 First insights into the structural features of Ebola virus methyltransferase activities
First insights into the structural features of Ebola virus methyltransferase activities
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

- Altmetric
The Ebola virus is a deadly human pathogen responsible for several outbreaks in Africa. Its genome encodes the ‘large’ L protein, an essential enzyme that has polymerase, capping and methyltransferase activities. The methyltransferase activity leads to RNA co-transcriptional modifications at the N7 position of the cap structure and at the 2′-O position of the first transcribed nucleotide. Unlike other Mononegavirales viruses, the Ebola virus methyltransferase also catalyses 2′-O-methylation of adenosines located within the RNA sequences. Herein, we report the crystal structure at 1.8 Å resolution of the Ebola virus methyltransferase domain bound to a fragment of a camelid single-chain antibody. We identified structural determinants and key amino acids specifically involved in the internal adenosine-2′-O-methylation from cap-related methylations. These results provide the first high resolution structure of an ebolavirus L protein domain, and the framework to investigate the effects of epitranscriptomic modifications and to design possible antiviral drugs against the Filoviridae family.
INTRODUCTION
The Mononegavirales order includes the Ebolavirus genus that comprises viruses with linear, negative sense, non-segmented, single-stranded RNA genomes (referred here as NNS viruses). Ebola virus is among the deadliest viruses of the order. Indeed, the recent outbreaks in West Africa (2013–2016) and in the Democratic Republic of the Congo (DRC) between August 2018 and June 2020 caused at least 11 000 and >2200 deaths, respectively (https://www.who.int/csr/disease/ebola/situation-reports/archive/en/, https://www.who.int/emergencies/diseases/ebola/drc-2019). Fruit bats are considered the main reservoir of this zoonotic virus. In rare circumstances, Ebola virus can be transmitted to human and non-human primates. Human-to-human transmissions mainly rely on direct contacts with biological fluids of infected patients that lead to the virus dissemination in the populations (1) (https://www.cdc.gov/vhf/ebola/history/distribution-map.html). After 2–10 days of incubation, Ebolavirus infection can cause haemorrhagic fevers that is fatal in almost 50% cases for Sudan virus disease and up to 80% for Zaire virus disease. Although the rVSV-ZEBOV-GP and Ad26-ZEBOV vaccines have shown good efficacy in limiting the past Ebola outbreak occurring in DRC (2018–2020) (2), effective antiviral drugs and therapies are still lacking.
The Sudan virus (SUDV) belongs to the Ebolavirus genus. Its genome of about 19 kb encodes seven proteins: the nucleoprotein (NP), VP35, VP40, glycoprotein, VP30, VP24 and ‘large’ protein L (3,4). The L protein drives virus replication by performing all the enzymatic activities required for genome replication, transcription and mRNA capping, and polyadenylation. Unlike the canonical eukaryotic pathway, viral mRNAs are co-transcriptionally capped by a non-canonical capping reaction in which the nascent viral mRNA binds covalently to a conserved catalytic histidine residue of the polyribonucleotidyltransferase (PRNTase) of the L protein cap domain (5). The PRNTase binds to and transfers a GDP molecule to the 5′ phosphate of the covalently bound RNA, forming the cap structure (GpppN1). The cap is subsequently methylated by the methyltransferase (MTase) domain at the 2′-OH position of the first nucleotide (N1) ribose and at the N7 position of the cap guanosine (cap-1, mGpppNm) (5–7). N7 methylation of the cap structure is required for viral mRNA translation into proteins by allowing mRNA recognition by the translation initiation factor eIF4E (8). The 2′-O-methylation of N1 protects the viral mRNA from the detection by cytoplasmic sensors belonging to the retinoic acid-inducible gene-I (RIG-I)-like receptor family (9). Thus, mis-capped RNAs can be detected by RIG-I (9,10) that in turn induces a cascade of intracellular events leading to interferon expression. In addition to its role during RNA transcription, the L protein also ensures genome replication when the NP protein concentration is increased. The pleiotropic activities of the L protein suggest that these different enzymatic activities are timely regulated to ensure the different specific functions required for virus replication and transcription.
Multiple sequence alignments revealed that the Mononegavirales L proteins contain six conserved regions (CRI to CRVI) located in the RNA-dependent RNA polymerase (RdRp) domain (CRI to CRIII) (11,12), the Cap or PRNTase domain (CRIV & V) (13), and the MTase domain (CRVI) (14,15). Negative staining electron microscopy experiments on the related NNS vesicular stomatitis virus (VSV) L protein revealed that the RdRp and Cap domains interact with each other and form a ‘donut-like’ structure, followed by three flexible globular domains corresponding to the connector domain (CD), the MTase domain, and a small C-terminal domain (CTD) (16). Recently the structure of several mononegavirus L proteins was determined by cryo-electron microscopy (17–20). For some of them, such as the respiratory syncytial virus L protein (21), the C-terminal region (CD+MTase+CTD) is not clearly defined, suggesting a conformational rearrangement of the L protein between the replication and transcription conformations (20).
The carboxy-terminal region of the L protein contains the conserved MTase domain upstream to the CTD. The MTase domain of Mononegavirales viruses has a Rossmann fold with a canonical S-adenosylmethionine (SAM) binding site (22). The MTase domain contains a typical 2′-O-MTase catalytic tetrad (K-D-K-E) and a GxGxG motif characteristic of the SAM-binding site, whereas the CTD shows no conserved signature (15). However, these MTase domains apparently lack the cap-binding site observed in most viral MTases (6,17,20). Conversely, the MTase and CTD are associated and form a narrow RNA-binding groove enriched in basic amino acids close to the catalytic site (6). The role of the CTD was recently investigated by biochemical studies showing that the RNA-binding properties and MTase activities of SUDV MTase depend on the presence of this domain (23). Cap methylations driven by the VSV MTase occur first at the ribose 2′-O-position of N1 followed by guanine-N7 methylation of the cap structure (7). The 2′-O-methylation of N1 hides the viral mRNA from detection by cytoplasmic sensors belonging to the retinoic acid-inducible gene-I (RIG-I)-like receptor family (9). N7 methylation of the cap structure is required for viral mRNA translation into proteins by allowing mRNA recognition by the translation initiation factor eIF4E (8). The MTase catalytic activity has been confirmed for other Mononegavirales (human metapneumovirus, hMPV, and SUDV) (6,24). Besides this shared functional feature, the hMPV MTase can methylate uncapped RNAs on the 2′-OH of the first transcribed nucleotide (6), and the SUDV MTase carries an additional activity of internal adenosine-2′-O-methylation of adenosine residues (i.e. on nucleotides internal to the RNA sequence) (24). The role of such post-transcriptional RNA modifications is not known in the context of ebolavirus infection. However, similar epitranscriptomic RNA modifications have been described in the RNA of other viruses, such as Zika virus (ZIKV), Dengue virus (DENV), and HIV (25–27), suggesting their involvement in the regulation of host–pathogen interactions. For instance, HIV recruits the cellular MTase FtsJ3 that catalyses internal adenosine-2′-O-methylation of the viral RNA genome, promoting the host defence subversion by hindering viral detection by the RIG-like receptor Melanoma Differentiation-Associated protein 5 (MDA5) (25). These findings suggest pleiotropic functions of L-associated MTase activities that catalyse different post-transcriptional modifications of viral RNA to regulate the virus life cycle and the early antiviral response.
In this work, we solved the crystal structure of the SUDV MTase to elucidate the structural and functional interplay of these activities.
MATERIALS AND METHODS
Cloning and expression
Codon-optimized SUDV MTase+CTD (residues 1713–2211) synthetic genes (Biomers) were cloned in the pET14b vector for production of the recombinant protein in bacteria. The MTase domain without the CTD (residues 1744–2046) and the mutated proteins were produced by directed mutagenesis to introduce double stop codons or single mutations, respectively. All constructs were obtained using SUDV MTase+CTD as a template, primers carrying the specific mutation, and the DNA polymerase PfuTurbo (Ambion) for PCR amplification. PCR products were purified using the Wizard SV PCR Clean-Up System (Promega). Transformed T7 Iq Express Escherichia coli cells (New England Biolabs) were cultured at 30°C until OD600 nm = 0.6 was reached. Then, temperature was shifted to 17°C and isopropyl β-d-1-thiogalactopyranoside (IPTG, Euromedex) was added (final concentration of 20 μM). The next day, bacteria were spun down (8000 × g at 4°C for 20 min) using a Sorval Lynx 6000 centrifuge before storage at −80°C. For SUDV MTase+CTD and mutated SUDV MTase+CTD, dry pellets were stored at −80°C. For SUDV MTase, 1L of bacterial culture was resuspended in 40 mL of lysis buffer (300 mM NaCl, 50 mM Tris (pH 8.0), 10 mM imidazole, 0.25 mg/ml lysozyme, 0.1% Triton X100 and 1 tablet of EDTA-free antiprotease cocktail (Roche) before storing them at −80°C.
Purification of the recombinant proteins
Wild type (WT) and mutant MTase+CTD proteins were purified as previously described (24). Briefly, bacterial pellets were lysed in lysis buffer (50 mM Tris pH 8, 150 mM NaCl, 5% glycerol, 30 mM imidazole, 1 mM PMSF, 100 μg/ml lysozyme, 1 μg/ml DNase, 0.1% Triton X100) supplemented with the detergent mix BugBuster (Merck Millipore). After clarification (18 000 × g, 4°C, 30 min), lysates were incubated with the CoNTA resin (Thermo Fisher). MTase+CTD proteins were eluted with 50 mM Tris pH 8, 150 mM NaCl, 5% glycerol, 1 M arginine and then concentrated and stored in 50% glycerol at −20°C. For the MTase, pellets were thawed at room temperature, and 1 mM PMSF, 10 μg/ml DNase, 20 mM MgSO4 were added. After incubation at 4°C for 30 min, cells were sonicated and clarified by centrifugation (18 000 × g, 4°C, 30 min) and a tablet of EDTA-free antiprotease cocktail (Roche) per 50 ml of lysate was added. Proteins were then purified by immobilized metal affinity chromatography (IMAC) on 5 ml His-trap columns (GE Healthcare). Proteins were eluted with 300 mM NaCl, 50 mM Tris (pH 8.0), 250 mM imidazole, and then loaded on Superdex S75 16/60 (GE Healthcare) equilibrated with 10 mM Tris (pH 8.0) and 150 mM NaCl.
Generation of VHH against the SUDV MTase domain
A healthy llama (Llama glama from Ardèche lamas, France) was immunized (1 injection/week for 5 weeks) with 0.8 mg purified SUDV MTase, produced as previously described (24) and stored in 50 mM Tris (pH 8.0), 150 mM NaCl, 5% glycerol. Lymphocytes were isolated from blood samples obtained five days after the last injection. The cDNA synthesized from purified total RNA by reverse transcription was used as template for PCR to amplify the sequences corresponding to the variable domains of the heavy-chain antibodies. PCR fragments were then cloned into the phagemid vector pHEN4 (28) to create a VHH phage display library. VHH selection and screening were performed as described previously (29).
VHH expression and purification
Selected nanobodies were cloned in the pHEN6 plasmid that contains the N-terminal pelB periplasmic signal sequence in frame with a VHH expression cassette and a C-terminal 6His tag for detection and purification. Nanobodies were expressed in WK6 bacteria cultured in Terrific Broth medium (AthenaES) supplemented with 100 μg/ml ampicillin and 0.1% glucose at 37°C until OD600 nm = 0.5–0.8. Expression was then induced by addition of 1 mM IPTG and growth was continued at 28°C overnight.
Periplasmic proteins were extracted according to Skerra et al. (30). Bacteria were pelleted by centrifugation at 3500 × g at 4°C for 15 min. The pellet was resuspended in 9 ml cold TES buffer (0.2 M Tris–HCl (pH 8.0), 0.5 mM EDTA, 0.5 M sucrose) per litre of culture and kept on ice for 1 h. Periplasmic proteins were removed by osmotic shock by addition of 13.5 ml of cold TES diluted four times with water. After 1–2 h on ice, the suspension was centrifuged at 21 700 × g at 4°C for 30 min. VHH were purified by IMAC using Ni-NTA resin (Thermo Fisher Scientific). After sample loading at 4°C for 1 h, contaminants were eliminated from the resin with wash buffer (50 mM phosphate buffer (pH 8.0), 300 mM NaCl, 10% glycerol), and proteins were eluted by step gradient using wash buffers containing 50–250 mM imidazole. Fractions containing the purified nanobody were concentrated to 1 ml on Amicon Ultra-MW10000 filters (Millipore). Finally, size exclusion chromatography (SEC) of VHH fragments was performed using a Superdex 75 16/60 column equilibrated with 10 mM Tris (pH 8.0) and 150 mM NaCl.
Purification of the SUDV MTase-VHH heterodimer
Monomeric MTase fractions were pooled and complexed to purified VHH (1:1.5). After 1h incubation at 4°C, the complex (MTase and VHH) and free VHH were separated by SEC using Superdex S75 16/60 (GE Healthcare) equilibrated with 10 mM Tris (pH 8.0) and 150 mM NaCl. Purified complexes (MTase and VHH) were concentrated to 7 mg/ml and stored at 4°C.
Crystallogenesis and X-ray crystallography
Concentrated complexes (7 mg/ml) were crystallized by vapor diffusion at 20°C using a 96-well sitting drop plate (SWISSCI 3 Lens Crystallization Microplate). Crystals grew spontaneously within 48 h by equilibrating 300 nl of protein with 100 nl of 0.1 M Tris–HCl (pH 8.0), 0.1 M NaCl, 8% (w/v) of PEG 8000 as F5 solution of the ProPlex HT-96 screen (Molecular Dimensions).
Crystals were cryo-protected with reservoir solution with 20% PEG 200 before flash freezing in liquid nitrogen. X-ray diffraction was performed on the beamline Proxima1 at the Soleil synchrotron. Data from crystals were collected at λ = 1.28242 Å. Datasets were processed individually and analysed with the autoPROC toolbox (31). A weak anomalous signal around 4 Å was detected for four crystals. Dataset merging with AIMLESS allowed enriching the anomalous signal at low resolution corresponding to the S scatter (32). The structure was solved by combining the molecular replacement and anomalous signal methods, using PHASER (33). The original placement of a VHH structure homologue (PDB: 5IMO presenting 70% sequence identity) allowed obtaining a partial map and the anomalous difference map helped to calculate the initial electron density map and align the sequence that identifies the MTase. Density was modified with PARROT (34), and auto-building with BUCCANNEER (35) allowed extending the model that was then manually built with Coot (36) and refined with BUSTER (37). The final model had a Rwork = 19.6% and a Rfree = 23.4%, and its good stereochemistry was confirmed with MolProbity (38). Using this model, a molecular replacement was performed on a single data set and the structure was refined with BUSTER up to 1.8 Å. Data collection and refinement statistics are listed in Table 1.

| Data processing | MTase Ebola SUDV (S) | MTase Ebola SUDV (native) |
|---|---|---|
| Wavelength (Å) | 1.282 | 1.282 |
| Space group | P62 2 2 | P62 2 2 |
| – a, b, c (Å) | 153.98, 153.98, 105.41 | 153.76, 153.76, 105.35 |
| –α,β,γ(°) | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 |
| Resolution range (Å) | 76.87–2.00 | 76.88–1.84 |
| (2.05–2.00) | (1.907–1.84) | |
| Total no. of reflections | 8 916 413 (355 358) | 2 566 705 (257 401) |
| No. of unique reflections | 49 877 (3627) | 63 699 (6280) |
| Completness (%) | 100 (99.7)* | 99.98 (100) |
| Multiplicity | 94.1 (50.5)* | 40.3 (41) |
| I/σ(I) | 30.5 (3.5) | 13.74.54 (2.14) |
| Rmeas | 0.2135 (1.601) | |
| CC1/2 | – | 0.99 (0.83) |
| Wilson B-factor (Å) | – | 32.08 |
| Structure solution & refinement | ||
| No. of reflections, working set | – | 63691 (6280) |
| No. of reflections, test set (%) | – | 3164 (311), 5% |
| R-cryst | – | 0.1782 (0.2455) |
| Rfree | – | 0.2009 (0.2663) |
| No. of non-H atoms | – | 3571 |
| - Protein | – | 3070 |
| - Ligand | – | 73 |
| - Water | – | 428 |
| R.m.s. deviations | ||
| - Bonds (Å) | – | 0.01 |
| - Angles (°) | – | 1.51 |
| Average B-factors (Å2) | – | 39.00 |
| - Protein | – | 36.5. |
| - Ligand | – | 56.05 |
| - Water | – | 54.12 |
| Ramachandran Plot | ||
| - Favoured (%) | – | 97.92 |
| - Allowed (%) | – | 1.82 |
| - Outliers (%) | – | 0.26 |
| Clashcore | – | 5.15 |
| PDB code | – | 6YU8 |
Comparison of domains, modeling of missing loops and structural analysis
Homologous structures were searched with DALI (39) and 3D-fold (40) search to retrieve MTase structures. Comparison with the VSV and hMPV MTase structures (PDB: 4UCZ and 5A22, respectively) allowed confirming the coiled structure that corresponds to the missing loop. To ensure the complete continuity of the main chain and the proper surface analysis, the two missing loops (regions 1764–1775 and 1795–1808) were modelled. The structural analysis and reference files for modelling were prepared with CHIMERA (41) and the missing loops were modelled with MODELLER 9.23 (42). Surface electrostatics was calculated with APBS (43). Sequences and interface were analysed with ENDSCRIPT (44).
Synthesis of RNA substrates
RNA oligos were chemically synthesized, as previously described, on solid support using an ABI 394 oligonucleotide 171 synthesizer (45). RNA elongation was performed with 2′-O-pivaloyloxymethyl phosphoramidite ribonucleotides and 2′-O-methyl phosphoramidite ribonucleotides (Chemgenes). Then, the 5′-hydroxyl group was phosphorylated, and the resulting H-phosphonate derivative was oxidized and activated into a phosphoroimidazolidate derivative to react with guanosine diphosphate (Gpp) to produce GpppRNA. After deprotection and release from the solid support, GpppRNAs were purified by IEX-HPLC and their purity (>95%) was confirmed by MALDI-TOF spectrometry. N7 methylation of the purified Gppp-RNAs was performed by incubation with human N7 MTase (46).
MTase activity assay
Methyltransferase activities were assessed using a radioactive filter-binding assay as previously reported (23). Briefly, 4 μM of protein was mixed with 1 μM of purified synthetic RNAs (list of RNAs in Supplementary Table S1), 10 μM of SAM and 0.5 μM of 3H-SAM (Perkin Elmer) in 50 mM Tris–HCl at different pH to evaluate the different MTase activities, as previously described. Specifically, Martin et al. highlighted that SUDV MTase activities are influenced by the pH of the reaction, with optimal pH values of 7.0 for the cap-N7 MTase activity, of 8.0 for the cap-2′-O-MTase, and of 8.5 for internal methylation (24). After 3 h at 30°C, reactions were stopped by 10-fold dilution in water, and samples were loaded onto DEAE filtermats (Perkin Elmer) using a Filtermat Harvester (Packard Instruments). After two washes with 10 mM ammonium formate pH 8.0, two washes with water, and a last wash with ethanol, filters were soaked with liquid scintillation fluid to measure the 3H-methyl transfer to the RNA substrates using a Wallac MicroBeta TriLux Liquid Scintillation Counter13. For statistical analysis, it was assumed that the different experimental groups were independent, and data followed a Gaussian distribution with the same variance. Two-way ANOVA and multiple comparisons Dunnett test were used (Prism) to evaluate differences between groups. The level of significance for α = 0.05 is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
RESULTS
X-ray structure of the SUDV MTase domain in complex with a VHH
The purified SUDV MTase was incubated with a single-chain camelid antibody (VHH, nanobody) and the complex was recovered after size exclusion chromatography (Supplementary Figure S1A). The heterodimeric complex was crystallized, and the crystal structure determined at 1.8 Å resolution (Table 1) using molecular replacement coupled with single wavelength anomalous diffraction (MR-SAD) based on sulphur as anomalous scatterer (Figure 1A). The crystal structure belonged to the space group P6222, with the following cell dimensions: a = b = 153.76 Å, c = 105.35 Å, and α = β = 90°, γ = 120°. It contained twelve heterodimers in the unit cell. The crystal higher order topology shows that the structural assembly forms a compact mesh crossed by large hexameric solvent channels in which a side is ∼60 Å long and the diagonal is ∼107 Å (Supplementary Figure S2A). Rotation by 90° of the unit cell showed that the VHH filled other solvent channels, thus stabilizing the crystal structure (Supplementary Figure S2B).

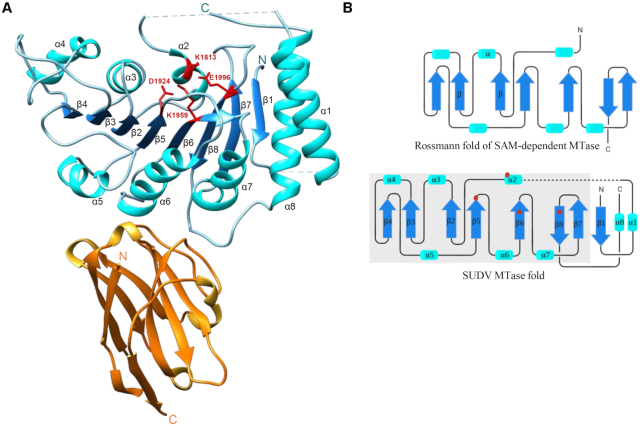
SUDV MTase structure resolved by X-ray crystallography. (A) Crystal structure of the Sudan ebolavirus (SUDV) methyltransferase (MTase) domain (blue) in complex with a VHH (orange). The N- and C-terminal extremities are indicated by the N and C letters, respectively, in both proteins. The catalytic residues K1813, D1924, K1959 and E1996 of the SUDV MTase are highlighted in red. The SUDV MTase structure shows the presence of eight β-sheets alternated with eight α-helices and two undefined loops (residues K1764 to P1775 and residues V1795 to S1808, dashed lines). The VHH adopts a classic β-sandwich fold structure composed of eight strands connected by flexible loops. Two loops (residues G55 to A61 and residues A100 to Y109) and a turn (residues R27 to R31) are involved in the interaction with the SUDV MTase domain, at the opposite side of the catalytic pocket. (B) Topological organization of the SUDV MTase domain (bottom) and of the canonical Rossmann fold of a SAM-dependent MTase (top). The MTase Rossmann fold is defined by a β-α-β motif (here β2-α3-β3) that contacts the SAM methyl donor. The overall strand/helix architecture is boxed in gray in the SUDV MTase fold representation. Helices are depicted as cyan barrels, β-strands as blue arrows, and coils as black lines. The N- and C-terminal extremities are indicated by N and C, respectively. Red points correspond to catalytic residues within the Rossmann fold secondary structure. This representation highlights the additional features at the N-terminus (1 β-strand and 1 α-helix) and C-terminus (1 α-helix) of the SUDV MTase.
The MTase domain presented a Rossmann fold (Figure 1A and B), typical of most MTases that catalyse the methyl transfer from SAM (6). The domain contained 8 β-strands of which β2 to β8 were part of the core of the Rossmann fold and adopted the classical parallel strand organization except for β8, which is antiparallel and sandwiched between the antepenultimate and penultimate strands (Figure 1B). This central β-sheet was surrounded by six α-helices. The 2′-O-MTase catalytic tetrad (K1813/D1924/K1959/E1996) and the GxGxG SAM-binding motif (G1833-X-G1835-X-G1837) were localized in the Rossmann fold secondary structure (Figure 1A and B and Supplementary Figure S3A and B). Besides the Rossman core, the first β-strand was antiparallel to the penultimate strand (β7), and two long α-helices (α1 and α8) interacted with each other and the first strand (Figure 1A and B).
Superimposition of the SUDV and hMPV MTase structures revealed high structural conservation (RMSD: 2.0 Å) despite the low sequence identity of these proteins (≤10%). The linker regions between β1 and α1 (residues 1764–1775), and α1 and α2 (residues 1795–1808) were not built due to lack of density and probable high flexibility (Figure 1A). The equivalent α1–α2 segment is present in the structure of other NNS viruses (hMPV, VSV, RABV) where it is stabilized by the CTD and shows the same structure: a long loop punctuated by a short α helix (α'). Thus, we decided to model the missing part (highlighted in green in Supplementary Figure S3A).
The VHH is a β-sandwich composed of 8 strands connected by flexible loops (Figure 1A). The VHH antigen interface bound to the MTase on a single epitope formed by the bottom part of three helices (α4, α5, α6) and on the opposite side of its catalytic site. This is consistent with the results of the MTase activity test performed in the presence of different VHH concentrations. The VHH recognized the MTase and MTase+CTD domains of SUDV L protein, but did not inhibit the MTase activities of SUDV MTase + CTD (Supplementary Figure S4A and B). This suggests that the VHH does not alter MTase folding and its catalytic site. VHH bound to the MTase domain through an extended surface composed of several hydrophobic residues located in two side loops (residues 55–61 and residues 100–109) and a turn (residues 27–31) close to the N-terminus, favouring protein stability. The overall binding interface area was ∼833.6 Å2 (Supplementary Figure S5).
The structural and functional interplay of SUDV MTase activities
To identify functional sites in the SUDV MTase, the hMPV MTase structural model was used, and the previous nomenclature was conserved to localize the SAM-binding (SAMP), RNA-binding (SUBP) and nucleoside-binding (NSP) pockets.
As co-crystallization and soaking experiments with SAM or SAH did not give any result, a SAM molecule was modelled in the SUDV structure by superimposing the hMPV MTase domain to localize residues involved in SAMP (Figure 2A). As previously observed for hMPV (residues 1718–1729), the SUDV MTase presented a long flexible loop between β3 and α4 that participates in SAMP (residues 1854–1875). However, this loop adopted an ‘open’ conformation in the SUDV structure where no SAM molecule was co-crystallized. Conversely, in hMPV, this loop showed a ‘closed’ conformation, clamping the SAM substrate in SAMP (Figure 2B). As the SAMP sequence is largely conserved (especially the GxGxG motif) (14,15), several residues were mutated (E1834A, G1835S, G1837S) to assess their roles (Figure 3A). We next mutated the MTase and determined the effect of such mutations on the MTase activity by functional binding assay. As the MTase domain cannot recruit RNA in the absence of the C-terminal domain (CTD) (23), the functional MTase+CTD protein (from amino acid 1750 to amino acid 2126, SUDV L protein numbering) was used to perform structure-guided functional studies. By using GpppGm(Am)-SUDV12, mGpppA(Am)-SUDV12 or mGpppGm-SUDV12, we could evaluate cap-N7 MTase, cap-2′-O-MTase or internal-2′-O-MTase activities, respectively (Supplementary Table S1). The mutation of the two glycine residues (G1835S, G1837S) led to the complete loss of the three MTase activities, as described previously for other viral MTases (47,48). Conversely, the E1834 mutation increased MTase activity (Figure 3A). This suggests that this residue may participate in the MTase reaction turn-over. Moreover, mutation of residues T1854 and L1855 in the flexible loop close to SAMP (Figures 2A and 3B) led to complete loss of MTase activities, advocating for their involvement in SAM binding, as described in hMPV (6).
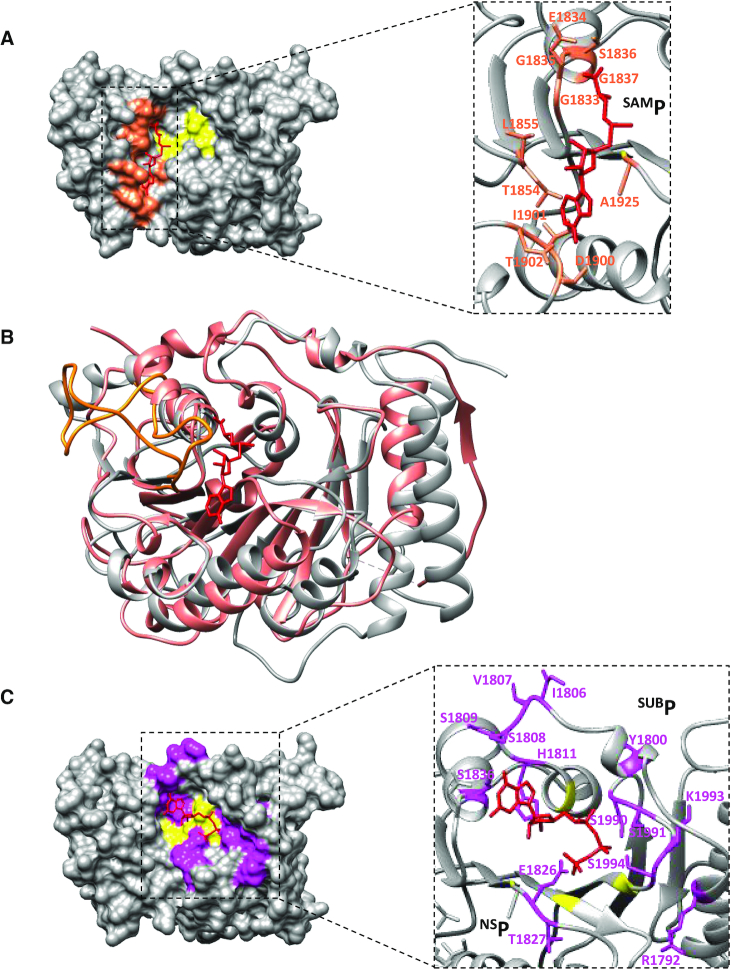
Identification of conserved functional pockets. (A) Modeling of the S-adenosylmethionine (SAM) molecule (red) within the SUDV MTase structure based on the hMPV MTase structure co-crystallized with SAM. Residues involved in the SAM-binding pocket (SAMP) are in orange, and catalytic residues in yellow. (B) Superimposition of the SUDV MTase domain (gray) with the hMPV L MTase domain (PDB: 4UCI, salmon), with the SAM molecule in red. The flexible loop participating in SAMP is highlighted in orange in the hMPV and SUDV MTase structures. The hMPV loop adopts a ‘closed’ conformation, clamping the SAM substrate in SAMP, whereas the SUDV loop shows an ‘open’ conformation. (C) Modeling of a GTP molecule (red) within the SUDV MTase structure based on the hMPV MTase structure co-crystallized with GTP. Residues involved in the substrate-binding pocket (SUBP) as well as the deep hydrophobic cavity (NSP) are in pink and catalytic residues are in yellow.
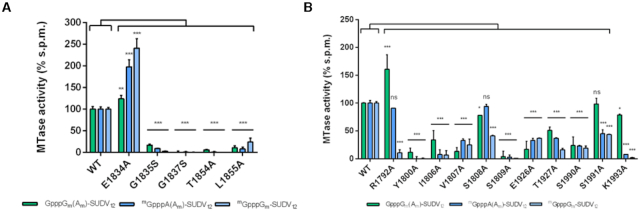
Single-mutation analysis of the SAM-binding and RNA-binding pockets. (A) MTase activity of SUDV WT and mutated MTase+CTD in the SAMP. The G1835S, G1837S, T1854A and L1855A mutations led to complete loss of the tested activities (cap-N7, cap-2′-O and internal A-2′-O-methylations). Mutation E1834A promoted the three methylation activities. (B) MTase activity of SUDV WT and mutated MTase+CTD in the SUBP and NSP. The Y1800A and S1809A mutations in the putative RNA-binding groove led to loss overall MTase activities (cap-N7, cap-2′-O and internal A-2′-O-methylations). Other mutations, such as I1806A, V1807A, T1927A and S1990A possibly involved in RNA binding, showed a significant reduction of all MTase activities. Several mutations in SUBP resulted in the uncoupling of the different MTase activities. The S1991A and K1993A mutations led to a drastic reduction (approximately by 50%) and almost complete loss of 2′-O-MTase activities (cap and internal A-2′-O-MTase activities), respectively, but not of the N7 MTase activity. The S1808A and R1792A mutations similarly impaired only the internal A-2′-O-MTase activity, but not the MTase activities associated with cap synthesis. Data are the mean ± standard deviation (n = 3); *P< 0.05, **P< 0.01, ***P< 0.001 (two-way ANOVA and multiple comparison Dunnett test, WT versus mutation).
Similarly, a GTP molecule modelled in SUDV MTase using the structure the hMPV MTase in complex with GTP, was employed to define SUBP (Figure 2C). This approach led to the identification of two loops that participate in SUBP of the SUDV MTase (Figure 2C). The first one, from residue 1803 to residue 1811, is quite variable within Mononegavirales and contains charged residues instead of the hydrophobic residues found in the hMPV MTase loop (residues 1663–1670). Most mutations in this loop (I1806A, V1807A and S1809A) and Y1800A caused extensive loss of function, like in hMPV (6) (Figures 2C and 3B). However, the S1808A mutation resulted in the specific decrease of the internal adenosine-2′-O-MTase activity (almost 50% reduction), whereas cap-dependent activities were conserved (Figure 3B). These observations suggest that this loop plays a key role in the different MTase activities, and that the central position of this serine in SUDV might participate in the internal adenosine-2′-O-MTase activity. The second loop, between residues 1988 and 1995 of the SUDV MTase and immediately followed by the catalytic residue E1996, was superimposed to that of hMPV despite sequence differences (residues 1940–1947). In this second loop, the S1990A mutation led to a broad loss of function, whereas the S1991A and K1993A mutations variably affected the different MTase activities. Indeed, these mutations induced ∼50% and almost 100% reduction of cap and internal 2′-O-MTase activities, respectively, whereas the N7 MTase activity was preserved. (Figure 3B). Mutation of the corresponding lysine to alanine in hMPV led to a slight decrease of 2′-O-MTase activity, while guanine-N7 MTase activity was marginally impacted (6). This result suggests that residues in this conserved loop are crucial for RNA positioning and might contribute to regulate the 2′-O-MTase activity.
Finally, the hMPV structure showed a deep hydrophobic cavity where the adenosine moiety of SAM or ATP (NSP) can bind to (Figure 2C). This pocket harboured the catalytic residue D1924. In the SUDV MTase, it was lined by the 1924–1933 loop that is assumed to adopt a ‘closed’ position. This loop was slightly longer than those of hMPV and VSV and enriched in charged and polar residues. Mutations of the conserved residues E1926A and T1927A led to an overall decrease of SUDV MTase activities (Figure 3B). Notably, the T1927A mutation slightly uncoupled the internal adenosine-2′-O-methylation from cap-N7 methylation and, to a lower extent, cap-2′-O-methylation from cap-N7 methylation (Figure 3B). These results indicate that E1926 and T1927 might contribute to the catalytic pocket stability and that T1927 might also participate in the groove for RNA positioning, as proposed for SUBP.
An additional α-helix in SUDV MTase domain
Comparison of the Mononegavirales MTase structures revealed that the SUDV MTase contains an additional α-helix (α1), close to its N-terminus (Figure 4). This helix is replaced by a long flexible loop in the VSV and parainfluenza 5 virus (PIV5) MTases, and by a short helix in the RABV MTase (Figure 4C). In hMPV MTase structure, this segment was not built, suggesting that this region is highly flexible. Alignment of different NNS MTase domains based on the RABV, VSV, hMPV and SUDV MTase structure superimposition did not reveal any equivalent structure of this long α1-helix within the viral order, supporting the hypothesis that this additional helix is filovirus-specific (Supplementary Figure S6). However, the DENV2 and ZIKV MTases (Figure 4C) present an α-helix that can be superimposed to the SUDV MTase α1. Flavivirus MTases can catalyse internal adenosine-2′-O-methylations (25–27). This supplementary α-helix contains charged residues forming a large positive groove next to the active site that could participate in RNA accommodation. Sequence and structure alignment revealed a conserved arginine residue (purple) in the α1 region of filoviruses and in the long flexible loop that overlaps with this region in VSV (Supplementary Figure S6). Its mutation to alanine (R1792A) led to uncoupling of the cap methylation (N7 and 2′-O-MTase activities) and internal adenosine-2′-O-methylation activities (Figure 3B). This suggests that the conserved arginine in filovirus MTase α1 participates in the internal adenosine-2′-O-MTase activity.
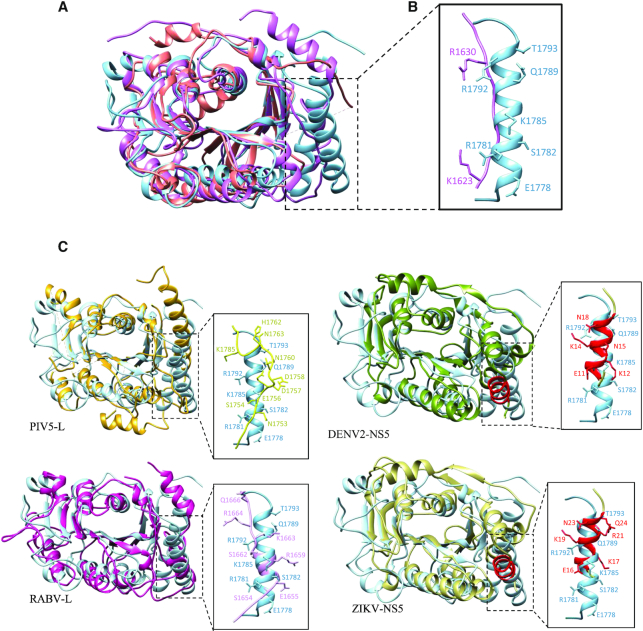
Structural comparison with other viral MTase domains. (A) Superimposition of the Sudan ebolavirus (SUDV) methyltransferase domain (MTase, sky blue) with the human metapneumovirus (hMPV) L MTase domain (PDB: 4UCI, salmon) and vesicular stomatitis virus (VSV) L protein (PDB: 5A22, purple). Compared with the other mononegaviruses, the additional N-terminal α-helix in SUDV MTase (α1) is not found in the hMPV and VSV MTase structures. (B) Close-up of the α1 homologous region in VSV superimposed to SUDV α1 in which the positively charged and polar residues are identified. This region was not resolved in the hMPV MTase structure, suggesting high flexibility. (C) Structural comparison with other viral MTase domains. Superimposition of SUDV MTase (sky blue) with the parainfluenza 5 virus L protein (PIV5-L, PDB: 6V85, top left, yellow), rabies virus L protein (RABV-L, PDB: 6UEB, bottom left, dark pink), dengue 2 virus non-structural protein 5 (DENV2-NS5, PDB: 5ZQK, top right, dark khaki) and Zika virus NS5 (ZIKV-NS5, PDB: 5M5B, bottom right, light khaki) MTase domains. The supplementary N-terminal α-helix in SUDV MTase (α1) was found also in the DENV2 and ZIKV NS5 MTase structures (red). A close-up of the α1 homologous regions in all structures is represented on the left of each superimposition with the positively charged and polar residues identified.
DISCUSSION
In this study, we described the structure of the SUDV MTase domain at a resolution of 1.8 Å in complex with a VHH using X-ray crystallography. As the L protein is the most conserved protein of the Mononegavirales order (12), we compared structures by superimposing SUDV MTase to other viral MTase domains, such as hMPV, RABV and VSV. This comparison suggests that the SUDV MTase domain is correctly folded and its structure is independent from other L protein domains (17). The SAMP, SUBP and NSP pockets, responsible for SAM, RNA and nucleoside binding (6) and previously described in other Mononegavirales MTases, are structurally and functionally conserved in SUDV.
These structural and functional analyses of the SUDV MTase domain contribute to drawing a comprehensive model of ebolavirus MTase activity regulation. This model relies on RNA accommodation in the active site because there is only one active site and only one co-substrate pocket for multiple MTase activities. Two noticeable surface regions are visible on the enzyme surface (Figure 5). The first region includes residues around the catalytic tetrad (i.e. in SAMP and NSP and the loop 1803–1811 of SUBP) that are critical for the different MTase activities. The second region includes the amino acid 1987–1995 loop and the α1-helix and creates a groove to recruit specifically RNA for 2′-O-methylation. It is thus possible to postulate a model where cap-N7 methylation occurs when RNA is accommodated by the first region, positioning the cap directly into the active site. Conversely, the cap-2′-O and internal adenosine-2′-O methylations are catalysed when RNA binds to the second region, projecting 2′-OH of nucleotide ribose into the catalytic site. The biochemical characterization of the MTase+CTD domains of SUDV revealed an original MTase activity profile for a virus of the Mononegavirales order (24). Comparative analysis showed a structural divergence between SUDV MTase and other mononegavirus MTases, and structural juxtapositions highlighted an additional secondary structure (α1-helix) close to its N-terminus. The NS5 MTase domain of flaviviruses (DENV and ZIKV), which catalyses internal A-2′-O-methylations, presents a homologous helix (26,27). The presence of positively charged and polar residues in this α-helix suggests that this structure may participate in the internal adenosine-2′-O-methylation. To evaluate this hypothesis, the R1792 residue in SUDV MTase was mutated to alanine. This mutation led to reduced internal adenosine-2′-O-methylation, but did not affect the cap-dependent MTase activities. Similarly, mutation of the corresponding lysine in DENV MTase (K14) abrogates the internal MTase activity (26). These observations support the hypothesis that this α-helix participates in internal 2’-O-MTase activity, possibly by specifically accommodating the RNA substrate to allow the internal SUDV MTase activity.
Structural mapping of residues involved in SUDV cap and internal methylations. Mapping of single mutations in the SUDV MTase domain (grey), coloured according to their involvement in cap-N7 methylation (left, residues in green), cap-2′-O-methylation (middle, residues in dark blue), and internal A-2′-O-methylation (right, in dark blue). The catalytic site (K1813-D1924-K1959-E1996) is coloured in pink and a SAM molecule (yellow) was modelled according to its putative position for the different methyltransferase activities.
The existence of these methylations in ebolavirus RNAs remains to be demonstrated. The detection of 2′-O-methylations is mainly based on high-throughput sequencing (49,50) and reverse transcriptase assays (51), and requires sufficient amount of viral RNA. Using these methods, a recent study mapped 17 2′-O-methylated sites in the RNA genome of HIV that regulate the activity of the cellular MTase FTSJ3 (25). This study also highlighted the role of internal methylations in the subversion of the host defences by limiting viral detection. Unlike HIV, EBOV may employ its own MTase to catalyse the internal 2′-O-methylation. The development of replicons or reverse genetic systems that can be manipulated outside BSL4 could be of great interest to identify the ebolavirus RNA sequences targeted by its own MTase (52). Indeed, we can hypothesize that the EBOV MTase activity is regulated during the replication and transcription steps through specific structural rearrangements as observed for parainfluenza viruses. The structure of the parainfluenza virus 5 (PIV5) L protein highlighted different conformations of the MTase and CTD domains that might allow switching between transcription and replication (20). The model proposed in this previous study suggests that only the transcription conformation allows efficient MTase activity, leading to mRNA methylation (20). Additional in vivo studies are needed to evaluate the methylation patterns of filovirus RNA genomes and to determine whether these epitranscriptomic modifications occur in specific conditions in infected cells.
In conclusion, this study describes the first high resolution structure of the ebolavirus MTase and the functional determinants that explain the three distinct RNA methyltransferase activities. Altogether, this study opens a new path toward a better understanding of viral internal methylation mechanisms, paving the way to decipher their role in innate immunity and to develop filovirus-specific drugs.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession number 6YU8.
ACKNOWLEDGEMENTS
The authors acknowledge SOLEIL for the synchrotron radiation facilities and would like to thank the beamline team of Proxima-1 for their assistance during data collection. We thank Sarah Attoumani and Anaïs Gaubert for technical support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Délégation Générale pour l’Armement (DGA) [2009.34.0038]; Aix-Marseille Université PhD fellowship (to B.M.); C.V. was funded by the National Research Agency ANR under the program ANR Rab-cap [ANR-16_CE11_0031_01]; French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-05-01]; C.V., B.M., B.Co. and E.D. designed and performed experiments, using material prepared by F.D. and J-J.V.; F.F. solved the crystal structure; V.Z. participated in protein purification and crystallogenesis; A.D. identified nanobodies against SUDV MTase; C.V., B.M., B.Ca, F.F., B.Co. and E.D. analysed the data and wrote the paper. Funding for open access charge: ANR [ANR-16_CE11_0031_01].
Conflict of interest statement. None declared.
REFERENCES
1.
3.
4.
5.
6.
7.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.