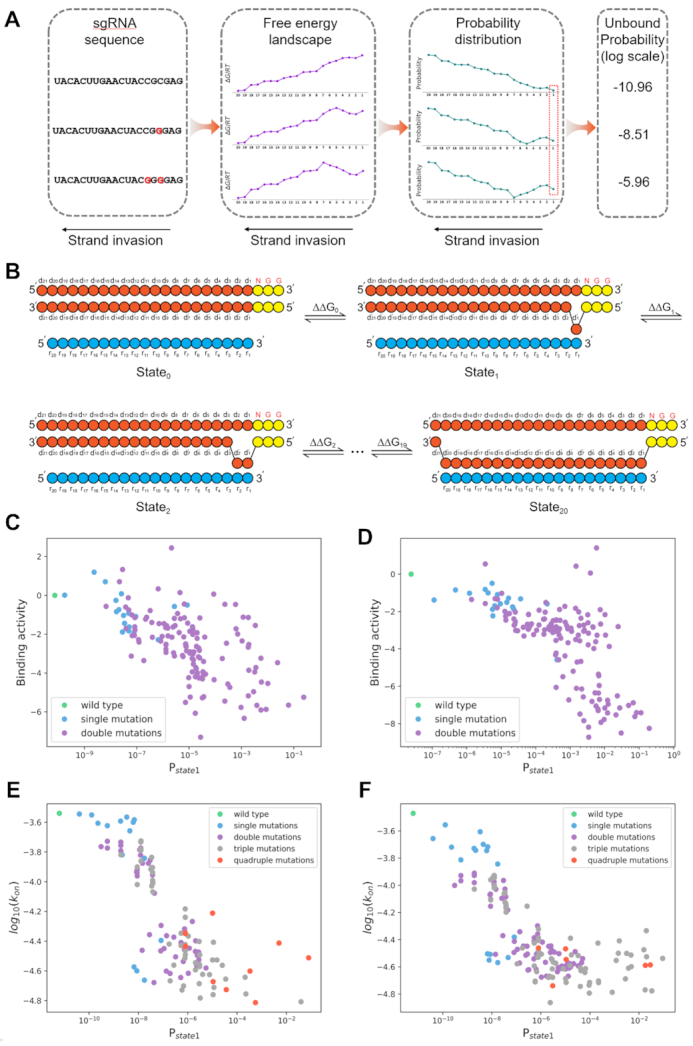
Thermodynamic properties of base pairing coupled with the process of R-loop formation achieved high performances in explaining binding activities. (A) Schematic diagram of the thermodynamic model. For a given sgRNA sequence, the free energy landscape during strand invasion is calculated via the nearest neighbor approach, then the probability of each state can be derived via treating each state during strand invasion as a Markov chain (see Methods). The unbound probability can thus be quantified and is reverse to the binding activity. (B) The model of strand invasion process. During association, the melting between DNA:DNA and the base pairing between DNA:sgRNA happened simultaneously from PAM-proximal region and extend to PAM-distal region; this process is also reversible. Each transition was considered in thermodynamic equilibrium, the probability of each state could be calculated using thermodynamic parameters (see Methods). (C, D) The binding activities derived from experimental data were negatively correlated to the probabilities of dissociation state (Pstate1, see Methods) for both (B) library r0 (n = 150, Spearman correlation coefficient = -0.64) and (C) library r349 (n = 152, Spearman correlation coefficient = –0.71). (E, F) The in vitro mismatched sgRNA association rates were highly correlated to the probabilities of dissociation state (Pstate1, see Materials and Methods) for (E) 10 nM dCas9, replicate 1 (n = 128, Pearson correlation coefficient = –0.79) and (F) 10 nM dCas9, replicate 2 (n = 168, Pearson correlation coefficient = –0.75).
Besides, it should be noted that R-loop is formed inside the dCas9 protein, thus interactions between dCas9 and nucleotides should not be neglected. Previous structural evidence also revealed that negatively charged sgRNA:DNA heteroduplex is accommodated in a positively charged groove (15). Additionally, non-bonding interactions including hydrophobic interactions and hydrogen bonds also existed between Cas9 and nucleic acids (15). Indeed, in the absence of this compensation term describing the interaction between dCas9 and nucleotide, the prediction of the model can only give moderately consistent results compared with the experimental dataset (Supplementary Figure S10). We therefore added a compensation term in each state transition to represent those interactions between dCas9 and nucleotides. This term was treated as a constant and optimized through a grid-search approach (see Materials and Methods), which was finally determined to be -3,000 J/mol/base. This is the only parameter in the model that is trained from our own data.
Using this compensation term and previously reported thermodynamic data of base pairing (34,35,42), we calculated the probability of dissociation for each calculable sequence. Thus, this model derives the binding activity of the dCas9 protein exclusively from the thermodynamic property of base (un)pairing and interaction energy between dCas9 and nucleotides during strand invasion. The results showed that there were strong correlations between the activity scores measured experimentally and the derived probabilities for both single mutants and double mutants in library r0 (Spearman correlation coefficient = –0.64, Figure 5C) and r349 (Spearman correlation coefficient = –0.71, Figure 5D). The generalization capacity of this model was also confirmed by other datasets. The probabilities of dissociations derived from the model were highly correlated to the association rates (kon) of mismatched sgRNAs measured in vitro (21) (Figure 5E, F, Pearson correlation coefficient = –0.79, –0.75, respectively), even for triple (or more) mismatches. Our model can also account for another dataset measuring the binding activities of mismatched sgRNA in vivo (40) (Supplementary Figure S11, Pearson correlation coefficient = 0.51), whereas a higher variance may be due to the noise brought from their experiment and data pre-treatment (see Supplementary note 1 for detailed discussions). It is noteworthy that, to the best of our knowledge, this biophysical model achieved the state-of-the-art performances in explaining binding behaviours using the thermodynamic parameters (34,35,42) from independent sources. This model hence demonstrated that nucleotide thermodynamics can account for the observed mismatch effects on dCas9 binding to a big extent. As this framework is rooted deeply in the real biophysics process during the interaction between Cas protein and nucleotide, it has potential to be adopted to understand the activity of other CRISPR systems at molecular level. This model was further applied to design sgRNAs with tailored activities at website: http://www.thu-big.com/sgRNA_design/Quantitative_CRISPRi_Design/.
We note that the currently available dataset of thermodynamic properties for mismatched base pairs between DNA and RNA is incomplete. This directly leads to the fact that we can only simulate a small fraction (∼9.2%) of all mutants whose mismatches have available thermodynamics data. In order to overcome this drawback, we proposed that the unavailable thermodynamic stabilities for DNA::RNA mismatches can be regarded as trainable parameters in the model. In this line, we can extend the abovementioned computational framework to theoretically all mismatches in sgRNAs. Hence, we designed a convolutional neural network that possessed the same calculation method as the thermodynamic model described above, but treated these thermodynamic parameters as trainable variables and activity scores as targets (see Materials and Methods, Figure 6A). We used the mean squared error as the loss function and updated parameters through backpropagation. As a result, this network also achieved good performances in two datasets (Figure 6B, C). Critically, the trained parameters were correlated with the experimentally determined nearest neighbor parameters (34,35,42) (Supplementary Figure S13). This result supports our hypothesis that the mismatched sgRNA activity dataset contains rich information about the thermodynamic stability of DNA::RNA base (un)pairing, and reversely, the binding activity of CRISPR-dCas9 system was determined by thermodynamic features of the sequence.
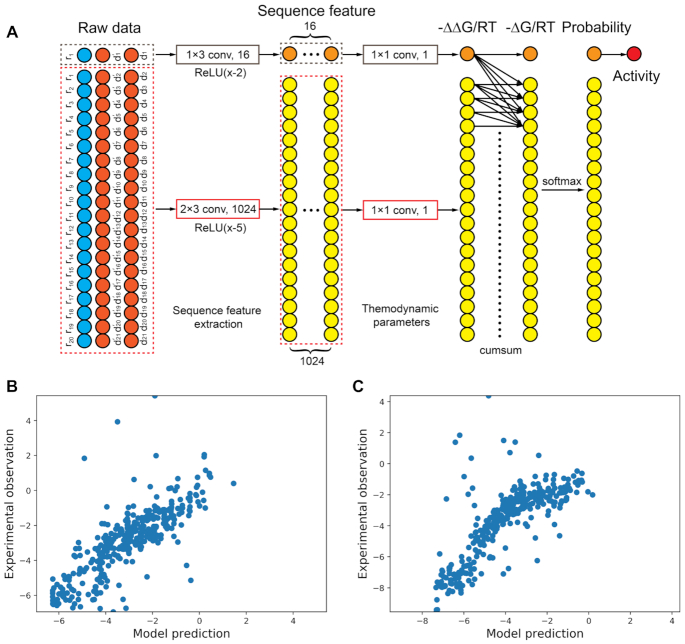
Convolutional neural network based on the thermodynamic model can well fit to experimental data. (A) Structure of the convolutional neural network, in which thermodynamic parameters were treated as trainable variables. Through this network, the one-hot encoded sequences can be transformed into binding activity by calculating probabilities of dissociation. Parameters were updated through backpropagation using mean squared error as the loss function. (B, C) Data were randomly split into training dataset containing 80% of raw data and test data set containing the rest 20% of data. The network achieved good performances in fitting test dataset for both (B) library r0 (n = 341, r2 = 0.61) and (C) library r349 (n = 344, r2 = 0.56).
DISCUSSION
In this work, we developed a pooled screening approach that can massively profile the dCas9 binding activities. Besides the synergetic effect in the seed region, we also found that the effect of multiple mismatches out of the seed region is almost additive; and this simple principle can guide the effort to rationally design gRNAs with attenuated activities quantitatively. Furthermore, inspired by that the relatively stable mismatch type of dDrG only showed a moderate impairment on dCas9 binding, we realized that both mismatch position and type, while the later one is less addressed in previous works, needs to be considered systematically in a general framework to quantitatively predict the effect of mismatch on binding activity. Hence, we constructed a thermodynamic model that can effectively predict the binding affinities by taking literature reported nearest neighbor parameters (34,35,42) of nucleotide thermodynamics into the process of strand invasion. Different from models on cleavage activities (27–30), our model regards binding (strand invasion) as a reversible process and focus on the probability of unbound state, whereas those models considering cleavage assume a critical irreversible cleavage step after the gating nucleotide is reached (27–30). Thus, in our model, it is the overall free energy landscape, consisting of every possible intermediate state during strand invasion, together determines the probability of the unbinding state (see Methods). Furthermore, to deal with the inadequate thermodynamic parameters of DNA/RNA mismatches, we built a convolutional neural network based on the thermodynamic model that also leads to good performances in activity prediction. In conclusion, this work can help to mechanically understand the sequence-determinants of CRISPR-dCas9 system at the molecular level, on the other hand, can guide quantitatively designing of CRISPR–dCas9 system with tailored activity.
The synergetic effects in seed identified here probably roots in the energy compensation carried out by dCas9 protein along with the strand invasion process. During binding, the energy compensation enables this system going through one mismatch in the PAM-proximal region. While for two mismatches in seed, the energy barrier cannot be adequately compensated thus block the strand invasion at the very beginning, resulting in a bigger chance for dCas9 to leave the current DNA target. Here, the effect of multiple mismatches can be amplified, giving rise to the synergetic effect. Moreover, if the mismatches are out of the seed region, as sgRNA has already comes to the end of the strand invasion process. Multiple mismatches here, together with the decreasing free energy during the early stage of strand invasion, resulting in a local energy minimum right before these mismatches; and thus render them exhibit less capacity to repel the dCas9 protein. On the other hand, there is also evidence showing that attenuating the interactions between Cas9 and nucleic acids, which results in less energy compensation during strand invasion, could improve the specificity (25,43,44).
One potential point to further improve this model is the energy compensation term related to the interaction between dCas9 and nucleotide substrates during strand invasion. Regarding the energy compensation as a constant in this work is straightforward, but may be not such meticulous since the interactions between dCas9 and nucleotides should vary across the strand invasion process, even for the same nucleotides at different sites. We propose that this problem can also be fixed by the CNN approach, via training these energy terms as convolution kernels, just like the nearest neighbor parameters in the current model. Similarly, this framework may enable us to quantitatively determine other essential but unavailable thermodynamic parameters for CRISPR-dCas9 system using high-throughput screening data, which allows predicting activities not only for off-target sequences, but also for on-target sequences in a ‘white-box’ manner. Crucially, a comprehensive exploration of sequence and mismatch space using unbiased high-throughput screening is needed to support such data-driven method. By this means, we can dramatically extend the applicability of this framework to ultimately construct general models for CRISPR/Cas system in future works.
In addition to elucidating the binding mechanism, the results of this work are also expected to lead to some useful applications. A straightforward idea is to design sgRNAs for the prokaryotic microorganisms, in terms of either sgRNA (library) without any potential off-target, or tailored designed sgRNA (library) with mismatches to carry customized activities; both of which are important for large-scale CRISPRi screen in bacteria to investigate the relation between gene function and its expression level (8,45–48). In particular, we note that sgRNA library with rationally designed mismatches to carry customized knockdown activity, by acting as unique identities, can be easily adopted into NGS-based high-throughput pooled screen, enabling investigating phenotypic readout of tuning thousands of bacterial genes in parallel (47,48). This is extremely hard to do by simply modulating the expression level of dCas9 by arrayed screen, when thousands of genes need to be investigated. We believe such large-scale pooled screen is paramount for bacterial functional genomics, in terms of both understanding and engineering complex bacterial genome and the genetic basis for the derived phenotype of practical interest. Moreover, the synergetic effects in seed indicated that the CRISPR-dCas9 system had the sensitivity to detect single-nucleotide polymorphisms (SNPs), as the binding activity could be quantitatively customized via inserting mutation(s) in seed to amplify differences. A similar strategy was also applied in CRISPR-Cas13a system (49), indicating that these synergetic effects might be a shared characteristic among a wide range of CRISPR-Cas systems.
DATA AVAILABILITY
Raw data of CRISPR screening for the tiling library has been deposited onto the NCBI Short Read Archive with accession number BioProject: PRJNA644791. The code and plasmid maps related to this work can be accessed via Github (https://github.com/fenghuibao/CRISPR_mismatch_analysis).
ACKNOWLEDGEMENTS
We would like to thank Prof. George Guoqiang Chen for his kind gift strain E. coli s17-1. We also thank Prof. D. Lu and members from the C. Zhang and H. Xing laboratories for critical discussions of this work. We thank T. Xiang for his help in building the web application.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2018YFA0901500 to C.Z.]; National Key Scientific Instrument and Equipment Project of NSFC [21627812 to C.Z.]; Postdoctoral innovation support plan from the China Postdoctoral Science Foundation (to T.W.); postdoctoral fellowship from the Tsinghua–Peking Joint Center for Life Sciences (to T.W.). Funding for open access charge: National Key Research and Development Program of China [2018YFA0901500].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.