 Ligand-dependent tRNA processing by a rationally designed RNase P riboswitch
Ligand-dependent tRNA processing by a rationally designed RNase P riboswitch
Nucleic Acids Research



- Altmetric
We describe a synthetic riboswitch element that implements a regulatory principle which directly addresses an essential tRNA maturation step. Constructed using a rational in silico design approach, this riboswitch regulates RNase P-catalyzed tRNA 5′-processing by either sequestering or exposing the single-stranded 5′-leader region of the tRNA precursor in response to a ligand. A single base pair in the 5′-leader defines the regulatory potential of the riboswitch both in vitro and in vivo. Our data provide proof for prior postulates on the importance of the structure of the leader region for tRNA maturation. We demonstrate that computational predictions of ligand-dependent structural rearrangements can address individual maturation steps of stable non-coding RNAs, thus making them amenable as promising target for regulatory devices that can be used as functional building blocks in synthetic biology.
INTRODUCTION
A fast adaption to changing environmental conditions or availability of nutrients is essential for bacteria to survive. They utilize versatile regulatory mechanisms to adjust gene expression for adaptation of their metabolism. For instance, transcription factors, when bound to metabolites, can modulate the action of RNA polymerase. Furthermore, various metabolites can directly interact with structured elements of mRNAs and trigger a structural rearrangement that affects transcription or translation of the target gene (1–3). Such regulatory RNA structures are known as cis-regulatory elements or riboswitches and are frequently found in the 5′-UTR of bacterial mRNAs.
Natural riboswitches are composed of a highly conserved aptamer region closely linked to an expression platform. Specific binding of the ligand to the aptamer induces a structural reorganization of the regulatory platform that, as a consequence, modulates gene expression in a ligand-dependent manner (4–7). This modular composition makes it possible to generate synthetic riboswitches that utilize artificial aptamers. Various in vitro-selected RNA aptamers that bind to theophylline (8), tetracycline (9) or neomycin (10) were successfully coupled either to natural (reprogrammed) or in silico predicted regulatory sequences (11,12) and such artificial riboswitches were engineered for diverse in vivo applications (13,14). They include synthetic RNA elements to control gene expression in response to varying external pH levels (15) or logic gates in a more complex context (16–19). Moreover, synthetic riboswitches are utilized as reporter molecules for intracellular metabolites, like coenzyme B12 (20,21), as fluorescent biosensors (22,23) or for the design of inducible CRISPR systems (24). Here, we describe the computational design and biochemical characterization of a ligand-dependent RNA device exerting a direct control of RNase P-mediated tRNA processing.
Identified in all kingdoms of life, RNase P represents a ubiquitous tRNA processing activity that specifically removes the 5′-leader of tRNA precursors (25,26). In most organisms, cleavage activity results from the RNA subunit of an RNase P ribonucleoprotein, representing one of the best studied ribozymes (27,28). Alternatively, the reaction can be catalyzed by protein-only enzymes (29–31). While the processing reaction catalyzed by various RNase P enzymes is well characterized, a direct riboswitch-based regulation of the cleavage site accessibility has not been demonstrated. Instead, tRNA 5′-end processing was controlled in an RNase P-independent way, where a theophylline-dependent hammerhead aptazyme removed the 5′-leader sequence from a precursor-tRNA (32). While the released tRNA was functional in translation, it did not represent a bona fide mature tRNA as they are found in vivo, because the ribozyme cleavage resulted in a 5′-OH group instead of the monophosphate that tRNAs processed by RNase P carry at their 5′-end. A similar aptazyme was used to release a tRNA precursor carrying a single-stranded leader sequence that subsequently was cleaved by RNase P, resulting in a mature tRNA with a 5′-phosphate group (33). While equally functional, this approach represents an indirect control of RNase P-mediated tRNA processing, as it regulates the production of a pre-tRNA that is then further processed. In the primary transcript, however, the tRNA structure is disrupted, since its 5′-part is included in the expression platform. Hence, the ligand-interaction does not immediately control the tRNA 5′-end maturation and does not provide new insights in the processing mechanism of RNase P.
For RNase P-mediated tRNA 5′-processing, it is known that length as well as sequence composition of the tRNA 5′-leader contribute to substrate recognition and binding affinity of the enzyme (34–37). In an in vitro experiment, Lin et al. used a pool of randomized leader sequences to investigate the impact of the leader on RNase P binding (35). Based on minimum free energy (MFE) calculations, the authors concluded that substrate affinity decreases when the distal sequences base-pair with the proximal 5′-leader region. Based on these findings, we use a computational approach to design a regulatory RNA device with an expression platform that only includes the 5′-leader but not the downstream tRNA sequence. Modelling the accessibility of this leader region allows to analyze the structural influence of the respective sequence on the RNase P-dependent cleavage reaction.
The mechanism of the designed riboswitch confirms the assumption of Lin et al. (35) and offers new insights about the structural requirements in the leader region essential for efficient RNase P processing. Our approach allows to regulate RNase P activity depending exclusively on the accessibility of the 5′-leader without influencing the downstream tRNA sequence and structure. Instead of an aptazyme, a sequestor hairpin was devised that masks or presents the single-stranded 5′-leader sequence of a tRNA precursor in a theophylline-dependent way. While these constructs are functional in vivo as well as in vitro, we identified a single base pair in the sequestor hairpin that has a strong impact on the regulatory potential of the riboswitch, allowing to modulate the processing efficiency of the adjacent tRNA molecule.
MATERIALS AND METHODS
In silico prediction of RNase P riboswitches
The design model in this contribution is based on the idea that RNase P readily processes a single-stranded tRNA 5′-leader sequence and that this cleavage process is blocked when the leader is masked in a double-stranded region, i.e. a sequestor hairpin. To implement this model, the following set of components was assembled into a novel RNA device (Supplementary Figure S1): The theophylline aptamer TCT8–4 (8) was defined as a sequence-structure constraint. To model theophylline binding in the thermodynamic equilibrium by the soft-constraint framework of the ViennaRNA package (38), the binding motif, , and the experimentally determined binding energy (8) were specified (Supplementary Figure S1A). The sequence directly following the aptamer (Supplementary Figure S1B) was kept from a theophylline-dependent transcription-regulating riboswitch (39), while the 3′-part of the original terminator hairpin was randomized and its length varied between 8 and 16 bases in order to create a switchable sequestor hairpin (Supplementary Figure S1C). A structure constraint was set to ensure that base pairing between the aptamer 3′-region and the randomized sequence is still possible. The last seven nucleotides at the 3′-end of the sequestor hairpin were defined as tRNA 5′-leader sequence of Escherichia coli's suppressor tRNATyr supF (Supplementary Figure S1C). To facilitate tRNA functionality, we used the complete sequence as constraint (Supplementary Figure S1D). Furthermore, a structural constraint for the closing stem of the tRNA was specified (Supplementary Figure S1D), as this sub-structure is known to be recognized by RNase P and is therefore essential for the cleavage process. We applied RNAblueprint (40,41) to sample sequences compatible with the combined set of sequence and structure constraints (Supplementary Figure S1E).
To assess the quality of a sampled sequence, scores were estimated following a similar procedure as described in (42) and utilizing the hard- and soft-constraint framework (43) of the ViennaRNA package. By default, only secondary structures containing Watson–Crick and wobble G–U base pairs were taken into account. Switching ability of a generated sequence x was assessed by formulating the leader accessibility score. In brief, the free energy of a constrained structure ensemble and the free energy of the complete/unconstrained structure ensemble was used to estimate the conditional probability
of observing sequence x in a conformation where all seven nucleotides of the tRNA leader region are unpaired. R is the gas constant of and temperature T was set to .To estimate the conditional probability , the energy of every structure that contains the correct binding motif incurs a bonus energy of (8,44) to model binding to the ligand. To assess how likely it is to observe a paired leader in the absence and an unstructured leader in presence of theophylline both probabilities were combined into the leader accessibility score
The value of the score lies between 0 and 1 and approaches 1 when sequence x implements the intended design model.
The range of possible leader accessibility scores for sequences compatible with the given structure constraints of different riboswitch lengths varied. We therefore defined the relative leader accessibility score
where max and min are the observed best and worse score for a specific riboswitch length, respectively.The leader accessibility score does not take proper and independent tRNA folding into account. However, for efficient RNase P processing, the correctly folded tRNA closing stem is essential (45–47). We therefore calculated the probability of this stem in the unconstraint ensemble and the probability in the ensemble of structures that bind ligand. Both probabilities were combined into the tRNA score
This score measures that both probabilities differ only marginally with and without ligand and becomes 1 when the stem is present under both conditions.
Additionally, we measured through the tRNA independent fold score whether the riboswitch and the tRNA sub-sequences are folding into distinct structural elements. This score was calculated by predicting the ensemble free energies and of the riboswitch subsequence and the tRNA subsequence, respectively. If the riboswitch and the tRNA fold independently, the sum of the individual ensemble free energies is equal to the ensemble free energy of the full-length construct. Thereby the tRNA independent fold score
becomes one. A complete and sortable list of all calculated in silico scores for the investigated as well as additional best and worse scoring constructs of different lengths is available online (http://www.bioinf.uni-leipzig.de/supplements/20-001).Cloning of riboswitch candidates
Riboswitch candidates fused to supF tRNA were generated by overlap extension PCR and inserted into plasmid pBAD by site-directed mutagenesis according to Domin et al. (19). Correct sequence of resulting individual clones was verified by sequencing.
Quantitative analysis of c3GFP activity
For each individual riboswitch construct, three different colonies of overnight grown E. coli TOP10 cells containing plasmid pACYC/tetM with the c3GFP reporter ORF and an amber stop codon at codon position 39 as well as the riboswitch-carrying plasmid pBAD/AmpR were diluted to an OD600 of 0.06 in 5 ml LB medium. Cells were incubated at 37°C in the presence of 50 μg/ml ampicillin, 10 μg/ml tetracycline, 0.01% arabinose (w/v) and 0 or 2 mM theophylline for 4.5 h. After cooling on ice for 15 min, cells were diluted to an OD600 of 0.8 and washed with 1 ml 1× phosphate-buffered-saline (PBS) buffer. The fluorescence intensity was measured using a fluorescence spectrometer (JASCO Deutschland GmbH, model FP-8500, mode: emission, ex bandwidth 2.5 nm, em bandwidth: 5 nm, response: 1 s, sensitivity: medium, ex wavelength: 397 nm; emission at 510 nm) and determined relative to the supF positive control.
Northern blot analysis
Eschrichia coli TOP10 pre-cultures carrying a riboswitch-bearing plasmid and plasmid pACYC/tetM were diluted to an OD600 of 0.06 in 10 ml LB with 50 μg/ml ampicillin, 10 μg/ml tetracycline, 0.01% (w/v) arabinose and 0 or 2 mM theophylline and grown at 37°C for 3.5 h. Total RNA was isolated by TRIzol extraction (Thermo Fisher Scientific). Ten μg of total RNA or 20 ng of in vitro transcript (control) were mixed with 3x RNA loading dye (140 mM Tris-borate pH 8.3, 8 mM Na2EDTA, 0.2% (w/v) bromophenol blue, 0.2% (w/v) xylene cyanol, 1 g/ml urea) to a 1× concentration and loaded on a denaturing 10% urea-polyacrylamide (PAA) gel. Gel run was performed at 200 V. Northern blot was performed as described (48) with minor deviations. For hybridization, the membrane was incubated for 16–20 h at 42°C in 6× SSC buffer containing 0.1% SDS and 16 nM of radioactively 5′-labeled DNA oligonucleotide complementary to the replaced region in supF-h tRNA hybrid (5′-GGCATACGCCAACGGTTTTAGAGACCGTCC-3′). After detection of the hybrid tRNA by autoradiography, the membrane was stripped with 0.5% SDS at 60°C for 2 h. To measure the amount of 5S rRNA for normalization, re-hybridization was performed using a specific probe for 5S rRNA (5′-TTCTGAATTCGGCATGGGGTCAGGTGG-3′) (49). The amount of processed tRNA was normalized based on 5S RNA signals using ImageQuant TL 8.1 (Cytiva) and determined relative to the positive control supF-h. The 5′-labeling of the probes was performed with T4 polynucleotide kinase according to the manufacturer (New England Biolabs). Unincorporated nucleotides were removed using NucAway™ Spin Columns (Thermo Fisher Scientific).
Expression and purification of C5 protein
Protein expression was performed as previously described for C5 protein (50,51). Overnight grown E. coli JM109 cells were incubated at 37°C in 2 × 250 ml LB medium containing 50 μg/ml ampicillin until an OD600 of 0.6 was reached. Protein expression was induced by IPTG addition to a final concentration of 1 mM followed by 3 h incubation at 25°C. After harvesting the cells, the pellet was stored at −80°C for further use. For purification, the pellet was thawed on ice, and subsequent steps were performed at 4°C. Cell lysis was achieved by sonication (70%, 8 × 15 s, interval, pause 30 sec in between; Sonopuls HD 2070, Bandelin) in buffer A (50 mM Tris–HCl pH 7.9, 10 mM Mg(C2H3O2)2•4H2O, 0.25% (v/v) tween 20, 1 M NH4Cl). The suspension was centrifuged at 30 600 g at 4°C for 30 min. After filtration of the supernatant (pore size 0.44 μm) and pre-equilibration of the HisTrapFF (volume 1 ml) with 5 column volumes (CV) of 90% buffer A and 10% buffer B (10 mM Tris–HCl pH 8.0, 500 mM imidazole) at a flow rate of 1 ml/min, the solution was applied with 0.5 ml/min to the column via ÄKTA pure system (Cytiva). The column was washed with 10 CV 90% buffer A and 10% buffer B followed by 5 CV of buffer A. For elution of the protein, 8 CV of buffer B were used. The protein-containing fractions were pooled and the final protein concentration was determined according to Bradford. Glycerol was added to a final concentration of 40 % and 100 μl aliquots were frozen in liquid nitrogen and stored at −80°C.
In vitro run-off transcription of riboswitch constructs and M1 RNA
DNA templates for T7-based in vitro transcription were generated by PCR using pBAD plasmids carrying the individual riboswitch constructs. The forward primer included the sequence of the T7 promoter. Amplified transcription templates were purified by phenol/chloroform extraction and ethanol precipitation. Riboswitch constructs were transcribed under standard conditions in the presence of NTPs (4 mM each) (52). M1 RNA was transcribed in 40 mM Tris–HCl pH 8.0, 8 mM MgCl2, 25 mM NaCl, 2 mM spermidine, 5 mM DTT. Transcripts were purified by denaturing polyacrylamide gel electrophoresis, gel extraction and ethanol precipitation. For internal labelling, α-[P32]-ATP was added to the transcription reaction.
RNase P holoenzyme kinetic analysis
RNase P holoenzyme was assembled by pre-incubation of C5 protein and M1 RNA in a 2:1 ratio for 5 min at 37°C in the reaction buffer (50 mM HEPES pH 7.4, 100 mM NH4Cl, 10 mM MgCl2) (53,51). Radioactively labeled tRNA substrates were incubated for 1 min at 90°C with or without 30 μM theophylline in the same buffer. After assembling for 5 min at room temperature (RT) in the reaction buffer, RNase P holoenzyme was added to the substrate to final concentrations of 10–150 nM substrate and 2 nM enzyme (2 nM M1 RNA, 4 nM C5 protein). The incubation was performed at 37°C in the reaction buffer for 8 min. Cleavage reaction was stopped by addition of 3x RNA loading dye (140 mM Tris-borate pH 8.3, 8 mM Na2EDTA, 0.2 % (w/v) bromophenol blue, 0.2 % (w/v) xylene cyanol, 1 g/ml urea) to a 1× concentration. Reaction products were separated by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography (Supplementary Figure S3). Signals of cleavage products were quantified using ImageQuant TL 8.1 (Cytiva). Michaelis-Menten parameters were determined using GraphPad PRISM. All in vitro activity tests were performed in at least three independent experiments. Parameters including standard deviation and R2 values are shown in Table 2.
Structure analysis of in vitro transcribed riboswitches
5′-Labeled RNA was denatured at 90°C for 1 min and cooled down to room temperature for at least 5 min for refolding. Probing was performed as described (54,55) in the absence or presence of 30 μM theophylline for 40 h at room temperature in a 20 μl volume. Reactions were stopped using 20 μl of 2× colorless RNA loading dye (10 M urea, 1.5 mM EDTA), and 10 μl were loaded on a 10% PAA gel.
Quantification and statistical analysis
Statistical parameters and calculations are reported in the Figures and Figures Legends.
RESULTS
RNase P riboswitch candidates derived from synthetic transcription-regulating riboswitches are not functional
For efficient and correct substrate recognition by RNase P in bacteria, the sequence of the tRNA 5′-leader is one important feature (56–59). However, the influence of its secondary structure is rarely analyzed. Screening of different 5′-leader sequences in vitro led to the hypothesis of a decreased binding affinity of highly structured 5′-leaders (35). Based on this hypothesis, we developed a riboswitch to control tRNA 5′-maturation (Figure 1A).

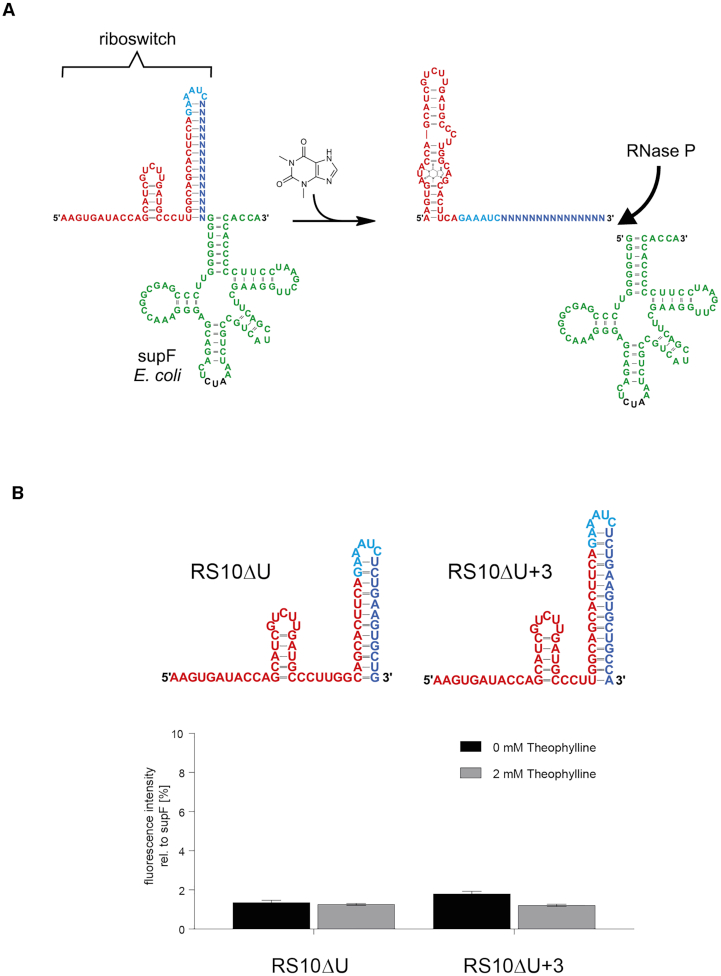
(A) Design principle of a riboswitch controlling tRNA 5′-end maturation. In the riboswitch, the upstream leader region consists of the theophylline aptamer (red) followed by a designed sequence (blue) that forms a hairpin structure with the 3′-part of the aptamer. The loop-forming region (cyan) is taken from a designed transcription-regulating and theophylline-dependent riboswitch (39). This hairpin sequesters the single-stranded leader region upstream of the E. coli suppressor tyrosine suppressor tRNA supF (green). As a result, RNase P cannot access the cleavage site of the tRNA precursor. Upon interaction with theophylline, the aptamer folds into its binding-competent state, leading to a disruption of the sequestor hairpin. The 5′-leader becomes single-stranded and is now accessible for RNase P cleavage. In the 3D structure, theophylline interacts with both loop regions (106). For reasons of clarity, the ligand is positioned in the lower loop of the secondary structure, symbolizing the bound state of the aptamer. (B) RNase P-riboswitch constructs in unbound state based on the terminator hairpin of a transcription-regulating riboswitch RS10 (39). In the left structure RS10ΔU, the sequestor hairpin is taken from the original terminator structure of RS10. In the right structure RS10ΔU+3, the sequestor hairpin was extended by three base pairs. Structures were predicted using RNAfold (38). Bar diagram: Fluorescence intensities are displayed as relative values to the positive control supF. Release of mature suppressor tRNA is monitored by read-through efficiency of a corresponding amber stop codon at position 39 in the mRNA of a plasmid-encoded c3GFP-based reporter construct. The obtained values represent only background fluorescence, indicating that neither of these constructs showed a theophylline-dependent switching behavior. Results show the mean ±SD of three independent experiments.
In a first attempt, RS10ΔU was constructed by keeping the sequence of one of our best performing theophylline-dependent transcription-regulating riboswitches (19,39) including its terminator hairpin but removing the downstream U-stretch (Figure 1B). A tRNA sequence follows immediately downstream of RS10ΔU (Supplementary Table S1). The deletion of the U-stretch inactivates the terminator function and transforms the remaining terminator hairpin into a sequestor hairpin because its 3′-terminal part represents the 5′-leader region of the tRNA precursor. This construct is expected to be processed by RNase P only if the 5′-tRNA leader is single-stranded (Figure 1A, B). It was shown for Bacillus subtilis that increasing the length of a single-stranded 5′-leader results in a high cleavage efficiency (34). Accordingly, RNase P processing should be enhanced if at least nucleotide positions -1 to -4, located immediately upstream of the tRNA, are single stranded. The 5′ part of the sequestor hairpin is part of the TCT8–4 theophylline aptamer sequence (8) that represents the sensor and is therefore immutable, as changes in this region likely affect ligand binding. To increase flexibility for subsequent designs, we further extended RS10ΔU by three bases complementary to the aptamer region, resulting in RS10ΔU+3 (Figure 1B).
The theophylline-dependent release of a mature tRNA was monitored using a reporter system based on stop codon suppression. The tRNA under riboswitch control was suppressor tRNATyr supF, bearing an anticodon 5′-CUA-3′ that recognizes the amber stop codon UAG (60). Plasmid pBAD, carrying the individual riboswitch constructs under an inducible arabinose promoter (araP) and plasmid pACYC containing an araP controlled c3gfp reporter with an amber STOP codon at position 39 were introduced into E. coli TOP10. After aminoacylation by tyrosyl-tRNA synthetase, the correctly processed supF tRNA should decode the stop codon in the c3gfp mRNA, leading to the expression of the encoded c3GFP. The activity of the protein was quantified through determination of the fluorescence intensity (excitation: 397 nm; emission: 510 nm). As positive control, a pBAD plasmid version was used where the complete riboswitch construct was replaced by an unstructured 9 nt long sequence closely related to a frequently used supF precursor construct (61,62). After transcription, the corresponding tRNA precursor is readily processed by RNase P, resulting in efficient expression of c3GFP.
TOP10 cells carrying the plasmid constructs RS10ΔU or RS10ΔU+3 were incubated in the absence or presence of theophylline (2 mM). With values of 1 to 2 % compared to the positive control, the resulting fluorescence intensity of both constructs was very low and indicated that these constructs were not functional as RNase P sensitive switches in in vivo experiments (Figure 1B).
Riboswitches that regulate transcription are kinetically controlled, i.e. the terminator hairpin formation rate needs to be sufficiently fast and within a rather short time frame. In contrast, RNase P regulation is not limited to such a timing, since RNase P is only able to recognize its product after the full transcription of the tRNA, including its 3′-CCA end displaying an important substrate recognition feature (63). Assuming the kinetically controlled sequestor hairpins of RS10ΔU and RS10ΔU+3 are too stable and therefore not functional under (nearly) equilibrated conditions, these elements must be modified in order to become switchable. Ligand binding stabilizes the aptamer part and should thereby disrupt the sequestor hairpin, leaving the leader region single-stranded and freely accessible for RNase P binding. Accordingly, a functional RNase P-sensitive riboswitch should be sufficiently modelled by thermodynamic folding predictions incorporating ligand binding.
RNase P riboswitches based on thermodynamic folding predictions exhibit an efficient regulation of tRNA maturation in E. coli
To implement this model, we conducted a computational design approach, developed the leader accessibility score, varied the length of the sequestor hairpin and randomized its 3′-part. The developed optimization score ensures that the probability of the leader region being single stranded is high in presence of theophylline and low in its absence, but is only comparable for designed sequences of the same length, as the number of possible base pairs that the randomized sub-sequence can form varies with every nucleotide. As ligand interaction contributes to the free energy of the aptamer structure, theophylline binding was included in our model with experimentally derived parameters for the binding energy and the binding motif (8,44). Optimizing the sequence of the 3′-part of the sequestor hairpin according to the leader accessibility score might allow for unintended base pairs between the riboswitch and the downstream tRNA sequence. In our model, only the riboswitch should undergo conformational changes, but the downstream tRNA element should be unaffected. We applied two secondary design goals to compare, sort and filter candidates with optimal leader accessibility scores of different lengths. The tRNA independent fold score estimates the number of secondary structures in the thermodynamic ensemble that form undesired base pairs between the riboswitch and the tRNA sub-sequences, see Materials and Methods. If this fraction is high, i.e., the independent fold score becomes zero, an unintended misfolding is assumed. The tRNA closing stem is known to be essential for efficient RNase P processing (45–47). Therefore, the tRNA score estimates the fraction of structures containing the tRNA closing stem in the thermodynamic ensemble as well as in the ligand-bound ensemble. If this fraction is high, i.e. the tRNA score is close to one, proper tRNA folding independent of the presence of the ligand is assumed, see Materials and Methods, Table 1. Based on these parameters, RS10ΔU and RS10ΔU+3 are both predicted as non-functional.

| Construct | In vivo | In vitro | In silico | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Switch Length | c3GFP Assay | Northern blot | Kinetics | In-line probing | Leader access. score | tRNA score | Indep. fold score | Relative leader access. score |
| RS10 based switches | |||||||||
| RS10ΔU | 60 | − | ND | ND | ND | 0.025 | 0.996 | 0.832 | 0.08 |
| RS10ΔU+3 | 63 | − | ND | ND | ND | 0.000 | 1.000 | 0.944 | 0.00 |
| Leader accessibility score optimized switches | |||||||||
| RP-RS A | 63 | +++ | ND | + | + | 0.484 | 0.992 | 0.932 | 0.95 |
| RP-RS B (RP-RS A-GC) | 63 | − | ND | ND | − | 0.477 | 0.991 | 0.935 | 0.93 |
| RP-RS C | 59 | ++ | ND | ND | ++ | 0.016 | 0.997 | 0.829 | 0.64 |
| Loop-closing base pair mutants | |||||||||
| RP-RS A-AU | 63 | + | ND | ND | + | 0.469 | 0.991 | 0.932 | 0.92 |
| RP-RS A-GA | 63 | +++ | ND | ND | +++ | 0.512 | 0.996 | 0.931 | 1.00 |
| RP-RS A-UG | 63 | + | ND | ND | + | 0.468 | 0.999 | 0.933 | 0.91 |
| RP-RS C-GC | 59 | − | ND | ND | − | 0.013 | 0.997 | 0.836 | 0.52 |
| RP-RS C-AU | 59 | + | ND | ND | + | 0.016 | 0.997 | 0.829 | 0.65 |
| RP-RS C-GA | 59 | +++ | ND | ND | +++ | 0.023 | 0.997 | 0.825 | 1.00 |
| RP-RS C-UG | 59 | + | ND | ND | ++ | 0.015 | 0.997 | 0.830 | 0.59 |
| Hybrid tRNA variants | |||||||||
| RP-RS A-h | 63 | +++ | ++ | ND | ND | 0.480 | 0.995 | 0.935 | 0.94 |
| RP-RS B-h | 63 | + | + | ND | ND | 0.476 | 0.988 | 0.938 | 0.93 |
| RP-RS C-h | 59 | +++ | ++ | ND | ND | 0.016 | 0.997 | 0.865 | 0.64 |
To investigate the functional capability of our computationally predicted riboswitches, we selected three candidate constructs for in vivo analysis (Figure 2A and Supplementary Table S1). RP-RS A and RP-RS B were the high scoring candidates with respect to the leader accessibility score and can be seen as optimized versions of the non-functional RS10ΔU+3, as the sequestor hairpins are of the same length. RP-RS C has a comparably short sequestor hairpin but is high scoring for its length, and both tRNA related scores indicate proper folding. The latter is not the case for designs with shorter sequestor hairpins, see online supplementary table (http://www.bioinf.uni-leipzig.de/supplements/20-001).
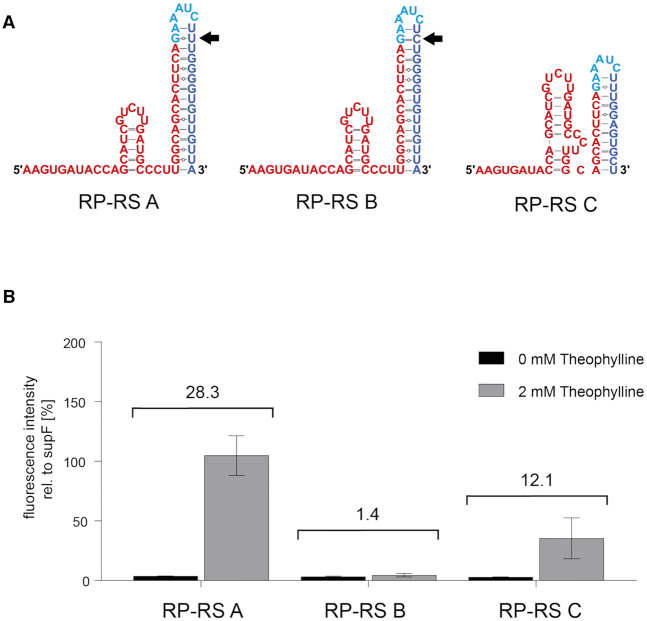
RNase P riboswitch activity in E. coli TOP10 cells. (A) Secondary structures of riboswitches with in silico predicted expression platforms (blue). In the unbound state (without ligand interaction), the designed candidates RP-RS A, RP-RS B and RP-RS C carry different sequestor hairpins consisting of 15 (RP-RS A, RP-RS B) or 11 base pairs (RP-RS C). The sequestor hairpins of RP-RS A and B differ only in a single base pair (indicated by the black arrow). (B) In vivo activity tests of RP-RS A, RP-RS B and RP-RS C in absence (black bars) and presence (grey bars) of 2 mM theophylline. Fluorescence intensities are displayed as relative values to the positive control supF. While RP-RS B is in a permanent OFF state, RP-RS A leads to a mean activity of 104%, comparable to the positive control. RP-RS C also exhibits a ligand-specific response in tRNA maturation. However, this activation is less pronounced compared to RP-RS A and leads to a relative mean fluorescence intensity of 35% in the read-out system. Results show the mean ±SD of three independent experiments.
RP-RS A, B and C showed considerable differences in theophylline-dependent activation of the c3GFP reporter gene relative to the positive control supF (Figure 2B). Cells expressing supF tRNA under control of RP-RS A show the highest levels of fluorescence, comparable to the positive control (104.8 ± 16.5%). Background expression in the absence of theophylline was very low (3.7 ± 0.2%), resulting in a highly significant 28-fold response ratio. RP-RS C was also responding to the presence of the ligand, leading to a 12-fold induction (2.9 ± 0.1% uninduced versus 35.4 ± 17.2% induced). RP-RS B, however, showed only a low expression level with no clear increase (3.2 ± 0.5 % uninduced versus 4.6 ± 1.5% induced), although it was predicted with a similar leader accessibility as RP-RS A (Table 1). The fact that RP-RS C was active despite a 10-fold lower leader accessibility score is not surprising, as a direct comparison of this score is only possible for constructs of the same length. The relative leader accessibility score is again not able to explain the observed difference of RP-RS A and RP-RS B, but clearly indicates that RP-RS C might implement a functional construct.
While these results clearly indicate an efficient and accurate ligand-dependent tRNA maturation for RP-RS A and RP-RS C, the actual data are a combination of tRNA maturation, aminoacylation and subsequent translation of the c3GFP reporter. Hence, a direct analysis of the maturation of the suppressor tRNA was performed based on a northern blot approach. For visualization of supF tRNA, an oligonucleotide probe hybridizing to the tRNA was required. As the supF sequence is identical with the endogenous tRNATyr (except the suppressor anticodon), such a probe would recognize both suppressor as well as endogenous tRNA for tyrosine, leading to considerable background signals. Therefore, riboswitches RP-RS A-h, B-h and C-h were constructed where the original supF tRNA was replaced by a hybrid tRNA supF-h that is sufficiently different to be selectively recognized by a hybridization probe. To this end, anticodon stem and most of the variable loop region of supF were replaced by corresponding parts of Thermus thermophilus tRNATyr (Figure 3A). The resulting tRNA chimera contained all identity elements for proper aminoacylation by tyrosyl-tRNA synthetase, including three base pairs in the variable loop (64–66). To investigate whether RP-RS A-h, RP-RS B-h and RP-RS C-h riboswitches are active and produce a functional hybrid tRNA in E. coli, c3GFP fluorescence levels were determined (Figure 3B). Similar to the original riboswitch constructs, RP-RS A-h and RP-RS C-h showed a ligand-dependent response (8-fold and 11-fold, respectively), while RP-RS B-h showed a low (4-fold) activation (4.3 ± 0.9% to 15.6 ± 12.7%). Based on its high standard deviation, this riboswitch shows only a minimal switching activity. Compared to the original constructs RP-RS A to C, the background signal of all constructs in the absence of theophylline was increased. In the presence of theophylline, RP-RS A-h showed a fluorescence intensity (125.4 ± 37.0%) comparable to RP-RS A (104.8 ± 16.5%; Figure 2B). Yet, due to the higher background, the determined activation rate was lower (8-fold). RP-RS C-h showed a fluorescence intensity (94.9 ± 16.0%) higher than RP-RS C (35.4 ± 17.2%), comparable to the positive control supF, RP-RS A and RP-RS A-h, even though the leader accessibility score remained unchanged (Table 1). Since the background in the OFF state was also increased, a switching ratio of 11-fold remained. Taken together, the hybrid constructs RP-RS A-h and RP-RS C-h exhibit a switching behavior that is similar to RP-RS A and C, while RP-RS B-h is also almost inactive. These data indicate that the introduction of the hybrid tRNA did not interfere the riboswitch functionality.
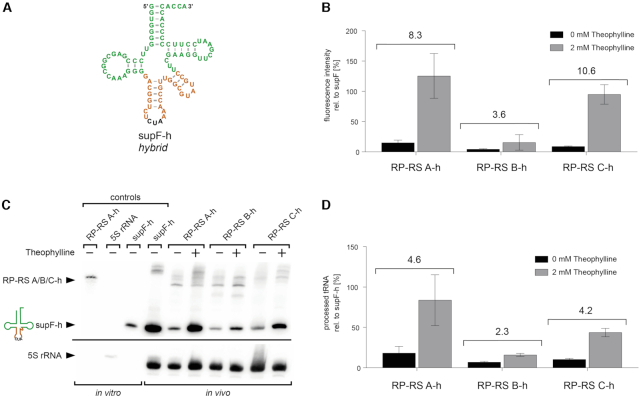
Riboswitch-controlled tRNA maturation and amber codon suppression. (A) For specific detection of processed tRNA in a northern blot analysis, supF tRNA was replaced by a hybrid construct supF-h, where anticodon stem and variable arm represent the corresponding region of Thermus thermophilus tRNATyr. (B) As in the original constructs, RP-RS A-h and RP-RS C-h show a ligand-dependent response and release a functional supF-h tRNA that leads to expression of c3GFP. RP-RS B-h shows only a minimal activation. Northern blot (C) and its quantification (D) show that RP-RS A-h and RP-RS C-h turn on tRNA maturation in the presence of theophylline by factor 5, while RP-RS B-h only leads to a 2-fold activation. The additional band above the precursor (only clearly visible for the positive control supF-h and RP-RS A-h in presence of theophylline) likely represents a complex of processed, uncharged tRNA supF and protein RelA. Controls: run-off transcripts of RP-RS B-h, 5S rRNA and mature supF-h, serving as size markers. Hybridization signals of 5S rRNA were used as loading control. The amount of cleaved tRNA was normalized based on the 5S rRNA signal and determined relative to the positive control supF-h. Quantification results show the mean ±SD of three independent experiments. The response ratios of individual constructs are indicated in the bar graphs (B, D).
In the northern blot analysis, a specific signal corresponding to the mature supF-h tRNA is visible in the lanes representing RP-RS A-h and RP-RS C-h constructs incubated with theophylline (Figure 3C). An additional slowly migrating band appears in the lanes for supF-h and the induced RP-RS constructs. Proteinase K treatment prior gel loading eliminated this signal (Supplementary Figure S2), identifying it as an RNA–protein complex. According to Kushwaha et al., this band very likely corresponds to a complex formed by interaction of the processed tRNA with the stringent response factor RelA that has a very high affinity to uncharged tRNAs (67).
The amount of mature tRNA was quantified relative to the corresponding 5S rRNA signals (Figure 3D). For comparison, the tRNA/5S rRNA ratio of the positive control supF-h was set to 100%. In the ON state, the processed tRNA amount of RP-RS A-h (83.8 ± 31.5% induced versus 18.2 ± 8.2% uninduced) is comparable to the positive control, whereas tRNA maturation in RP-RS C-h was less efficient (43.6 ± 5.2% induced versus 10.3 ± 1.5% uninduced), resulting in a 5- and 4-fold activation, respectively. With values of 15.9 ± 1.8% (induced) and 6.9 ± 1.1% (uninduced), RP-RS B-h lead to a 2-fold activation with a low amount of mature tRNA in presence of theophylline (Figure 3D).
A comparison of tRNA and protein expression levels of RP-RS A-h to RP-RS C-h shows a qualitative rather than a quantitative concordance in their activities. Signal amplification by a repeated participation of the expressed tRNA in translation is likely, as the amount of tRNA influences the rate of protein synthesis (68–70). However, a clear quantitative correlation of tRNA levels and amount of c3GFP protein is not expected since various factors like mRNA structure, cell stress, tertiary complex formation, aminoacylation and others have an impact on the translation efficiency in vivo (71–76). In summary, the data indicate that RP-RS A/A-h and RP-RS C/C-h are theophylline-sensitive riboswitch constructs and exercise a direct regulation of RNase P-catalyzed tRNA maturation, although a linear correlation of tRNA amount and fluorescence intensity is not observed.
Riboswitch RP-RS A regulates tRNA cleavage efficiency in vitro
To verify that the riboswitches indeed regulate RNase P activity by controlling the accessibility of the 5′-leader cleavage site, the processing reaction catalyzed by the E. coli RNase P holoenzyme (catalytic M1 RNA moiety combined with the C5 protein subunit) was investigated in vitro. As demonstrated by the Altman lab, the in vitro transcript of M1 RNA is an efficient and accurate ribozyme that exhibits high catalytic activity on tRNA precursor transcripts (28,77). However, the protein subunit is involved in the accurate substrate recognition and the catalytic efficiency, especially through its direct interactions with the 5′-leader of the precursor (34,36,37,78).
As a positive control, a naturally occurring well characterized precursor tRNA was chosen. Construct nat-supF is composed of supF tRNA fused to the native 43 nt leader from E. coli tRNATyr (79–81). Radioactively labelled run-off transcripts of RP-RS A and nat-supF were used to investigate the kinetic parameters of RNase P cleavage in vitro (Figure 4). Since cellular uptake of theophylline is limited, the endogenous ligand concentration is probably much lower than in the medium (2 mM). Accordingly, a reduced theophylline concentration of 30 μM was chosen for the in vitro analysis, corresponding to a 100-fold excess of the aptamer's kD value of 320 nM (8). The reaction was performed with substrate concentrations ranging between 10 and 150 nM of RP-RS A and nat-supF using buffer conditions that are compatible with both enzyme activity and ligand binding by the aptamer. Thus, the hairpin structure is probably less stably folded as calculated under these conditions. Reaction products were separated on denaturing polyacrylamide gels, visualized by autoradiography (Supplementary Figure S3). Signal intensities were quantified and used to determine kinetic parameters kcat and KM (Figure 4, Table 2).
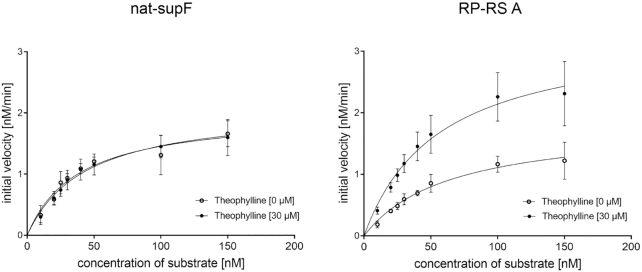
Kinetic analysis of RNase P holoenzyme activity. In vitro cleavage of RP-RS A (right) was compared to supF with the naturally occurring 5′-leader (nat-supF; left) (80). Substrate concentrations from 10 to 150 nM were incubated with the holoenzyme (final concentration 4 nM C5 protein, 2 nM M1 RNA) in presence or absence of 30 μM theophylline in at least three independent experiments. The processing of nat-supF is not affected by the presence of theophylline. In contrast, RP-RS A shows a ligand-dependent increase in tRNA maturation, indicating that the binding of theophylline leads to a single-stranded leader region in the transcript that is accessible for RNase P. Michaelis-Menten parameters of this reaction are summarized in Table 2.
| Construct | Theophylline (30 μM) | K M (nM) | k cat (min−1) | R 2 |
|---|---|---|---|---|
| nat-supF | − | 38.39 ± 7.64 | 1.01 ± 0.08 | 0.85 |
| + | 43.45 ± 7.21 | 1.05 ± 0.08 | 0.87 | |
| RP-RS A | − | 63.16 ± 12.55 | 0.90 ± 0.08 | 0.89 |
| + | 56.42 ± 9.71 | 1.68 ± 0.14 | 0.85 |
For nat-supF, higher KM (38.39 ± 7.64 nM) and lower kcat (1.01 ± 0.08 min−1) values were observed in absence of theophylline compared to prior analysis (79,80) and likely result from slightly different buffer conditions. The addition of theophylline did not affect the cleavage of the positive control (KM: 38.39 ± 7.64 nM and kcat: 1.01 ± 0.08 min−1 versus KM: 43.45 ± 7.21 nM and kcat: 1.05 ± 0.08 min−1). Compared to nat-supF, KM values of RP-RS A in the absence (63.16 ± 12.55 nM) as well as in the presence of the ligand (56.42 ± 9.71 nM) are elevated to some extent. However, for RP-RS A, kcat was clearly increased from 0.90 ± 0.08 min−1 to 1.68 ± 0.14 min−1, indicating an increased cleavage efficiency of RNase P. Taken together, the riboswitch construct controls in vitro an RNase P-mediated precursor cleavage in a ligand-dependent way, corroborating the in vivo data on theophylline-induced tRNA processing in our riboswitch constructs.
A loop-closing base pair modulates sequestor hairpin stability and riboswitch functionality
While RP-RS A shows a 28-fold ligand response ratio, RP-RS B is nearly inactive (response ratio of 1.4). Yet, both constructs are almost identical in their sequence and differ only in a single base pair 43–50 in the sequestor stem, where RP-RS A carries a G–U pair, while in RP-RS B, a G–C interaction is present (Figures 2A and 5). As this base pair is located close to the loop at the tip of the sequestor hairpin, it probably contributes not only to the overall structural stability, but also has an impact on the closing of the hairpin loop (82). Thus, the influence of this base pair on hairpin stability and riboswitch functionality was investigated. The original G–U pair of RP-RS A was replaced by A–U (RP-RS A-AU), representing a Watson–Crick base pair less stable than the G–C interaction of RP-RS B. In addition, a G–A mismatch (RP-RS A-GA) as well as a U-G (RP-RS A-UG; inverse to the original G–U) were introduced (Figure 5). The resulting RP-RS A variants were recombinantly expressed in E. coli TOP10 and theophylline-induced fluorescence intensity was quantified (Figure 5B). RP-RS A-GA showed a high ligand-dependent response, similar to the original RP-RS A construct carrying the G43-U50 pair. RP-RS A-AU and RP-RS A-UG also responded to theophylline and induced tRNA processing and c3GFP expression. The induction, however, was somewhat weaker in the case of RP-RS A-UG and clearly weaker in the case of RP-RS A-AU than that of the original RP-RS A.
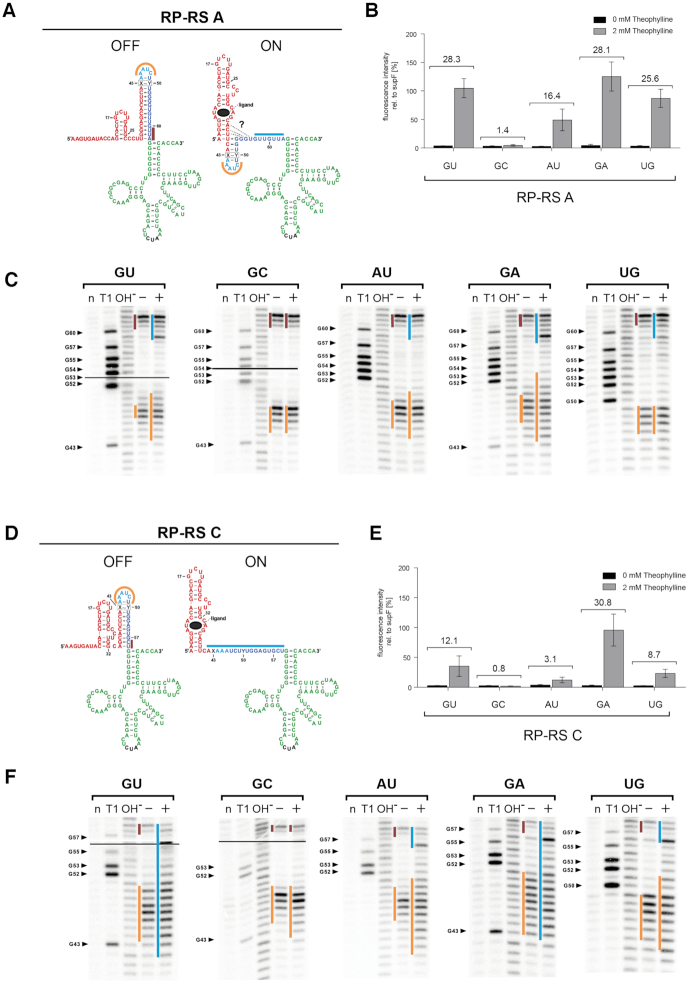
Functional and structural characterization of RP-RS A and C variants. (A) and (D) Constructed secondary structures (fusion of the predicted expression platform with the supF tRNA structure) of ON and OFF states of RP-RS A and RP-RS C. The theophylline binding aptamer is represented in red, the designed 3′-part of the sequestor hairpin is in blue, the suppressor tRNA in green with the anticodon in black. Bars in orange, brown and blue represent sequences that were mapped to be single-stranded in the corresponding structures. The base pair 43–50 in the sequestor stem is indicated as X-Y in a box. In the ON state, additional possible interactions of the blue sequestor part with the aptamer base are indicated by dashed lines and a question mark. (B) and (E) In vivo activity tests of RP-RS A and RP-RS C variants carrying different X43-Y50 base pairs. In the presence of 2 mM theophylline (grey bars), the GC variants are in a permanent OFF state, showing no tRNA maturation and, consequently, no GFP expression. All other constructs show a theophylline-induced activation of the reporter gene. Candidates with rather weak X43-Y50 interactions (G–A) exhibit a higher response ratio than those with more stable base pairs (G–U, A–U, U–G). (C) and (F) In-line probing patterns of RP-RS A and RP-RS C variants, respectively. Only the designed part of the sequestor stem is presented, in-line probing of the full transcripts is shown in the Supplementary Data (Supplementary Figure S4). Black lines in the two panels on the left indicate the border of two exposure screens. All constructs except the G43-C50 variant show a ligand-dependent opening of the sequestor. In the uninduced state (absence of theophylline indicated by ‘–‘), all constructs show fraying of the basal three base pairs of the hairpin. Yet, tRNA maturation is not possible, as this region is not sufficient to give RNase P access to the cleavage site. In the GC mutants, the structure does not change upon the addition of theophylline. All other candidates show a structural rearrangement that corresponds to a certain extent to their response ratio shown in (B) and (E). Lanes: (n) no reaction; (T1) incubation with RNase T1; (OH−) hydrolysis ladder; (−/+) incubation in in-line probing buffer for 40 h in absence (−) or presence (+) of theophylline. Response ratios of the tested riboswitch variants are indicated in the bar graphs (B, E).
To determine whether the different activities of the variants are linked to differences in theophylline-induced refolding, in vitro transcripts of individual riboswitch constructs were subjected to structural investigation by in-line probing (55) in the presence and absence of the ligand. The probing data show a high correlation of in vitro induced refolding and in vivo functionality (Figure 5B and C). From the resulting in-line cleavage pattern, the formation of the theophylline-dependent aptamer structure can be inferred in all constructs and corresponds to the reported shape (Supplementary Figure S4) (83). In the absence of theophylline, the formation of the predicted sequestor hairpin immediately upstream of supF tRNA can also be deduced in all constructs (Figure 5C and F). In RP-RS A-GU and RP-RS A-GA, the presence of the ligand induced an opening of the sequestor hairpin, so that the proximal region of the leader sequence was single-stranded and accessible for RNase P, correlating with efficient processing (Figure 5C). In contrast, RP-RS A-AU and RP-RS A-UG showed a less pronounced refolding into a single-stranded proximal leader region, which is in agreement with the prior results based on fluorescence measurements. Only 4–5 unpaired residues were detected, while the rest of the sequestor hairpin remained double-stranded and obviously interfered with RNase P accessibility to a certain extent. Accordingly, the in vivo response to theophylline was reduced. Yet, and in contrast to RP-RS A-GC (corresponding to the original RP-RS B), these constructs still represent functional and theophylline-responsive riboswitches. The calculated leader accessibility scores for the investigated RP-RS A mutants ranged between 0.468 and 0.512, corresponding to high scoring sequences of this length (see Table 1). The G–A mutant with a value of 0.512 showed the highest accessibility in vitro and in vivo. All RP-RS A constructs were predicted to switch. However, the calculated scores allow only a general prediction based on the overall stability of the designed constructs but cannot determine the effect of a single base pair in these structures.
Interestingly, our second active construct RP-RS C also carries a G–U base pair at position 43–50 in the loop closing region of the sequestor hairpin (Figure 2A). To investigate whether the nature of this base pair is equally important for RP-RS C functionality and represents a general feature of our constructs, we introduced the same base pair variants of RP-RS A in RP-RS C (Figure 5D). Similar to its RP-RS A-GC counterpart, RP-RS C-GC is completely inactive and does not respond to theophylline, showing only background fluorescence intensity (Figure 5E). RP-RS C-GA is comparable with the positive control supF, more active than the G–U counterpart and represents full functionality, as it was also shown for the respective RP-RS A variant. RP-RS C-AU and RP-RS C-UG show a reduced but well detectable theophylline response that also corresponds to that of RP-RS A-AU and RP-RS A-UG, although at a lower efficiency (Figure 5B and E). Furthermore, in-line probings of these RP-RS C versions exhibit a certain correlation with their response ratios, which is also in accordance with the RP-RS A results (Figure 5F). In the presence of theophylline, RP-RS C versions with GU and GA positions at base pair 43–50 show a complete unfolding of the sequestor hairpin, while unfolding in RP-RS C-AU is only moderate. RP-RS C-UG, however, shows an unexpectedly high unfolding compared to its measured in vivo response. In RP-RS C-GC, the sequestor hairpin shows only a slight unfolding in the loop region, while the rest of the structure remains double-stranded, so that RNase P has no access to the cleavage site, rendering this variant completely inactive. For the RP-RS C variants, the leader accessibility score and especially its normalized version predict a theophylline-dependent switching that correlates with the experimental data to a certain degree. RP-RS C-GA is the best scoring sequence with a high switching activity, while RP-RS C-GC is predicted to be the worst (Table 1). However, as indicated above, prediction of the impact of an individual base pair in the sequestor stem is not possible, as the values for RP-RS C-GC (inactive) and RP-RS C-UG (moderately active) are very similar.
Taken together, the results show that for both RP-RS A and RP-RS C, ligand-dependent structural rearrangement is in line with the in vivo regulation of RNase P-mediated tRNA release. In this context, the interaction of base positions 43 and 50 play a crucial role for the responsiveness towards theophylline. A noticeable difference between RP-RS A and RP-RS C, however, is the fact that in the construct with higher response ratio (RP-RS A), the base of the sequestor hairpin consists of three rather weak base pairs (GU and AU) and is under all conditions fraying into single-strands of at least three nt length. In RP-RS C, this region is more stable, as it carries two GC and one AU base pair. Correspondingly, fraying is restricted to the terminal AU and the neighboring GC pair.
DISCUSSION
The theophylline aptamer TCT8–4 is one of the best characterized ligand-binding RNA structures (8). With its high selectivity and in vivo compatibility, it is frequently used to construct ligand-responding regulatory RNA devices (39,84–89). Here, we designed theophylline-dependent riboswitches regulating the processing of a specific precursor tRNA by directly controlling the accessibility for RNase P. The leader region is masked by the sequestor hairpin, and ligand binding induces a structural rearrangement that disrupts this hairpin, giving RNase P access to its cleavage position. Further, this design allows investigation of the impact of the tRNA 5′-leader structure on RNase P mediated processing (Supplementary Figures S4 and S5).
Modelling the accessibility of the 5′-leader of the tRNA sequence in absence and presence of ligand by developing the leader accessibility score successfully guided the selection process toward functional riboswitches. The inactive constructs RS10ΔU and RS10ΔU+3 based on the transcriptional riboswitch RS10 (39) showed very low scores of 0.025 and 0.000, respectively, indicating that a ligand-dependent switching activity is highly unlikely (Table 1). As the sequestor hairpin was originally designed as a robust and fast forming terminator hairpin, its high stability does not allow for a ligand-induced refolding once it is formed. Hence, the leader accessibility scores indicate that the two alternative structures of these constructs are not formed in a ligand-dependent thermodynamic equilibrium.
While sampling sequences compatible with the applied constraints (Supplementary Figure S1), only Watson–Crick and wobble G–U base pairs were included to generate perfectly stacking sequestor hairpins. In principle, it would be possible to allow for other base pairs or imperfect hairpins containing bulges and interior loops. However, the combinatorial complexity of the sampling process would increase and the accuracy of the applied energy model used for secondary structure prediction is currently limited to canonical base pairs only. For a better data interpretation of the riboswitch variants with different length, we calculated the relative leader accessibility score with respect to all compatible sequences observed during sampling and of sequences containing G–A as well as U–G mismatches at position 43–50 of the riboswitches (Table 1). For RP-RS C and its variants, the relative scores show an approximate correlation with a ligand-induced switching behavior. In the group of sequences with a length of 59 nts, particularly RP-RS C-GA displayed the best score and also the best response ratio based on c3GFP expression. RP-RS C-GC, in contrast, has the lowest relative score and shows no ligand-dependent switching. Comparing the scores of RP-RS C-UG and RP-RS C-GA with the in-line probing data, a clear quantitative correlation between the experimental activities and the structural rearrangement was not observed, as both riboswitches show a ligand-induced opening of the sequestor hairpin (Figure 5F). Their relative scores, however, clearly predict a better switching behavior for RP-RS C-GA (Table 1).
When focusing on RP-RS A and RP-RS B, constructs of identical length, the calculated scores do not show a strong correlation with functionality. The example of RP-RS B, which differs by a single G–C base pair at position 43–50 from the functional RP-RS A sequence, clearly indicates limitations of the applied in silico approach. According to all applied scores, both constructs should work equally well. In-line probing experiments as well as activity tests of RP-RS B show that the determined secondary structure does not match the predicted accessible leader sequence in presence of theophylline. Similar observations can be made for the shorter RP-RS C construct and its derivatives. While these values are based on thermodynamic calculations, structural investigations revealed additional features affecting riboswitch functionality that are not predicted by the accessibility scores. Base pair 43–50, located close to the top of the sequestor hairpin, has a strong impact on the switching efficiency, which could not be predicted by our in silico calculations. At this position, rather weak interactions (G–U, A–U, G–A, U–G) are functional, while stable G–C pairings completely abolish the ligand-dependent rearrangement of both RP-RS A as well as RP-RS C constructs (Figure 5). As hairpin formation proceeds in a zipper-like process starting at the loop region as a seed (90,91), it seems that these closing base pairs must be rather unstable to allow a ligand-dependent rearrangement. Hence, the thermodynamic stability of the sequestor hairpin seed region directly affects riboswitch functionality. In addition, structure probing of functional RP-RS A variants shows that a small hairpin containing the 43–50 base pair remains even in the ligand bound ON state (Figure 5). This observation indicates that predominantly the basis of the sequestor hairpin is required to be single-stranded in order to give access to the RNase P cleavage site, while the remaining short upper part of the hairpin represents no steric obstacle for tRNA processing. These data indicate that the current in silico model is not sensitive enough to capture all observed effects. While the applied scores are good indicators on the overall stability of a designed construct, they are not accurate enough to unambiguously quantify the effect of a single base pair exchange. This is presumably an inherent limitation of any model based on RNA secondary structures, which by definition cannot capture details of the 3D structure. Furthermore, RNase P-mediated cleavage efficiency also depends on the actual sequence and base composition of the leader region (35–37,92). However, the accessibility score does not include this parameter. Therefore, a linear correlation of the accessibility score and riboswitch activity with the current in silico model is not expected.
Compared to RP-RS A (28-fold), RP-RS C shows a less efficient response ratio (12-fold). One explanation is given by the base pair composition in the basal part of the sequestor hairpins. RP-RS A carries two G–U and one U–A base pair, while in RP-RS C, a C–G followed by G–C and A–U is found, leading to a higher helical stability as in RP-RS A. Consistent with this hypothesis is the fact that structure probing of all RP-RS A variants shows an efficient fraying of the three bottom base pairs of the hairpin even in the absence of theophylline (Figure 5C). Yet, the single-stranded state of these bases is not sufficient to give access to RNase P, and no mature tRNA is released. In contrast, RP-RS C carries the more stable closing interactions, and all variants show only weak fraying of the terminal two base pairs (Figure 5F). Obviously, the increased stability of the hairpin base has a considerable impact on the ligand-induced rearrangement of the riboswitch, leading to less efficient response of RP-RS C constructs.
Despite this hairpin fraying in the absence of theophylline, the low background of RP-RS A and RP-RS C can be explained by the observation that Bacillus subtilis RNase P requires at least four single-stranded proximal leader nucleotides for efficient catalysis with the tRNA precursor (34), and it is very likely that the E. coli enzyme has similar substrate requirements. The sequence composition of the 5′-leader also has an impact on the cleavage efficiency by RNase P. A crucial position for the E. coli enzyme is N(-1), immediately upstream of the tRNA sequence, where a uridine is the preferred base (56). Yet, RP-RS A carries a less preferred A-1 residue and still is cleaved efficiently. Besides this N(-1) position, additional substrate recognition features for RNase P in a pre-tRNA are known, including the 2′-hydroxyl in the acceptor stem, the D-stem loop and the 3′-CCA motif (93,94), so that the recognition is not restricted to N(–1). Furthermore, Niland et al. showed that N(–1) recognition is only important for cleavage by the isolated catalytic M1 RNA subunit, but not for the holoenzyme consisting of M1 and the protein unit C5 (36). In our constructs, these additional recognition elements for RNase P are present, as the structural analysis clearly shows a correctly folded tRNA in presence and absence of the ligand. Hence, the only determinant whether processing takes place or not is the accessibility of the 5′-leader that is regulated by the riboswitch.
While RP-RS A and RP-RS C led to a very low processing in the absence of theophylline, the hybrid constructs show a moderate background. It is possible that the tRNA hybrid sequence interferes and thus affects the functionality of these constructs. Secondary structure analysis through in-line probing showed theophylline-induced rearrangements comparable to the original constructs (Supplementary Figure S5), which agrees with their similar accessibility scores (Table 1). Additional interference can be also caused by tertiary interactions, which are not considered in the in silico calculations or the structural probing. Further, it has to be noted that the amount of mature tRNA does not necessarily correlate with the amount of produced protein (68,69). As the riboswitch constructs are plasmid-encoded, their transcription leads to rather high amount of pre-tRNA that is processed upon induction. This increased tRNA concentration likely leads to an oversaturation of the corresponding tyrosyl-tRNA synthetase, and the processed but uncharged supF tRNA molecules do not contribute to an increase in the c3GFP expression. As uncharged tRNAs are bound by RelA at high affinity (67), such a scenario is supported by the fact that the observed slow-migrating signal in the northern blot analysis is proteinase-sensitive (Supplementary Figure S2). Such a synthetase oversaturation can explain why a protein-based signal amplification does not match the amount of processed supF tRNA, as observed for RP-RS A-h and C-h.
This observation is also important to evaluate the c3GFP-based results. Here, the incubation time is a central parameter that immediately affects the read out. If the time points are chosen too long, a proper quantitation is difficult due to the oversaturation of expression system with the processed tRNA. For our constructs, 4.5 h were chosen as the incubation time to allow a direct comparison of all tested constructs. Only at this time point moderately active switches (RP-RS C) are detected, whereas RP-RS A seemed to be slightly oversaturated as indicated by the formed tRNA–protein complex (presumably RelA) and the high amount of processed tRNA in the northern blots (Figure 3). Thus, a read-out system for tRNA processing based on protein expression clearly allows to monitor the regulatory potential of individual constructs. A precise quantitation of the switching behavior, however, is rather difficult, as it is well-known that the correlation of tRNA, mRNA and protein levels is notoriously poor (95). Furthermore, as the read-out system is based on a suppressor tRNA, this product has to compete with release factor 1 (RF1) for codon interaction (96). As a certain amount of suppressor-tRNA is required to efficiently outcompete RF1 in translation of the reporter gene (97,98), it is conceivable that the varying amounts of supF tRNA produced by the individual riboswitches are differently efficient in this competition, resulting in non-linear levels of protein expression. Nevertheless, a clear theophylline-dependent tRNA maturation was visible at protein as well at the level of mature tRNA. The fact that the northern analysis shows only a small amount of unprocessed tRNA in the OFF state is not surprising, as it was frequently reported that unprocessed precursor tRNAs are rapidly degraded in E. coli (99–101,49,102).
The structural refolding observed by in-line probing analysis corresponds to the theophylline-induced in vivo switching behavior of our riboswitch constructs. Yet, we do not observe a linear correlation of the probing signals and the switching efficiency in RP-RS A and RP-RS C derivatives. For instance, structure analysis shows a rather similar hairpin opening in RP-RS C-UG and RP-RS C-GA (Figure 5F). However, the in vivo activation based on c3GFP expression is much lower for RP-RS C-UG. A reason for this discrepancy is probably the long incubation time used for in-line probings. To obtain distinct cleavage signals, RNA transcripts are usually incubated for up to 40 h (55), and the obtained cleavage signals represent an RNA structure at its thermodynamic equilibrium. Thus, parameters such as folding kinetics and time-dependent refolding events cannot be represented with this method. For instance, a region that shows repeated folding/unfolding over time will eventually lead to a cleavage signal, while this behavior might not affect riboswitch functionality in vivo. Furthermore, a slow hairpin opening would be visible in the structural investigation, but if this is not sufficiently fast for ligand binding in vivo, the uncleaved riboswitch transcript will be rapidly degraded as an unprocessed tRNA precursor molecule (103). Hence, the structural analysis offers valuable supportive data, but it cannot be integrated into a predictive framework for riboswitch functionality consisting of in vivo and in silico data.
Kinetic analysis of RP-RS A-h processing by RNase P holoenzyme indicated the postulated regulation of the substrate accessibility. Even in vitro, a theophylline-dependent activation of the cleavage efficiency was visible for RP-RS A, while processing of nat-supF was unchanged. The increased background could be caused by experimental in vitro conditions which had to be balanced between enzyme activity and correct aptamer folding. Thus, the hairpin structure is probably folded less stably as proposed under the tested conditions. While the activity of the holoenzyme (nat-supF KM: 38.39 ± 7.64 nM; kcat: 1.01 ± 0.08 min−1) is lower compared to prior kinetic studies involving the positive control supF (KM: 13 nM, kcat: 6.6 min−1, KM: 17 nM, kcat: 10.3 min−1) (79,80), our data clearly indicate a theophylline-dependent cleavage reaction only for the riboswitch construct, while the nat-supF precursor processing is not affected by the presence or absence of the ligand (Table 2). There are various possible reasons for the observed differences in the kinetic parameters compared to the data presented by Kirsebom et al. Besides differences in buffer compositions also the experimental setup, including enzyme preparation and holoenzyme reconstitution, can have an impact on the actual values. Nevertheless, our reconstituted holoenzyme is highly active under standard conditions, where the individual components M1 RNA and C5 protein show no activity (Supplementary Figure S6).
In summary, the kinetic data show that the presence of theophylline leads to a 1.9-fold increased cleavage efficiency in the riboswitch transcript in vitro. While this value is lower than the in vivo data, it clearly corroborates the ligand-dependent control of RNase P-mediated tRNA processing of the riboswitch constructs. The in vitro conditions differ from the in vivo situation in terms of enzyme/substrate ratio and concentration and can influence the rate of E/S complex formation and cleavage efficiency. In vivo, the riboswitch transcript competes for 5′-processing with endogenous pre-tRNA molecules, and it is likely that in this situation, RNase P does not interact with the ligand-free riboswitch, since natural tRNA precursors represent the preferred substrates. As a consequence, the in vivo situation likely results in a lower background signal compared to the in vitro experiment. Furthermore, it is known that additional proteins like the RNA chaperone Hfq bind to tRNA precursors (104) and can stimulate tRNA processing reactions in vitro (105). The presence of such factors in the cell might increase RNase P-mediated cleavage and, as a consequence, the response ratio in vivo versus in vitro, where such factors are missing. In addition, the in vitro cleavage reaction on RP-RS A shows a considerable background activity in the absence of theophylline (Figure 4). This background might result from a ligand-independent hairpin fraying of the riboswitch transcript that then gives access to the cleavage position. In vivo, Hfq-like proteins might suppress this fraying, resulting in lower background signals.
Interestingly, RP-RS A led to a somewhat higher kcat compared to the positive control nat-supF in presence of the ligand, underlining the fact that the identity of N(–1) is not the determining feature for an efficient cleavage by RNase P holoenzyme. Niland et al. showed that base pairing of the discriminator and the 3′-CCA with N(–1) and N(–2) can also be an antideterminant for efficient cleavage (37). For the positive control supF, such an interaction is possible, since U(–1) and G(–2) may interact with the discriminator A and the first C from the 3′-CCA-end, which is not the case for RP-RS A (contains A(–1) and U(–2)).
Taken together, it is possible to construct highly efficient riboswitches regulating RNase P processing by presenting and masking the 5′-leader region in a ligand-dependent way. These data indicate that RNase P-mediated 5′-processing of a precursor tRNA in vivo is strongly affected by the structure of the leader region, supporting the hypothesis by Lin et al. (35). In the design principle, the ligand-induced formation of the aptamer structure must be able to compete with the sequestor hairpin. Hence, this hairpin carries a series of G–U base pairs distributed over the whole helical structure that allows an easy and fast rearrangement. The seeding region involved in the closing of the hairpin loop also has to be structurally rather unstable, as the G43–C50 pair completely abolishes switching, while weaker interactions (A–U, G–U, U–G, G–A) allow for a ligand-induced precursor cleavage. The third important parameter is the base of the hairpin, where a slight fraying of the terminal three positions does not lead to background cleavage but also contributes to the structural rearrangement upon theophylline binding. The comparison of the fold-changes between experiments and predicted leader accessibility scores reveals the complexity of the underlying regulatory system. Besides sequence and structure of the 5′-leader, additional parameters can influence the switching efficiencies. Rapid degradation of the precursor-tRNA in vivo, the interaction of RNA binding proteins with the expression platform as well as a limited capacity of the translation machinery can have an impact on the observed ON and OFF states of the riboswitches and thus influence each experiment in a different way. Although an integration of all experimental approaches to predict riboswitch functionality is currently not possible, the combination of reporter gene expression, determination of mature tRNA amounts, in-line probing and in vitro kinetic analyses lead to a rather detailed picture of theophylline-dependent tRNA processing in our constructs.
DATA AVAILABILITY
A complete and sortable list of all calculated in silico measures for the investigated and additional best and worst scoring constructs of different lengths is available online (http://www.bioinf.uni-leipzig.de/supplements/20-001).
ACKNOWLEDGEMENTS
We thank Leif A. Kirsebom (Uppsala University, Sweden), Roland K. Hartmann (University of Marburg, Germany), Eric Westhof (University of Strasbourg, France) and Robert Serfling (Leipzig University, Germany) for valuable discussions. Furthermore, we thank Astrid Schön (Leipzig University, Germany) for plasmid pDW27_M1RNA, Karolin Wellner for plasmid pACYC/tetM_araC/c3GFP and Roland Hartmann (Marburg University, Germany) for plasmid pQE30_rnpA-HisTag. Additional thanks go to Tobias Friedrich and Barbara Klüver (both Leipzig University, Germany) for expert technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [MO 634/9-2, STA 850/15-2]; Deutsche Forschungsgemeinschaft (DFG); Leipzig University. Funding for open access charge: Deutsche Forschungsgemeinschaft (DFG); Leipzig University.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.