 Terminal deoxynucleotidyl transferase-mediated formation of protein binding polynucleotides
Terminal deoxynucleotidyl transferase-mediated formation of protein binding polynucleotides
Nucleic Acids Research



- Altmetric
Terminal deoxynucleotidyl transferase (TdT) enzyme plays an integral part in the V(D)J recombination, allowing for the huge diversity in expression of immunoglobulins and T-cell receptors within lymphocytes, through their unique ability to incorporate single nucleotides into oligonucleotides without the need of a template. The role played by TdT in lymphocytes precursors found in early vertebrates is not known. In this paper, we demonstrated a new screening method that utilises TdT to form libraries of variable sized (vsDNA) libraries of polynucleotides that displayed binding towards protein targets. The extent of binding and size distribution of each vsDNA library towards their respective protein target can be controlled through the alteration of different reaction conditions such as time of reaction, nucleotide ratio and initiator concentration raising the possibility for the rational design of aptamers prior to screening. The new approach, allows for the screening of aptamers based on size as well as sequence in a single round, which minimises PCR bias. We converted the protein bound sequences to dsDNA using rapid amplification of variable ends assays (RAVE) and sequenced them using next generation sequencing. The resultant aptamers demonstrated low nanomolar binding and high selectivity towards their respective targets.
INTRODUCTION
The adaptive immune system relies on intricate genetic variations to generate a wide range of antigen receptors in lymphocyte precursors (1). These precursors then mature into daughter cells containing one of these variant antigen receptors through somatic recombination. During maturation, these cells are screened based on their ability to selectively bind to cell antigens strongly and bind to their own cell antigens weakly. At the heart of the process of clonal selection in the development of lymphocytes, is the V(D)J recombination which involves DNA editing of the variable regions in both the exons of immunoglobulins and T-cell receptors of the developing lymphocyte (2). Terminal deoxynucleotidyl transferase (TDT) enzyme plays an important role in producing genetic variation in the variable region of the DNA by incorporating random nucleotides into the V, D and J exon regions of both B and T cells (3).
TdT is a unique polymerase enzyme as it is capable of catalysing the stepwise addition of random nucleotides without the need for a DNA template (4). As such there has been increasing interest in the use of TdT enzyme in the synthesis of oligonucleotides and DNA materials for supramolecular structures and for end labelling of DNA (5–7). There has also been a growing interest in using TdT in sensor development (8). Researchers recently proposed the use of TdT for the synthesis of high molecular weight polynucleotides and suggested that the TdT catalysed the poly-condensation of nucleotides via a living chain growth mechanism (9). This was demonstrated through the formation of polynucleotides with narrow size distributions owing to the fact that the polymerisation mixtures contained an initiator sequence composed entirely of a poly (T) sequence and dTTP nucleotides only, which reduces the probability of secondary structure formation in the elongating chain, which would otherwise inhibit the reaction. In contrast, the use of a mixture of dNTPs in TdT reactions can lead to DNA strands with broad size ranges due to different kinetic rates of incorporation observed for different dNTPs and the increased rate of secondary structures forming within the elongating DNA chains, which can also inhibit further chain growth. The size distributions of random sequences can also be significantly affected by varying both the time of reaction and the ratio of initiator to the dNTP monomer concentrations. Furthermore, the enzymatic properties of TdT can change when different divalent metal ions are used in each reaction (10). The presence of different divalent metal ions can lead to bias in the kinetics of incorporation of each nucleotide. Although TdT has been identified in a number of early vertebrates, the putative role of TdT before gene rearrangement is unclear (11). Previous studies have suggested that TdT was involved in DNA repair. TdT has also been identified as a phenotype immunological biomarker in the early onset acute lymphoblastic leukemia (12). The reason for this expression is suggested to be due to the hematopoietic immaturity of lymphoid cells (13). Since TdT is capable of producing ssDNA and RNA chains, and interactions of nucleic acid with proteins as can be found naturally in the form of transcription factors or toll like receptors, we therefore speculate as to whether nucleic acid/antigen interactions formed the basis of a common lymphocyte ancestor as a precursor to the adaptive immune system in vertebrates.
With this in mind, we set out to demonstrate whether TdT enzyme can impart protein antigen binding properties into nucleic acids, through the formation of diverse libraries of polynucleotides (aptamers). Through the development of a new non-evolutionary screening method, we demonstrated the in vitro formation of libraries of oligonucleotides using TdT, which display broad size distributions or variable sized DNA (vsDNA) and tailored apparent binding properties towards two model proteins respectively, namely human thrombin and human lactoferrin. Upon conversion of these sequences to dsDNA, and NGS analysis, identified individual polynucleotide candidates showed high binding and specificity to each target. The proposed non-evolutionary approach for the screening of protein binding aptamers allows for the rapid elution of protein binding polynucleotide candidates with increased avidity and the ability to tune the apparent binding when compared to classical evolutionary based screening methods, which rely on randomized selection. The use of TdT enzyme in the formation of the aptamer library also opens up the possibility for the development of larger polynucleotide based aptamers (<200 nt). The ability to tune each vsDNA library against a particular target is possible due to the ease of altering a number of conditions such as the ratios of each nucleotide and the initiator sequence in the pre-polymerization mixtures. Upon addition of TdT, we can also control the size distribution of the formed vsDNA libraries through varying the incubation time before termination of the reaction. This new method will ultimately allow us to move away from evolutionary based aptamer selection, which suffer from PCR bias and is both costly and time consuming. In addition, we discuss the the possibility that lymphocyte precursors containing TdT formed polynucleotide-based antigen receptors, which acted as the precursor for V(D)J recombination and the advent of immunoglobulin and T-cell receptors in the evolution of the adaptive immune system (14).
MATERIALS AND METHODS
Both the forward and reverse primers of sequences, 5′ATC AGT TCG AGC AGA TGA GC′3 and 5′CCA GAC TGC GAG CGT TTT TTT TTT-3′ respectively as well as a 10–100 nt oligonucleotide ladder, were purchased from IDT. Candidate polynucleotide sequences were also purchased from IDT using their Ultramer® technology including two scrambled sequences (SC01 and SC02), which correspond to the randomised sequence of the longest polynucleotide sequences tested. Terminal deoxynucleotidyl transferase (TdT), dNTPs, and human thrombin were purchased from Sigma Aldrich. SYBR gold stain was purchased from Thermo Scientific. 5x FIREPol® Master Mix and sample loading buffer were purchased from Solis Biodyne. The Buffer SB1: 50 mM Tris–HCl, 250 mM KCl and 7.5 mM MgCl2 pH 7.4 (5×) were used for all TdT catalysed library formation reactions and library selection steps. All oligonucleotide purification steps were performed using an oligonucleotide purification kit from Norgen Biotek. Purification of the PCR products was performed using a PCR clean-up kit purchased from Macherey-Nagel GmbH. All electrophoretic mobility shift assays (EMSA); (5–6%) and agarose gels (2%) were prepared in house. All PCR reactions were prepared in a dedicated laminar flow cabinet and all PCR experiments were performed on a T100 thermocycler (BioRad).
TdT mediated formation of ssDNA libraries
The TdT catalysed formation of the vsDNA libraries were achieved by preparing 400 μl solutions containing, 1–2 μM of the initiator sequence, 75–400 μM of dNTPs (dATP X μM, Y μM dCTP, Y μM dTTP and Z μM dGTP) in 1× SB1. Reactions were initiated by the addition of 1–2 U/μl of TdT and the entire mixture was allowed to incubate at room temperature for 0.5–2 h. Each reaction was terminated using 4 μl of 0.2 M EDTA or heat at 75°C for 10 min. The resultant libraries were purified and concentrated using the oligonucleotide clean-up kit (Machary Nagel) and eluted into 20–30 μl of elution buffer (5 mM Tris–HCl). The size range of the MIAs were monitored using a 5% denaturing acrylamide gel and visualised by staining with 1× SYBR gold stain.
Partitioning of thrombin and lactoferrin binding polynucleotides
A solution containing about 30 ng/μl of each resultant vsDNA library (7 μl) in 1× SB1 was refolded by heating the solution to 94°C and cooling at a rate of 0.5 °C s−1. The library was then incubated with 0.5 μM of human thrombin or 0.2 μM lactoferrin protein respectively for 1 h at room temperature. The resultant complex was then separated on a 5% native EMSA acrylamide gel. The DNA: protein complexes were visualised by staining with 1× SYBR gold stain. The complex band was extracted from the gel and transferred to a 0.5 ml tube punctured with a 20 gauge needle and placed into a 2 ml tube. The tube was centrifuged at 10 000 g for 10 min to crush the gel fragment. The resultant gel pieces were then incubated with 50 μl of nuclease free water at 37 °C for 2 h with lateral shaking. The liquid was transferred to the top of a filter tip and centrifuged again at 10 000 g for 10 min. The resultant solution was purified using the oligonucleotide clean-up kit (Machary Nagel) and eluted into 20–30 μl of elution buffer.
Rapid amplification of variable ends (RAVE)
In order to amplify and sequence the bound polynucleotides, A RAVE based assay was performed. A poly (A) tail was introduced at the 3′end of the polynucleotides using TdT tailing reaction. 20 μl solutions containing dATP, 10 mM Tris–HCl, 50 mM KCl and 1.5 mM MgCl2 buffer, the extracted bound polynucleotide sequences (10 μl) and TdT 1–2 U/μl were prepared and incubated for 0.5–2 h followed by termination of the reaction by heating the solution to 75°C for 10 min. The resultant product was used directly as the template in PCR. 20 μl solutions containing 1× master mix (10 μl), 0.1–0.5 μM (1 μl) of the forward and reverse primers and 1–5 μl of the DNA sequences with poly (A) template were prepared. PCR reactions were performed over 30–40 cycles consisting of a denaturing step at 94°C for 30 s, an annealing step at 45–65°C for 30 s and an extension step of 72°C for 30 s. The resultant dsDNA libraries were sequenced using a NovaSeq 6000 (Macrogen, South Korea). All sample libraries were prepared using an Illumina TruSeq DNA PCR free library construction (Insert 350 bp) prior to sequencing with a pairwise read.
Bioinformatic analysis of DNA libraries and binding affinity studies of candidate polynucleotide sequences
The sequences identified from NGS were analysed using MEME Suite to identify the binding motifs, UNAfold to determine the secondary structures, and G-mapper to predict their ability to form G-quadruplexes (15,16). Binding motifs Candidate sequences were chosen based on both their Gibbs free energy of folding, G-mapper score. Sequences were then resynthesized with a 5′ biotin tag and analysed for binding affinity using SPR. Binding motifs were identified by analysing the top 100 sequences by copy number using the MEME suite.
Binding affinity studies using surface plasmon resonance and EMSA
SPR studies were performed on a MP-SPR Navi™ 200 OTSO SPR instrument. Polynucleotide candidates were immobilized onto SPR chips prior to analysis. Firstly, a self-assembly monolayer was prepared by incubating bare gold chip into 5 mM of mercaptodecanoic acid 11-MUA into degassed ethanol for 24 h. The resultant chips were then washed using water and ethanol, dried using nitrogen and docked into the instrument. The instrument was primed with the run buffer SB1 prior to immobilization of the aptamer and the flow rate was set at 20 μl/min. For the immobilisation of the polynucleotide candidates, the chip surface was activated using an aqueous solution of mixture of NHS and EDC (80 μl). Streptavidin was injected (80 μl, 50 μg/ml) in 50 mM sodium acetate buffer pH 5.0 onto the activated chip surface. The unreacted activated ester groups were blocked by injecting 1M ethanolamine pH 8.3 (80 μl). 1 μM biotin tagged polynucleotides (80 μl) in SB1 were then injected on the analyte channel (channel 1), while scrambled polynucleotide sequences (SC01 for thrombin and SC02 for lactoferrin) were injected onto the reference channel (channel 2) respectively. Kinetic analysis was performed by priming the SPR instrument with 1× SB1 run buffer and setting the flow rate to 20 μl/min. 0–300 nM of each protein (80 μl) was injected on both channels and the relative response of the analyte after the removal of the response of the reference channel containing the corresponding scrambled sequence was recorded. The kinetic binding parameters were fitted against the relative response using either a 1 to 1 binding model or 2 to 1 binding model where two G-quadruplex motifs were predicted, using the trace drawer data analysis software. The binding affinities (KD) were determined from the association and dissociation rates where ka is the association rate and kd is the dissociation rate. All SPR experiments were performed in duplicate. The binding of LF02 and TH10T and SC01 and SC02 were also confirmed by a qualitative 5% EMSA using SYBR gold stain by titrating decreasing amounts of each protein against 1 μM of each aptamer sequence.
CD spectroscopy
Aptamers (10 μM) were suspended in SB1 and placed in 1 mm path length quartz cells. All CD spectroscopy experiments were performed on a J-1500 Circular Dichroism Spectrophotometer. An average of four scans from 310 to 210 nm were made at 100 nm min−1, with a 1 s response time and 0.1 nm bandwidth. The baseline signal was subtracted from each spectrum.
RESULTS
In this paper, we set out to develop a new non-evolutionary screening method for the elucidation of DNA sequences, which showed functional binding to proteins as described in Figure 1. The procedure involves generating a library of vsDNA using TdT. The resultant variable Size DNA (vsDNA) libraries were incubated with the target molecule and the bound vsDNA was separated from the unbound library using an electro mobility shift assay (EMSA). The bound vsDNA was converted to dsDNA and directly sequenced using next generation sequencing (NGS). Binding motifs were then identified and polynucleotide candidates were screened for binding affinities and specificity.

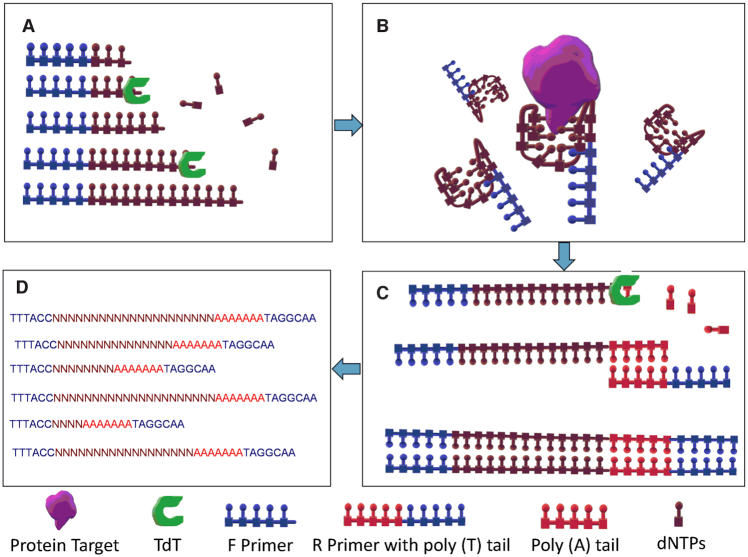
Overview of the selection size dependent single round selection of protein binding polynucleotide using vsDNA libraries; (A) TdT catalysed synthesis of a random ssDNA library in the absence of the target; (B) partitioning of protein bound DNA from unbound sequences; (C) TdT catalysed tailing reaction to incorporate a poly (A) tail and qPCR amplification to form a dsDNA and (D) next generation sequencing (NGS) to elucidate the sequences of the candidate polynucleotide sequences.
The synthesis of vsDNA libraries via TdT enzyme for the screening of protein binding polynucleotides
In this new method, we exploited the broad sizes distribution of polynucleotides in vsDNA libraries using TdT enzyme from pre-polymerization mixtures containing a mixture of dNTPs and the initiator sequence. Figure 2A shows that the size distributions of the resultant TdT catalysed vsDNA libraries correlate well with the time of reaction and allows us to easily control the size distributions of each library. These vsDNA libraries are characteristically visualised as a broad sized smear with the initiator sequence observed in excess. Libraries corresponding to a 30 min reaction resulted in a smear which falls within the range of 20–100 nt when compared to the ssDNA ladder while 1 and 2 h reactions resulted in libraries with much larger size distributions >100 nt. Both thrombin and lactoferrin were incubated with vsDNA mixtures respectively for at least 1 h in order to form stable vsDNA–target complexes. The extent of binding (apparent binding) of each vsDNA library against each target were analysed on a 5% EMSA native acrylamide gel stained with 1× SYBR gold along with the corresponding vsDNA libraries in the absence of thrombin (Figure 2B) and lactoferrin (Figure 2C) respectively. The EMSA gels also served as the partitioning method for separating complexes from the unbound library. The use of EMSA removes the necessity for a counter screening step against other closely related proteins. However, a counter screening step can be performed by incubating the vsDNA library with the protein conjugated stationary phase such as agarose beads or magnetic microspheres and retaining the unbound library.
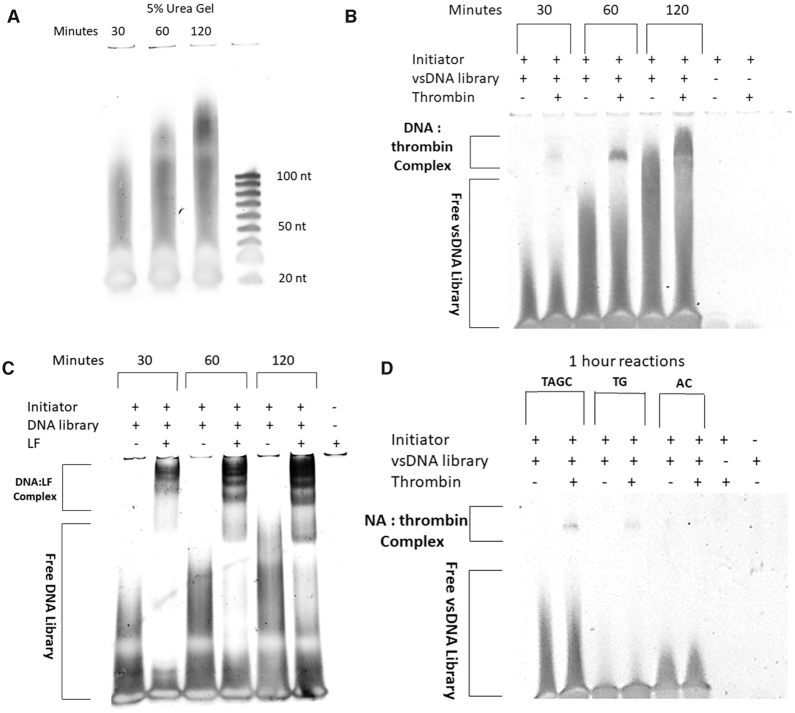
(A) 5% denaturing gel showing the effect of time on the size distribution of formed TdT catalysed vsDNA libraries; (B) 5% EMSA of each library incubated with 500 nM thrombin protein target at different reaction times; (C) 5% EMSA of each library incubated with 200 nM lactoferrin protein target at different reaction times and (D) the effect of nucleotide ratios on the formation of DNA:thrombin targets.
For both targets, the time of reaction correlated well with the intensity of the complex band, but with longer time periods, the resolution between the complex and unbound DNA library decreases as the size distribution of each library increases. Therefore, we selected 1 h as the time of reaction for all subsequent TdT catalysed reactions. To test our ability to tune the apparent binding affinities of our libraries against the targets, we formed vsDNA libraries, containing different mixtures of nucleotides. We synthesized libraries (Supplementary Table S1) containing the optimised TdT library containing ATCG (TVS1), TG (TVS2) and AC (TVS3) nucleotides respectively and incubated them with thrombin. Based on previous reports for the selection of thrombin binding aptamers, a 15 nt, composed entirely of G and T nucleotides and 29-mer DNA aptamer composed of a mixture of all four nucleotides were selected which bind to thrombin at two different binding sites respectively (17,18). These reported aptamers form G-quadruplex motifs upon binding to thrombin. To our surprise, a complex peak for both TVS1 and TVS2 containing just TG nucleotides and a mixture of ATCG nucleotides were observed while no complex band was observed for TVS3, consisting of AC nucleotides only (Figure 2D). Therefore, through control of time of reaction, primer to nucleotide ratios and the ratios of each individual nucleotide within our pre-polymerisation mixtures, we can tune the apparent binding of each library to a particular protein target and increase the fraction of bound DNA. This in turn allows us to narrow the range of sequences screened through pre-screening rational design of the vsDNA libraries. Optimised vsDNA libraries with a higher proportion of G and T were chosen for further screening against thrombin and vsDNA libraries containing a higher proportion of GT and C (LVS1) were used for the lactoferrin.
Next-generation sequencing of protein binding polynucleotide candidate sequences
After the extraction of the complexes bands from each EMSA gel and subsequent purification, the bound ssDNA was converted to dsDNA libraries and amplified for NGS analysis by adapting a rapid amplification of complmentary ends (RACE) assay (19).
This rapid amplification of variable ends assay (RAVE) involves firstly introducing A poly (A) tail to the 3′ end of the polynucleotide sequences via a TdT tailing reaction. This provided a template for the reverse primer during the PCR amplification step. The poly (A) tailing reactions were optimised by incubating the extracted libraries for both 30 min and 2 h reaction intervals. The successful incorporation of the poly (A) tail, amplification of the binding polynucleotide candidates and the optimal annealing temperature were confirmed by a 2% agarose gel (Figure 1S). PCR products for both the 30 min and 2 h incubation times resulted in the formation of the dsDNA PCR product although an increase in the yields of dsDNA was observed for the 2 h reaction. The 2% agarose gel showed a broad sized PCR product band corresponding between 100 and 250 bp respectively, which became more pronounced as less template and longer poly (A) tailing times were used. This suggests that we screened thrombin and lactoferrin binding candidate polynucelotides of a broad size range during the EMSA partitioning. The TdT tailing reaction further increases the overall size distribution of the DNA. In order to confirm this, we performed next generation sequencing (NGS) of the purified dsDNA products of the lactoferrin candidate sequences and the thrombin candidate sequences on a NovaSeq 6000. All sample libraries were prepared using an Illumina TruSeq DNA PCR free library construction (Insert 350bp).
Characterisation of the binding properties of polynucleotide candidate sequences
Candidate polynucleotide sequences for lactoferrin and thrombin screened from vsDNA libraries were analysed based on their binding motifs, size, secondary structure folding energies and G-quadruplex score. The binding motifs of both thrombin and lactoferrin based polynucleotides showed G-rich sequences suggesting that G-quadruplex motifs featured heavily in the binding motifs for both targets. For both thrombin and lactoferrin candidate pools (Figure 3A and B), the size distribution of sequences was wide, although low copy numbers were observed (Figure 3C and D). This may be due to the loss of some sequences, where the size of the Poly (A) tail is too large, meaning that the reverse read was cut to short before the random region could be properly sequenced or due to flipped sequences. Supplementary Table S2 and Supplementary Table S3 shows a list of polynucleotides sequences with the corresponding sizes and G-Score for both thrombin and lactoferrin. In some cases, the program identified more than one possible G-quadruplex motifs in the sequence, giving rise to the possibility of bivalent thrombin and sequences being found which correlates with previous attempts to engineer bivalent polynucleotides (20). The most promising candidate sequences for each target were chosen for further binding affinity studies using surface plasmon resonance (SPR). Sensorgrams for TH01, TH05, TH07 (Supplementary Figure S2A–C) and the truncated sequence TH10T (Figure 4A) were obtained through the injection of a range of concentrations (based on the preliminary KD measurements) for 240 s followed by 300 s of dissociation. The binding kinetics of each candidate polynucleotide sequence was determined from the normalised response of the analyte after subtraction of the response of the scrambled aptamer sequence reference channel and the sensorgrams were fitted using either 1:1 kinetic model or 2:1 kinetic model (Supplementary Figure S2D and Figure 4B and C). For thrombin binding, a number of sequences demonstrated low nanomolar binding using the 1:1 kinetic model, although minimal loss of binding affinity was observed when the sequences were truncated. The truncated lead sequence of TH10T showed the highest binding affinities with KD values of 8.5 nM, respectively. Both a 1:1 binding model and a 2:1 model were used for the kinetic analysis, as two possible binding sites on the polynucleotide for the thrombin were identified and demonstrated binding affinity values of 27 and 13 nM for each binding site. The binding of TH10T was further confirmed and compared to SC01 using a qualitative EMSA gel (Figure 4D and E). Thrombin showed a higher preference for binding to TH10T than SC01.
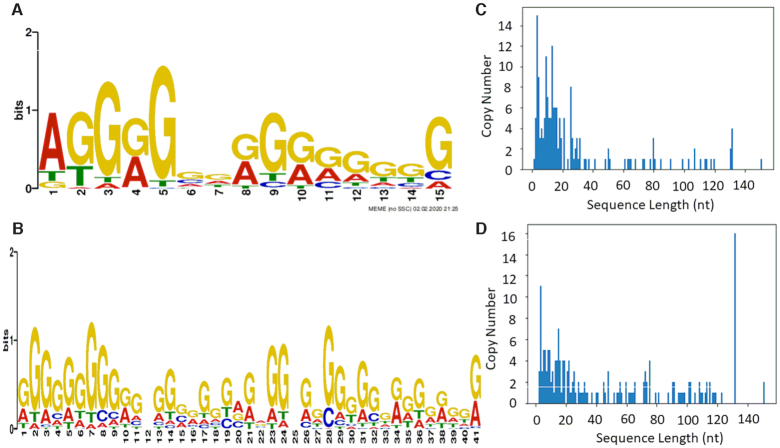
Summary of NGS data; MEME analysis of (A) sequenced thrombin polynucleotide candidates (E-value: 7.9e–28, 71 sites) and (B) lactoferrin polynucleotide candidates (E-value: 4.6e−71 128, 75 sites); (C) size distributions of random regions for sequenced thrombin sequences and (D) size distributions of random regions for sequenced lactoferrin sequences.
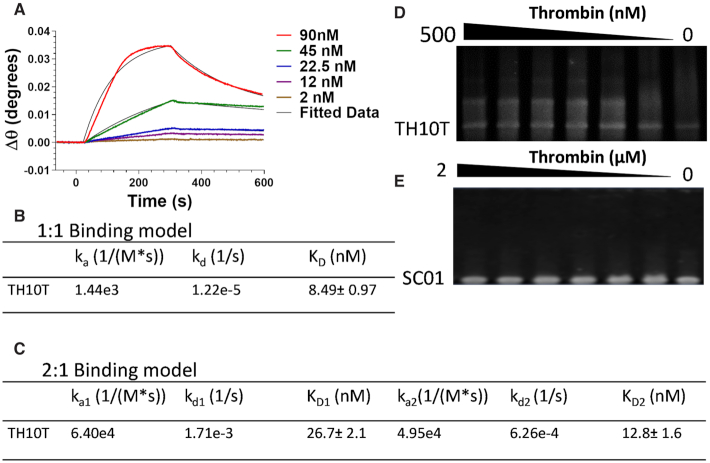
SPR Analysis of human thrombin binding towards (A) TH10T and (B, C) Kinetic parameters. 0–300 nM of each protein was injected on both channels (80 μl) and the relative response was determined by subtracting the reference channel response from the analyte channel. Run buffer: SB1, flow rate: 20 μl min−1. All SPR experiments were performed at room temperature and in duplicate; (D, E) Qualitative 5% EMSA of TH10T and SC01 towards Thrombin.
For lactoferrin, candidate sequences, sensorgrams for LF01, LF02, LF03, LF04 and LF07 (Supplementary Figure S3A–E) and the truncated sequence of the lead candidates LF02T (Figure 5A), LF03T and LF04T (Supplementary Figure S4A and B) were obtained in the same manner as described for thrombin. The truncated polynucleotides LF02T, LF03T and LF04T demonstrated low nanomolar binding with 1.4 nM, 5.5 and 5 nM respectively when fitted with a 1:1 kinetic model (Figure 5B-C). LF02T and LF04T were also fitted using a 2:1 model demonstrating binding affinities of KD1 of 30 pM and KD2 0.2 nM for LF02T and KD1 of 4.9 nM and KD1 8.3 nM for LF04T. These results confirmed comparable binding to typical antibodies raised against thrombin and lactoferrin. Candidates for both targets showed an increased avidity affect which may be due in large to the increase in size which agrees with previous reports on the dimerization of aptamers (21). The binding of LF02T was also assessed by EMSA and compared to SC02 (Figure 5D and E). Human lactoferrin showed a higher preference for binding to LF02T than for SC02.
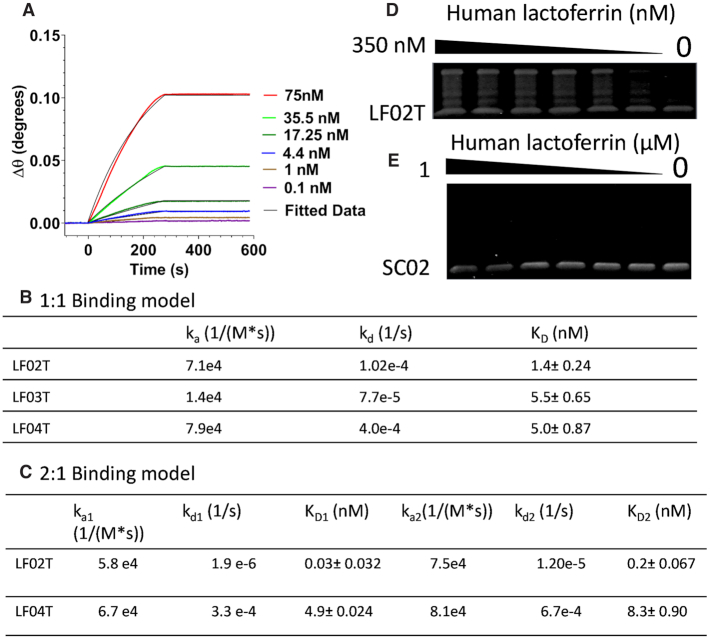
SPR Analysis of human lactoferrin binding towards (A) LF02T and (B, C) kinetic parameters of LF02T, LF03T and LF04T. 0–300 nM of each protein was injected on both channels (80 μl) and the relative response was determined by subtracting the reference channel response from the analyte channel. Run buffer: SB1, flow rate: 20 μl min−1. All SPR experiments were performed at room temperature and in duplicate; (D, E) Qualitative 5% EMSA of LF02T and SC02 towards human lactoferrin.
The lead candidate aptamer TH10T was tested for specificity using SPR. Concentrations of haemoglobin, fibrinogen and human serum album (HSA) were injected and sensorgrams were obtained based on the absolute SPR signal for both the analyte channel and reference channel (Supplementary Figure S5A-C). The data was fitted using a 1:1 binding model to obtain the binding affinities. Both haemoglobin and HSA demonstrated micromolar binding towards TH10T while fibrinogen showed a KD of 0.3 μM (Supplementary Figure S5D), suggesting that the inhibition effect of thrombin binding polynucleotides may be enhanced by inhibition of the substrate. The specificity of LF02T, LF03T and LF04T was tested against HSA as described for thrombin (Supplementary Figure S6A–C). HSA demonstrated micromolar binding towards LF02T, LF03T and LF04T and the corresponding reference channel containing SC02, confirming that high specificity for binding of each sequence towards human lactoferrin (Supplementary Figure S6D). Overall these results showed that protein binding polynucleotide sequences could be formed by TdT enzyme which displayed comparative binding performance to antibodies. CD spectroscopy studies were performed on the truncated aptamer sequences for both targets Figure 6A shows the CD spectra of the thrombin aptamer TH10T which showed a strong maxima signal at 265 nm characteristic of a parallel G-quadruplex. However, the broad size of the peak suggests the presence of a hybrid type G-quadruplexes containing both parallel and antiparallel G-quadruplexes as observed in some previous reports for the selection of thrombin binding aptamers (22,23). The CD spectra of LF02T, LF03T and LF04T (Figure 6B) also shows a maxima at about 265 nm although the asymmetric nature of the peak also suggests hybrid G-quadruplexes are present.
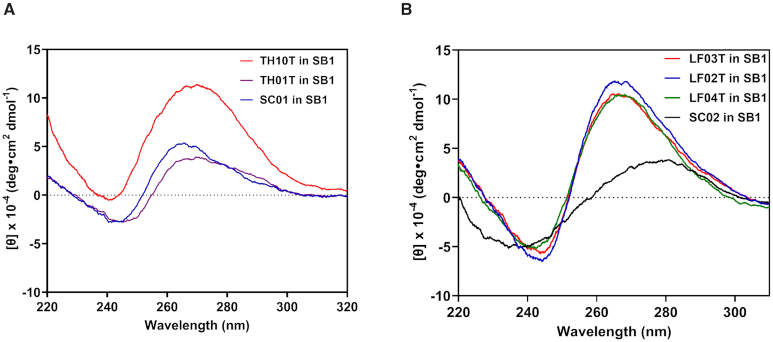
CD Spectra of (A) Thrombin binding aptamers and (B) Lactoferrin binding aptamers in SB1.
DISCUSSION
We demonstrated the use of TdT to form vsDNA libraries by incubating different mixtures of nucleotides, with an initiator sequence. The broad size distributions and nucleotide content of these libraries can be controlled based on a number of conditions including time of reaction, the ratio of the initiator sequence to the total nucleotide concentration and the ratios of each individual nucleotide. Previous in vitro selection studies for thrombin binding oligonucleotides from fixed length libraries have elucidated that final binding sequences bind to thrombin through the presence of G-quadruplex like motifs with the 19 nt TBA aptamer capable of binding to the exosite I site while HD22 binds to exosite II (17,18). This prior knowledge allowed us to rationally design vsDNA libraries before screening and increase stringency of screening of ssDNA sequences due to the ability to encourage certain G-quadruplex motifs through the addition of a higher proportion of dGTP and dTTP in our pre-polymerization mixtures. It is also worth noting that most successful protein binding oligonucleotides tend to have a greater proportion of guanosine bases, but since GC rich sequences tend to be difficult to amplify by PCR over several rounds of screening, these crucial sequences may be missed (24). Furthermore, the kinetics of polynucleotide formation via TdT also depends on divalent metal ion present in the buffers due to the difference in kinetic bias observed in TdT for the incorporation of nucleotides. In commercially available TdT kits, Co2+ is used as the divalent metal ion catalyst due to the increased kinetic properties incorporating dATP as poly(A) tailing. However, in this study, we chose Mg2+ metal ions due to the slower kinetics observed when compared to Co2+, which helped us gain better control of formation of our vsDNA libraries and tune the binding properties of each vsDNA library against each target respectively using EMSA.
We demonstrated that a significant population of vsDNA sequences could bind to both human thrombin and lactoferrin protein targets respectively. Through the use of EMSA gels, we were able to observe sequences binding both targets with a higher stringency compared to classical in vitro selection methods. This is due to the increased efficiency of our screening method, which screens both for the size of DNA strand and the sequence, and through the tuning of the apparent binding affinities. EMSA analysis also acts as our partitioning step prior to RAVE and NGS although several methods can be potentially used for this step. We demonstrated that the bound DNA can be converted to dsDNA for NGS by adapting the RACE assay. In order to achieve this we added a poly (A) tail to the 3′ end of the sequences and performed PCR. A broad size PCR product was observed in our gel, which is either a result of the broad sizes of bound DNA separated from the vsDNA library or the poly (A) tail. It is also worth noting that some primer dimer was formed. However, these sequences were subsequently eliminated during the NGS analysis.
Elucidation of the polynucleotide sequences showed a broad size distribution of sequences with high G-content due to the higher ratios of dGTP used in formation of the vsDNA libraries. Although, it's worth noting that a number of sequences were lost due to sequence switching. We used both mFold and QGRS Mapper web applications to determine the secondary motifs and 3D quadruplex motifs respectively. In comparison, to existing thrombin binding aptamers, there was very little similarity between our reported lead candidate TH10T. Although, smaller sequences show some similarities in containing TTGGTT. This may suggest that a large number of different G-quadruplex structures can actually bind to thrombin and binding may occur on the two different sites of thrombin or that PCR bias, may result in dominance of sequences with anti-parallel G-quadruplexes. Those sequences, which showed a highly negative free energy of folding and a high G-score, and represented a cross-section of different lengths of sequence were selected for further binding validation.
Candidate ssDNA sequences for both target proteins were resynthesized and validated for their ability to bind each protein target using surface plasmon resonance (SPR). Using this screening method, we can potentially screen for ssDNA sequences up to 200 nucleotides in length, which corresponds to maximum length of oligonucleotide that can be synthesized using IDT’s proprietary Ultramer® methodology (25). This is in comparison to fixed length libraries which are typically <100 nucleotides in length due to the phosphoramidite chemistry used to synthesize oligonucleotide. We demonstrated that both thrombin and human lactoferrin bound candidate polynucleotides with nanomolar binding which is comparable to the typical strength of interaction between antibodies and their corresponding antigens. In the case of the thrombin polynucleotides, TH10T was shown to have bivalent binding towards thrombin due to the two G-quadruplex motifs identified in the sequence, which correlates well with previous reports on the formation of bimolecular polynucleotides, although TH10T displayed a lower binding affinity towards thrombin (20,26). For Human lactoferrin the sequences LF02T, LF03T and LF04T all displayed binding in the low nanomolar range although, both a 2:1 model and 1:1 model were applied on the assumption that these sequences may also display bivalent interactions as observed with thrombin TH10T. The use of a 2:1 binding model was assumed to be independent, due to the non-equivalent binding sites on each aptamer ligand. To test the specificity of candidate sequences towards their perspective targets we measured the binding affinities of the sequences against a number of proteins found in blood plasma, or urine. In the case of thrombin, we chose haemoglobin, HSA and fibrinogen due to these proteins making up a significant proportion of proteins found in blood, while for lactoferrin, which is a biomarker for urinary tract infection, we used HSA as a major protein present in urine during urinary tract infection (UTI); (27). We found that for, TH10T, HSA and haemoglobin bound with a micromolar binding to TH10T while interestingly, fibrinogen demonstrated at least a 10-fold decrease in binding towards TH10T compared to thrombin. Furthermore, as the reference sequence displayed significant binding towards SC01, the absolute SPR signal was measured meaning that binding affinities reflected the total binding including the non-specific binding towards the sensor matrix. For human lactoferrin, HSA also demonstrated micromolar-binding affinities against LF02T, LF03T and LF04T and the corresponding SC02 sequence. Overall, these results confirm that TdT can be used to generate vsDNA libraries for screening towards protein antigen targets. It also confirms that the size of the DNA strand is a critical factor in the screening of oligonucleotides capable of binding protein targets in a non-evolutionary approach. It is also worth noting that this methodology can be adapted to other types of target such as small molecule targets and whole cells. In the case of small molecule targets, the use of TDT generated vsDNA libraries and lack of 3′ constant regions would be beneficial for increasing the likely hood of finding candidate aptamers with highly specific binding. In the case of whole cells, the use of this screening method can be adapted; if an appropriate partitioning, method is used. If needed, further rounds of screening can be performed either by ligating a constant region to 3′ end of the vsDNA library or by identifying the binding motifs adjacent to the 5’ initiator sequence and using this as the basis for the formation of new vsDNA library.
The demonstration of using TdT enzyme to form polynucleotides that can bind to proteins in vitro raises the abstraction that a precursor for clonal selection of antibodies and T-cell receptors in lymphocyte precursors may have been based on natural polynucleotide/antigen interactions prior to clonal selection of antibody receptors. Furthermore, the non-evolutionary manner in which we were able to generate oligonucleotides of different lengths via TdT and the significantly higher proportion of sequences, which were found to bind to the target may indicate that these precursor receptors could be formed using a mechanism similar to the so called shotgun immunity (1). However, the exact conditions for the generation of polynucleotide ligands in lymphocyte precursors remains to be determined. ssDNA and RNA sequences in the genome have been identified to bind naturally to targets which also strengthens the case for nucleotide based receptors (28,29).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Villum Fonden [00022912]; Marie Curie Co-Fund Action [713640]; NanoTRAINforGrowthII – INL Fellowship programme in nanotechnologies for nanomedicine, energy, ICT, food and environment applications; European Institute of Innovation & Technology (EIT) Health, project [20876] Funding for open access charge: European Institute of Innovation & Technology (EIT) Health, project [20876].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
15.
16.
17.
18.
20.
21.
22.
23.
24.
25.
26.
27.
28.