 Arabidopsis mTERF9 protein promotes chloroplast ribosomal assembly and translation by establishing ribonucleoprotein interactions in vivo
Arabidopsis mTERF9 protein promotes chloroplast ribosomal assembly and translation by establishing ribonucleoprotein interactions in vivo
Nucleic Acids Research



- Altmetric
The mitochondrial transcription termination factor proteins are nuclear-encoded nucleic acid binders defined by degenerate tandem helical-repeats of ∼30 amino acids. They are found in metazoans and plants where they localize in organelles. In higher plants, the mTERF family comprises ∼30 members and several of these have been linked to plant development and response to abiotic stress. However, knowledge of the molecular basis underlying these physiological effects is scarce. We show that the Arabidopsis mTERF9 protein promotes the accumulation of the 16S and 23S rRNAs in chloroplasts, and interacts predominantly with the 16S rRNA in vivo and in vitro. Furthermore, mTERF9 is found in large complexes containing ribosomes and polysomes in chloroplasts. The comprehensive analysis of mTERF9 in vivo protein interactome identified many subunits of the 70S ribosome whose assembly is compromised in the null mterf9 mutant, putative ribosome biogenesis factors and CPN60 chaperonins. Protein interaction assays in yeast revealed that mTERF9 directly interact with these proteins. Our data demonstrate that mTERF9 integrates protein-protein and protein-RNA interactions to promote chloroplast ribosomal assembly and translation. Besides extending our knowledge of mTERF functional repertoire in plants, these findings provide an important insight into the chloroplast ribosome biogenesis.
INTRODUCTION
The mitochondrial transcription termination factor (mTERF) proteins are tandem degenerate α-helical repeats proteins that are encoded by nuclear genomes of all eukaryotes except fungi (1). The mTERF family was named for its founding member, a human mitochondrial protein that promotes transcription termination in vitro (2). Each mTERF repeat spans ∼30 amino acids that fold into two consecutive antiparallel α-helices followed by a shorter α-helix perpendicular to the first one (3–5). The mTERF repeats stack together to form an elongated solenoid structure with a central groove capable of binding nucleic acids (5). mTERF proteins typically harbor an N-terminal organellar transit peptide and localize to mitochondria or chloroplasts and are considered to be putative organellar gene regulators (reviewed in 6). Whereas metazoans have three to four mTERF members, some plant genomes encode >30 mTERF proteins (1,7,8). The functions of mTERF proteins were first characterized in metazoans showing that they influence mitochondrial gene transcription, DNA replication and ribosome biogenesis (reviewed in 9,10). In plants, several of these genes are essential for embryo viability (11–13). Others have been linked to a variety of abiotic stress-responses (8,14–17) but how these genes trigger these responses in plants is not understood. Plant mTERFs are predicted to act in mitochondria or chloroplasts but knowledge about their roles in organelles is scarce. In fact, only five of the ∼30 mTERF proteins found in angiosperms have been connected to their gene targets and functions in organelles. In Arabidopsis, mTERF5 (also known as MDA1), mTERF6 and mTERF8 are chloroplast DNA binding proteins involved in the regulation of chloroplast gene transcription (18–20). mTERF5 stimulates the initiation of transcription of the psbE and ndhA genes (18,21), whereas mTERF8 and mTERF6 promote the termination of transcription of psbJ and rpoA, respectively (19,20). mTERF6 has additionally been reported to affect the maturation of trnI.2 but the reason for this effect remained unclear (22). Finally, mTERF15 and mTERF4 contribute to the RNA splicing of the nad2-3 intron in Arabidopsis mitochondria and group II introns in maize chloroplasts, respectively. Therefore, to date the functional repertoire of mTERFs in plant organelles concerns the regulation of gene transcription and intron splicing. In Arabidopsis, the mTERF9 gene (known as well as TWIRT1) encodes a chloroplastic mTERF protein that has been involved in the development of the shoot apical meristem (23) and the plant acclimation to high salinity (17,24) and photo-oxidative stress (25). However, the function of mTERF9 in chloroplasts was not further studied and the molecular basis underlying its physiological effects on plants is unknown. To answer this question, we examined the molecular defects in the mterf9 mutant and characterized the primary functions of mTERF9 in Arabidopsis. We show that mTERF9 is important for chloroplast ribosomal assembly and therefore, translation. We performed a comprehensive analysis of the RNA and proteins bound by mTERF9 in vivo and demonstrated its predominant interaction with the 16S rRNA and a large set of proteins required for the biogenesis of the small ribosomal subunit in chloroplasts. Our findings further reveal that mTERF9 can support direct interactions with both protein and RNA ligands which likely account for the protein function in the ribosomal assembly in vivo. Finally, we demonstrated that mTERF9 interacts physically with the CPN60 chaperonin complex in vivo suggesting a functional cooperation between these proteins in the chloroplast ribosome biogenesis and translation. This work expands the functional repertoire ascribed to plant mTERF proteins in translation and provides mechanistic insights into their in vivo functions in organellar gene expression.
MATERIALS AND METHODS
Oligonucleotides used in this study are listed in Supplementary Table S1.
Plant material
Arabidopsis thaliana ecotype Columbia (Col-0) and Nicotania benthamiana were used in this study. The T-DNA insertion mutant allele mterf9 (WiscDsLox474E07) was obtained from the ABRC Stock Center. Complemented mutants were obtained via Agrobacterium tumefaciens transformation of mterf9 homozygous plants. The binary vector (pGWB17) used for agro-transformation expressed the At5g55580 coding sequence in fusion with a 4xMyc C-terminal tag under the control of the CaMV 35S promoter. Transgenic plants were selected on Murashige and Skoog (MS) plates containing 25 μg/ml hygromycin. Experiments were performed using 7-day-old plants grown in vitro (1 × MS pH5.7, 0.5% sucrose, 0.8% Agar; 16 h light: 8 h dark cycles; 65–85 μmol photons m−2 s−1), 14-day-old plants grown on soil for chloroplast isolation or 4-week-old plants for protein pulse labelling experiments.
Subcellular localization of mTERF9
Nicotiana benthamiana leaves were infiltrated with Agrobacterium tumefaciens GV3101 carrying pMDC83:mTERF9 and pB7RWG2:RAP at an OD600 of 0.5 each. Protoplasts were prepared as described previously (26) and examined under a Zeiss LSM 780 confocal microscope. GFP was excited at 488 nm and emission was acquired between 493 and 556 nm. RFP and chlorophyll were excited at 561 nm and emissions were acquired between 588–641 and 671–754 nm, respectively.
Chlorophyll a fluorescence induction and light-Induced PSI absorbance changes
Chlorophyll a fluorescence induction kinetics and P700 absorbance changes at 820 nm were performed with leaves of 2-week-old WT, mterf9 mutant and complemented mutant plants grown on soil using a Dual-PAM-100 System (Walz, Effeltrich, Germany) (27). ΦPSI, ΦPSI NA and ΦPSI ND were expressed as described (28).
RNA analyses
Tissues were ground in liquid nitrogen and RNA was extracted with Trizol following manufacturer's protocol (Invitrogen™). RNA was further extracted with phenol-chloroform pH 4.3. Five microgram of Turbo DNase (Thermo Fisher) treated RNAs were used for Superscript IV reverse transcription with random hexamers. The resulting cDNA was diluted 20-fold for qPCR reaction. ACT2 (AT3G18780) and TIP41 (AT1G13440) were used as reference genes. For rRNA gel blotting, 0.5–1 μg of RNA (10 μg for other transcripts) was fractionated on 1.2% agarose–1% formaldehyde gel and blotted as described (29). Gene PCR products of 200–300 bp were labelled with 32P-dCTP following the prime-a-gene labeling kit instructions (Promega) and used as probes (Supplementary Data Set 2). Results were visualized on an Amersham Typhoon imager and data quantification was performed with ImageJ.
Protein analyses
For in vivo labeling of chloroplast proteins, leaf discs of Arabidopsis plants were incubated in 1 mM K2HPO4/KH2PO4 pH 6.3, 1% Tween-20, 20 μg/ml cycloheximide, 100 μCi 35S-methionine and vacuum infiltrated. Leaf discs were kept under light for 15 min, washed in water and frozen in liquid nitrogen. Proteins were extracted in Tris pH 7.5, 10% glycerol, 1% NP40, 5 mM EDTA, 2 mM EGTA, 35 mM β-mercaptoethanol, 1× EDTA-free protease inhibitor cocktail (Roche), and 200,000 cpm per sample were resolved on SDS-PAGE. After electrophoresis, the gel was stained in 50% methanol, 10% glacial acetic acid, 0.5 g/l Coomassie brilliant blue R-250 and vacuum dried before being exposed to a phosphorimager plate. Results were visualized on an Amersham Typhoon imager. For immunoblot analysis, total leaf proteins were extracted in the same buffer, resolved on SDS-PAGE and transferred onto PVDF membrane at 80 V for 1.5 h using the wet transfer. Anti-PsaD, -PetD and -RH3 antibodies were donations of Alice Barkan (University of Oregon). Anti-NdhL, -NdhB and -RbcL antibodies were donations of Toshiharu Shikanai (University of Kyoto) and Géraldine Bonnard (CNRS UPR2357), respectively. Other antibodies against chloroplast proteins were purchased from Agrisera and anti-Myc antibodies (clone 9E10) from Sigma-Aldrich.
Chloroplast isolation and fractionation
Chloroplasts were purified by density gradient and differential centrifugations as described previously (30). Chloroplasts were lysed in 30 mM HEPES–KOH pH 8, 10 mM Mg(OAc)2, 60 mM KOAc, 1 mM DTT, 1× EDTA-free protease inhibitor cocktail and 1 mM PMSF. Stromal (soluble) and thylakoid proteins were separated by centrifugation at 20 000 g for 10 min at 4°C.
Sucrose gradient fractionation
For the analysis of high-molecular-weight complexes by differential sedimentation, 0.25 mg of stromal proteins were fractionated on 10–30% linear sucrose gradient at 235 000 g for 4 h at 4°C as described (31). Proteins from each fraction were ethanol precipitated overnight at 4°C before their fractionation on SDS-PAGE. Polysome analyses were performed on leaf tissues as described (29). Briefly, 0.4 mg of leaf tissue was ground in 1 ml of cold polysome extraction buffer (200 mM Tris pH 9, 200 mM KCl, 35 mM MgCl2, 25 mM EGTA, 200 mM sucrose, 1% triton X-100, 2% polyoxyethylene-10-tridecyl ether, heparin 0.5 mg mL−1, 100 mM β-mercaptoethanol, 100 μg ml−1 chloramphenicol, 25 μg ml−1 cycloheximide) and the extract was cleared by filtration and centrifugation. Polysomes were treated or not with 500 μg ml−1 puromycin/500 mM KCl at 37°C for 10 min before adding 0.5% sodium deoxycholate. Insoluble material was pelleted at 16 000 g for 10 min at 4°C and soluble extracts were fractionated on linear 15–55% sucrose gradient at 235 000 g for 65 min at 4°C. For RNA isolation, 200 μl of sucrose gradient fraction was mixed with 400 μl of 8M guanidine–HCl to dissociate RNPs and RNAs were precipitated by the addition of 600 μl ethanol 100% and incubation at −20°C overnight. Proteins were precipitated as described above.
CoIP-MS
Two mg of stromal proteins treated or not with 100 μg/ml RNase A and 250 U/ml RNase T1 mix (Thermo Fisher) were diluted in one volume of Co-IP buffer (20 mM Tris pH 7.5, 150 mM NaCl, 15 mM MgCl2, 0.5 mM DTT, 3 mM ATP, 1 mM EDTA, 1% NP-40, 1× EDTA-free protease inhibitor cocktail, 1 mM PMSF) and incubated with 50 μl of anti-Myc Miltenyi magnetic beads at 4°C for 30 min on a rotator. Beads were washed in Co-IP buffer and eluted as recommended by the manufacturer. Eluted proteins were prepared as described (21,32). Briefly, proteins were precipitated overnight with 5 volumes of cold 0.1 M ammonium acetate in 100% methanol and digested with sequencing-grade trypsin (Promega) and each sample was analyzed by nanoLC–MS/MS on a QExactive+ mass spectrometer coupled to an EASY-nanoLC-1000 (Thermo Fisher). Data were searched against the Arabidopsis thaliana TAIR database with a decoy strategy (release TAIRv10, 27282 forward protein sequences). Peptides and proteins were identified with Mascot algorithm (version 2.5.1, Matrix Science, London, UK) and data were further imported into Proline v1.4 software (http://proline.profiproteomics.fr/). Proteins were validated on Mascot pretty rank equal to 1, and 1% FDR on both peptide spectrum matches (PSM score) and protein sets (Protein Set score). The total number of MS/MS fragmentation spectra was used to relatively quantify each protein (Spectral Count relative quantification). Proline was further used to align the Spectral Count values across all samples. The mass spectrometric data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository (33) with the dataset identifier PXD018987 and 10.6019/PXD018987.
For the statistical analysis of the co-immunoprecipitation proteomes, the mass-spectrometry data collected from three biological replicates of the experimental mTERF-Myc coIPs were compared to biological triplicates of control WT coIPs using RStudio v1.1.456 and the R package IPinquiry v1.2. The size factors used to scale samples were calculated according to the DESeq2 normalization method (34). EdgeR v3.14.0 and Stats v3.3.1 were used to perform a negative binomial test and calculate the fold changes and adjusted P-values corrected by Benjamini–Hochberg for each identified protein. The –log10(adj_P) and volcano plot graphs were calculated and drawn with Excel, respectively. The functional protein annotations were retrieved from the TAIR database (35) using the bulk data retrieval tool. The complete list of protein interactants and the number of peptides are provided in Supplementary Data Set 1.
Yeast two hybrid analysis
Coding sequences of mTERF9 or putative interacting partners were cloned into the bait vector pDHB1 or prey vector pPR3-N (Dualsystems Biotech) (36). The NMYZ51 yeast strain was co-transformed with bait and prey vectors using the PEG/LiOAc method (37). Co-transformants were selected on yeast synthetic and drop-out (DO) minus leucine (L) and tryptophan (W) agar medium. Positive colonies were sub-cultured in -WL DO liquid medium overnight. Overnight cultures were diluted to an OD600 of 0.3 to make the starting cultures and diluted by tenfold to 10−2. Five microliters of each dilution was plated on -WL DO agar medium or on DO medium minus leucine, tryptophan, histidine and adenine (-WLHA) supplemented with 3-aminotriazol to select protein interactions. 3-AT was used at concentrations of 1 mM to test mTERF9 interaction with ERA1 and mTERF9, 2 mM with PSRP2 and RPL1 and 40 mM for CPNB1 and CPNB3. The expression of bait and prey proteins in yeast were confirmed by immunoblotting on total yeast protein extracts. 5 ml of saturated yeast culture (OD600 = 3) was centrifuged at 800 g for 5 min. The pellet was resuspended in 200 μl of 2 M NaOH and incubated 10 min on ice. One volume of 50% TCA was added and the mixture was incubated for 2 h on ice and centrifuged 20 min at 16 000 g at 4°C. The pellet was dissolved in 200 μl of 5% SDS before adding 200 μl of protein loading buffer (25 mM Tris pH 6.8, 8 M UREA, 1 mM EDTA, 1% SDS, 700 mM β-mercaptoethanol, 10% glycerol) and incubated at 37°C for 15 min under agitation. Extracts were cleared by centrifugation for 5 min at 16 000 g at 20°C and the supernatant fractions were kept for immunoblot analysis. Ten microliters of protein fractions were resolved by SDS-PAGE and transferred to a PVDF membrane. Bait and prey proteins were immunodetected using antibodies against LexA and HA antibodies respectively, purchased from Sigma-Aldrich.
RNA immunopurification analysis
0.5 mg of stromal proteins were diluted in 450 μl of RIP buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1× EDTA-free protease inhibitor cocktail, 1 mM PMSF) and incubated with 50 μl of anti-MYC Miltenyi magnetic beads at 4°C for 30 min on a rotator. Beads were washed and eluted in RIP buffer supplemented with 1% SDS. Immunoprecipitated and supernatant RNAs were extracted with Trizol and further purified with phenol/chloroform. The RNA from the pellet and 3.5 μg RNA from the supernatant were fragmented and labeled with Cy5 (635 nm) and Cy3 (532 nm), respectively and hybridized on a tilling microarray (chip) covering the Arabidopsis chloroplast genome, as described in (38). Data were analyzed with GenePix Pro 7.0 software with local background subtraction method. The median of ratios of the background-subtracted pellet to supernatant signals were calculated and the super-ratios of the mTERF9 IP to control IP were plotted along the Arabidopsis chloroplast genome. RIP-chip data are provided in Supplementary Data Set 2. For qRT-PCR analysis, half of the input and IP RNAs were treated with Turbo DNase (Thermo Fisher) and cDNA synthesis and qPCR were conducted as described above.
Expression of recombinant mTERF9
The mTERF9 sequence coding for the mature mTERF9 (amino acids 45–496) lacking the chloroplast transit peptide was amplified by PCR on Arabidopsis cDNA and cloned into pMAL-TEV vector within BamHI and SalI restriction sites. The N-terminal MBP fusion protein (rmTERF9) was expressed in E. coli and purified by amylose affinity chromatography as described (21). The purity of the recombinant protein was visualized on SDS-PAGE and Coomassie Brilliant Blue staining. The band migrating at the expected size of rmTERF9 (∼96 kDa) and a comigrating band (∼60 kDa) were gel excised and analyzed by mass spectrometry (LC–MS/MS) to confirm their identity.
Northwestern blot analysis
Recombinant proteins were electrophoresed on a SDS-10% polyacrylamide gel and electroblotted to a PVDF membrane. After transfer, proteins were renatured by incubation of the membrane overnight at 4°C in renaturation buffer (100 mM Tris pH 7.5, 0.1% NP-40). Membranes were subsequently blocked for 10 min at 23°C in blocking buffer (10 mM Tris pH7.5, 5 mM Mg(OAc)2, 2 mM DTT, 5% BSA, 0.01% Triton X-100). Blocked membranes were hybridized for 4 h at 4°C in 5 ml of hybridization buffer (10 mM Tris pH7.5, 5 mM Mg(OAc)2, 2 mM DTT, 0.01% Triton X-100) containing 0.5 or 1 fmol of [α-32P]-labeled RNA probes. Membranes were washed 4 times in wash buffer (10 mM Tris pH 7.5, 5 mM Mg(OAc)2, 2 mM DTT) and exposed to a phosphorimager plate. Results were visualized on an Amersham Typhoon imager.
Accession numbers
The gene described in this article corresponds to the following Arabidopsis Genome Initiative code: At5g55580 (mTERF9). AGI codes of mTERF9 protein interactors can be found in Supplementary Data Set 1. The T-DNA mutant used was WiscDsLox474E07 (mterf9).
RESULTS
mTERF9 is a chloroplast nucleoid-associated protein required for plant growth
To characterize the molecular function of mTERF9, we analyzed the Arabidopsis mterf9 mutant that was previously reported to be affected in plant development (24). This mutant carries a T-DNA insertion in the fourth intron of the mTERF9/At5g55580 gene (Figure 1A). mTERF9 encodes a 496 amino acid protein harboring seven tandem mTERF motifs that are preceded by a predicted N-terminal chloroplast transit peptide (Figure 1A). We confirmed the mterf9 mutant phenotype at different developmental stages. mterf9 plants exhibited a pale leaf pigmentation and a slower growth phenotype compared to wild-type (WT), but remained fertile (Figure 1B). The introduction of a WT copy of the mTERF9 gene under the control of the CaMV 35S promoter into mterf9 fully restored the WT phenotype demonstrating that the mutant phenotype resulted from mTERF9 disruption. RT-PCR analysis confirmed the lack of mTERF9 full-length mRNA in mterf9 and its restoration in the complemented mterf9 plants (CP) (Figure 1C). The chlorotic phenotype displayed by mterf9 suggests a potential reduction of photosynthetic activity in the mutant. Therefore, the functional status of photosynthesis of the mutant was monitored using a pulse amplitude modulated system (Supplementary Table S2). In all respects, the complemented lines showed characteristics comparable to the WT. The mterf9 mutant displayed a decrease in photosystem II (PSII) activity as revealed by a reduced maximum quantum yield of PSII (0.70 versus 0,81; mterf9 versus WT) and an increased minimum fluorescence value (Fo) (Supplementary Table S2). Effective quantum yield of PSII measured in the steady state 5 min after induction was decreased from 0.73 in the WT to 0.58 in mterf9 whereas non-photochemical quenching was not affected. Overall, photosystem I (PSI) activity was reduced by one third in mutant plants as compared to the WT and no PSI donor side limitation could be detected. Instead, the quantum yield of non-photochemical energy dissipation due to PSI acceptor side limitation was reduced by about a half. The data indicate and confirmed a pleiotropic photosynthetic deficiency in the mterf9 mutant rather than a specific defect. To confirm the predicted chloroplast intracellular localization of mTERF9, we transiently co-expressed an mTERF9 protein fused to a C-terminal GFP with the chloroplast nucleoid-associated protein, RAP (39) fused to RFP in Nicotiana benthamiana leaves and examined leaf protoplasts by confocal microscopy (Figure 1D). The results revealed that the mTERF9-GFP fusion protein localizes to punctuated foci overlapping with the chloroplast chlorophyll autofluorescence and additionally, with the fluorescence of the nucleoid marker RAP-RFP as demonstrated by the degree of colocalization measured between the two fluorophores (Supplementary Figure S1). These results indicate that mTERF9 functions in chloroplasts of plant cells where it associates with the nucleoid.

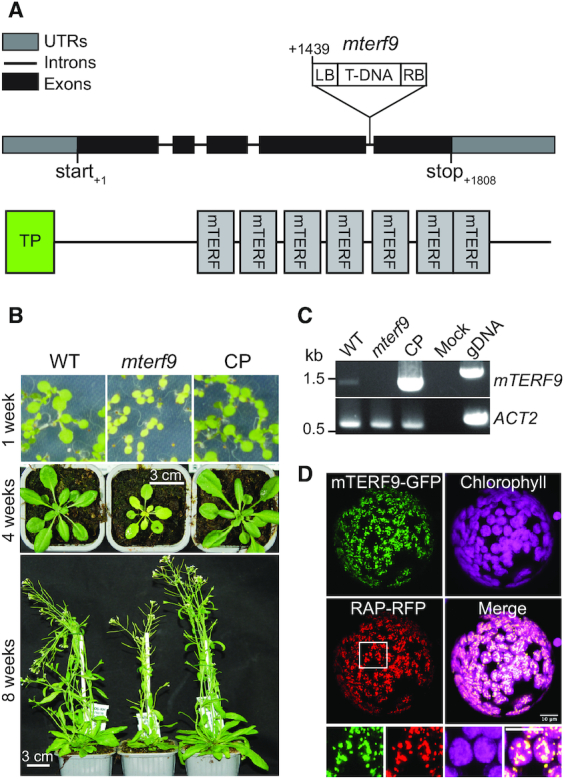
mTERF9 is a chloroplast nucleoid-associated protein required for plant development. (A) Schematic representation of the mTERF9 gene and protein with the position of the mterf9 T-DNA insertion. (B) Phenotypes of wild-type (WT), mterf9 and complemented (CP) plants grown in medium or soil at indicated growth stages. (C) RT-PCR analysis of mTERF9 expression in WT, mterf9 and complemented plants. Genomic DNA (gDNA) was used as positive control for PCR and ACTIN-2 (ACT2) serves as internal control for RT-PCR. (D) Subcellular localization of mTERF9-GFP and RAP-RFP fusion proteins in tobacco leaf protoplasts. Close-up views of the framed area are shown below. Scale bar: 5 μm.
mTERF9 deficiency impairs chloroplast protein accumulation and translation
The pale leaf and defective photosynthesis phenotypes displayed by the mterf9 mutant suggests an mTERF9 function related to chloroplast biogenesis. To investigate mTERF9 function in chloroplasts, we first analyzed the accumulation of representative subunits of chloroplast protein complexes by immunoblotting in the mutant. With the exception of the plastid-encoded RpoB and nuclear-encoded LHCB2, FBA, CPN60α1 and β1 proteins, the results showed a ∼50–75% decrease in the amount of chloroplast proteins tested in mterf9 compared to WT and CP plants (Figure 2A). We additionally confirmed the expression of the mTERF9 protein fused to a C-terminal 4xMyc tag in the CP plants and showed its dual-detection in both the stroma and membrane fractions of chloroplasts (Figure 2A and B). The global reduction of the amount of chloroplast protein complexes in mterf9 including the plastid ribosomal protein S1 (RPS1) suggests a possible defect in chloroplast translation and ribosome biogenesis. To confirm this, we investigated the de novo synthesis of chloroplast proteins by protein pulse-labeling with 35S-methionine. The results showed that the synthesis rates of RbcL and D1 proteins were lower in mterf9 with a respective ∼25 and 80% decrease relative to WT and CP plants, respectively (Figure 2C). Overall, the results indicated that the loss of mTERF9 activity impairs the accumulation of chloroplast proteins and translation.
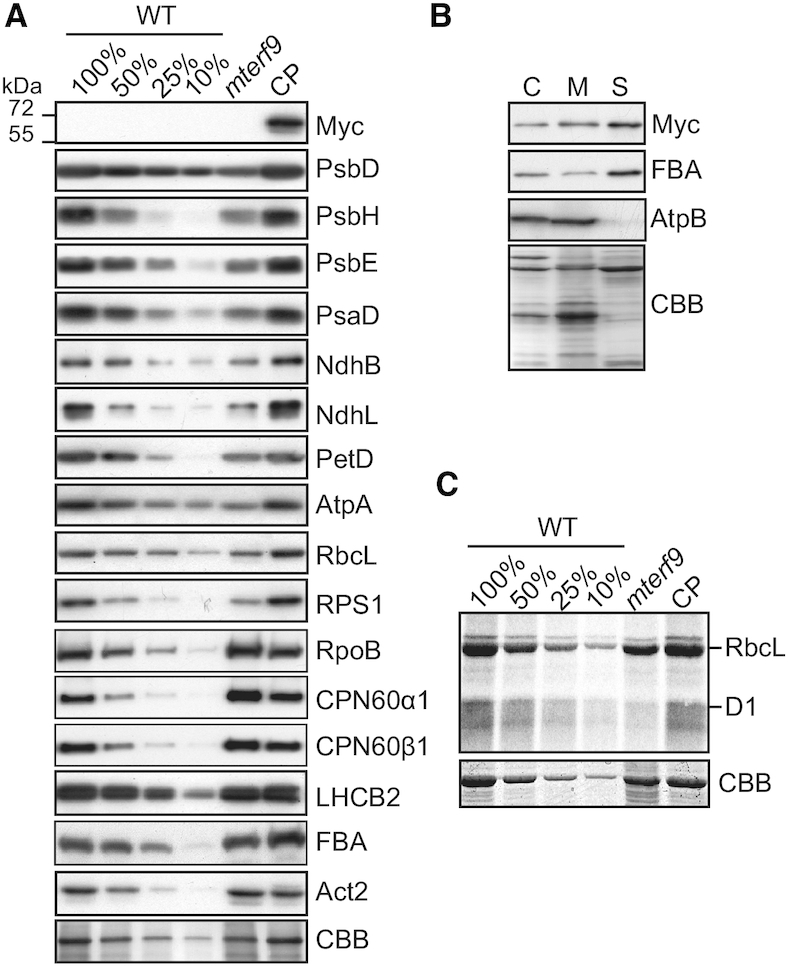
Chloroplast protein accumulation deficiency in mterf9. (A) Immunoblot analyses of leaf protein extracts with antibodies against mTERF9-Myc and subunits of the photosystem I (PsaD), photosystem II (PsbD, PsbH, PsbE,), Cytochrome b6f (PetD), NADH dehydrogenase-like (NdhB, NdhL), ATP synthase (AtpA), Rubisco (RbcL), light-harvesting complex II (LHCB2), chloroplast ribosome (RPS1), Plastid-encoded RNA polymerase (RpoB), chaperonin 60 (CPN60α1, CPN60β1) complexes and the stromal fructose-bisphosphate aldolase 1 enzyme (FBA). Immunoblots used 5 μg of leaf protein extracts and the corresponding dilutions except for NdhB and NdhL that used 15 μg of protein aliquots. A replicate membrane was stained with Coomassie Blue (CBB) to show equal protein loading. The PsbD, PsbH, PsbE, PsaD, NdhB, NdhL, PetD, AtpA, RbcL, RPS1 immunoblot images for the WT were previously reported (21) and are reproduced here with permission. (B) mTERF9 localizes to the stroma and chloroplast membranes. Isolated chloroplasts were lysed in hypotonic buffer and membrane and soluble protein fractions were separated by centrifugation. Chloroplast (C), soluble (S) and membrane (M) protein fractions were analyzed by immunoblotting using antibodies against Myc epitope, a stromal protein (FBA) and a membrane associated subunit of the ATP synthase complex (AtpB). The Coomassie Blue (CBB) stained membrane is shown. (C) In vivo chloroplast translation assays. Leaf discs from the indicated genotypes were pulsed-labeled with 35S-methionine and neosynthesized proteins were separated by SDS-PAGE and visualized by autoradiography. The CBB stained gel is shown below and serves as loading control.
mTERF9 deficiency causes reduced accumulation of the 16S and 23S rRNAs
Members of the mTERF family are predicted to control gene expression in organelles (6,9) and the loss of chloroplast translational activity in mterf9 can result from the altered expression of some chloroplast genes. To identify which genes were affected in mterf9, we measured chloroplast gene transcripts by qRT-PCR (Figure 3) and found that the steady-state levels of mRNAs were moderately increased (0 < log2FC < 2.5) or unchanged in the mterf9 mutant. The transcript overaccumulation in mterf9 was confirmed by northern blot analysis for selected genes using complementary probes against matK, ndhD, rbcL, ycf3 and rpoC1 transcripts (Supplementary Figure S2). In contrast, the 16S and 23S rRNAs, two RNA constituents of the chloroplast small and large ribosomal subunits were reduced compared to the WT and CP plants as observed by qRT-PCR (Figure 3). The rRNAs are unstable when not incorporated into the chloroplast ribosomal subunits and therefore, their reduction in mterf9 is indicative of a partial loss of chloroplast ribosomes content in the mutant. A global increase in the steady-state levels of chloroplast mRNAs has previously been reported in plants whose chloroplast translation is chemically or constitutively impaired (40,41). Therefore, the moderate increase of chloroplast transcripts in mterf9 is likely a secondary effect of reduced chloroplast translation. Some mTERF proteins have been involved in RNA intron splicing in plant organelles and a lack of splicing for some chloroplast genes can lead to translation impairment when these encode components that are important for the ribosome biogenesis. Thus, we additionally assayed the intron splicing efficiency for chloroplast genes in mterf9 relative to WT by qRT-PCR (Supplementary Figure S3). At the exception of a slight reduction for ycf3 intron 1, splicing was not significantly disrupted in mterf9. However, northern blot analyses using ycf3 strand specific probes were not consistent with the qRT-PCR results and showed very little, if any splicing defect in ycf3 intron 1 (Supplementary Figure S2). Instead, the northern blot results showed an overexpression of ycf3 pre-mRNAs as discussed previously. Therefore, neither the transcripts over-accumulation nor the intron splicing defects in mterf9 can explain the overall reduced accumulation of chloroplast proteins and translation in mterf9. By contrast, the observed decrease of the 16S and 23S rRNAs in mterf9 indicate that mTERF9 promotes the accumulation of chloroplast ribosomes, which is congruent with the global reduction of chloroplast-encoded proteins in mterf9.
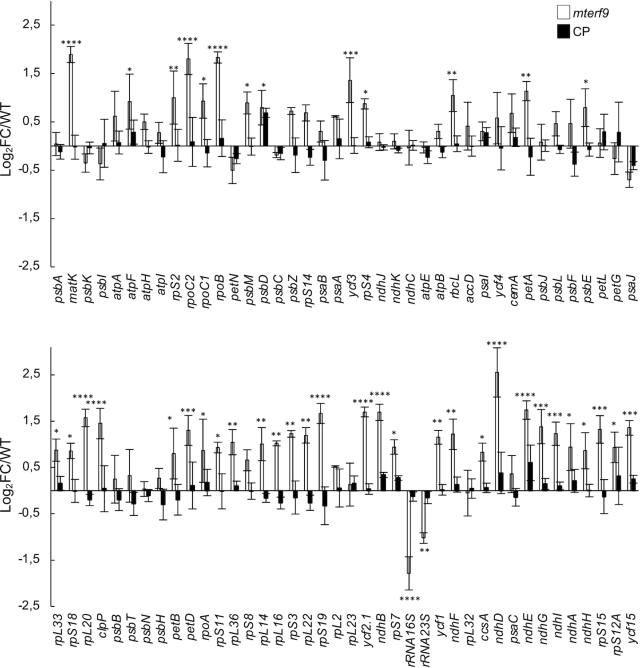
Steady state levels of chloroplast gene transcripts in Arabidopsis in mterf9 and CP plants. Transcript levels were determined by qRT-PCR and are displayed as the log2 fold change (FC) relative to WT for the mutant or the CP plants. Genes are ordered according to their genome positions. The nuclear ACT2 and TIP41 genes were used for data normalization. The values from three biological replicates performed each with technical triplicate were averaged per genotype and standard errors are indicated. ANOVA Dunnet's multiple test correction: *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.00005.
mTERF9 is required for the accumulation of 16S and 23S rRNAs
Chloroplast rRNA genes are organized in an operon and the 16S and 23S rRNAs are co-transcribed with the 4.5S and 5S rRNAs leading to RNA precursors that are subjected to a series of processing events (reviewed in 42) (Figure 4). For example, the 23S rRNA is internally fragmented at two ‘hidden breaks’, leading to the accumulation of seven distinct transcripts (Figure 4A). To further investigate and confirm the decrease of the 16S and 23S rRNAs in mterf9, RNA gel blot analyses were conducted in biological triplicates and signals were quantified (Figure 4 and Supplementary Figure S4) with probes designed to detect each rRNA and their processed forms (Figure 4A). The results confirmed the ∼50% reduction in the abundance of the processed 1.5 kb 16S rRNA in mterf9 compared to the WT or complemented plants (Figure 4B and C). RNA gel blot hybridization with three probes designed to detect the different fragments of the 23S rRNA revealed significant reduction of the 2.4, 1.3, 1.1 and 0.5 kb 23S rRNAs in mterf9 with a pronounced effect on the 2.4 kb isoform (∼60% reduction). In addition, the accumulation of the processed 4.5 and 5S rRNAs were not significantly affected in the mutant. The RNA gel blotting results confirmed the rRNA deficiencies in mterf9 and the importance of mTERF9 for the accumulation of the 16S and 23S rRNAs in vivo.
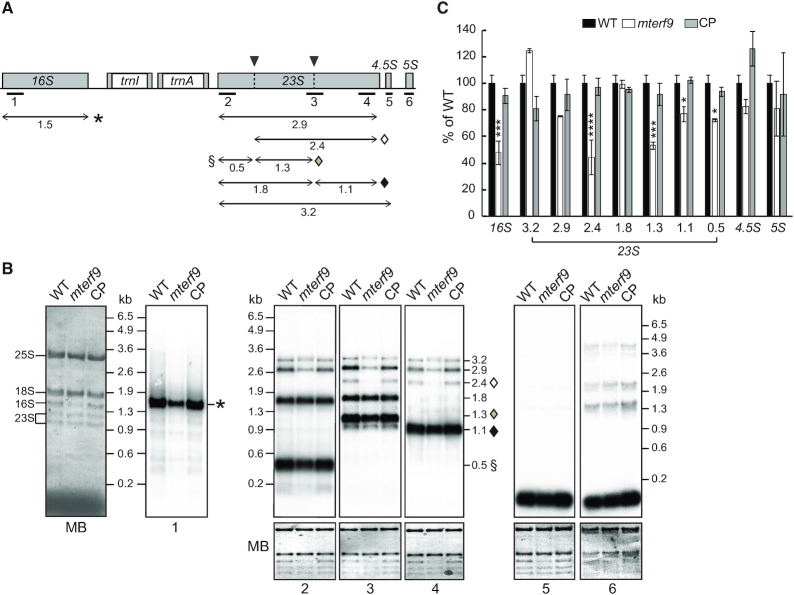
Defects in the 16S and 23S rRNA accumulation in mterf9 plants. (A) Schematic representation of the chloroplast rRNA operon. Exons and introns are represented by gray and white boxes, respectively. The positions of the probes used for RNA blot hybridization are indicated beneath the map. The 23S rRNA hidden breaks positions are shown above the gene with black arrowheads. The major accumulating transcripts for the 16S and 23S rRNA genes are mapped with arrows and their size is given in kb below. Transcripts specifically impaired in the mterf9 are indicated with symbols. (B) Total leaf RNA (0.5 μg) was analyzed by RNA gel blot hybridization using probes diagrammed in (A). An excerpt of the RNA membranes stained with methylene blue (MB) are shown to illustrate equal RNA loading, respectively. The positions of the cytosolic 25S and 18S rRNAs and chloroplastic 16S and 1.3 and 1.1 kb 23S rRNAs are indicated on the left side of the first MB stained membrane. (C) Relative quantification to WT in the accumulation of chloroplast rRNAs in mterf9 and CP plants. The average and standard error of three biological replicates is shown. Fisher's test: *P < 0.05; ***P < 0.0005; ****P < 0.00005.
mTERF9 associates with the ribosomal 30S subunit to promote ribosomal assembly and translation
The deficiency in the rRNAs accumulation in mterf9 points towards a reduction in the chloroplast ribosome content in the mutant and a possible defect in ribosomal assembly. The chloroplast 70S ribosome is composed of the small 30S and large 50S subunits that respectively contain the 16S and the 23S, 4.5S and 5S rRNAs. Preliminary immunoblotting analysis indicated a partial loss of RPS1, a protein of the 30S subunit (Figure 2A). We analyzed the sedimentation of the 30S and 50S ribosome subunits in mterf9, WT and CP plants by sucrose gradient sedimentation of stromal protein complexes (Figure 5A). The fractionation of the 30S and 50S ribosomal subunits on the gradient were monitored by immunoblotting using antibodies against RPS1, RPS7 and RPL33. In the WT and CP plants, RPL33 mostly sedimented in the last fractions of the gradient (fractions 10 to pellet), whereas RPS1 and RPS7 sedimented in the middle of the gradient (peak fractions 5 to 7 and 7 to 10, respectively). By contrast, in mterf9, RPL33, RPS1 and RPS7 sedimentation patterns were shifted to lower molecular-weight fractions, with a pronounced shifting for RPS1. These results demonstrate that the loss of mTERF9 function in Arabidopsis compromised the assembly of the chloroplast ribosome. Additional immunoblot analysis with an antibody against the Myc tag showed that mTERF9 co-sedimented predominantly with RPS1 and RPS7, indicating that it is found in particles of the same size than the 30S ribosomal subunit in chloroplasts. By contrast and in agreement with its in vivo association with the 50S ribosomal subunit (47), the chloroplastic RNA helicase, RH3 co-sedimented mostly with RPL33.
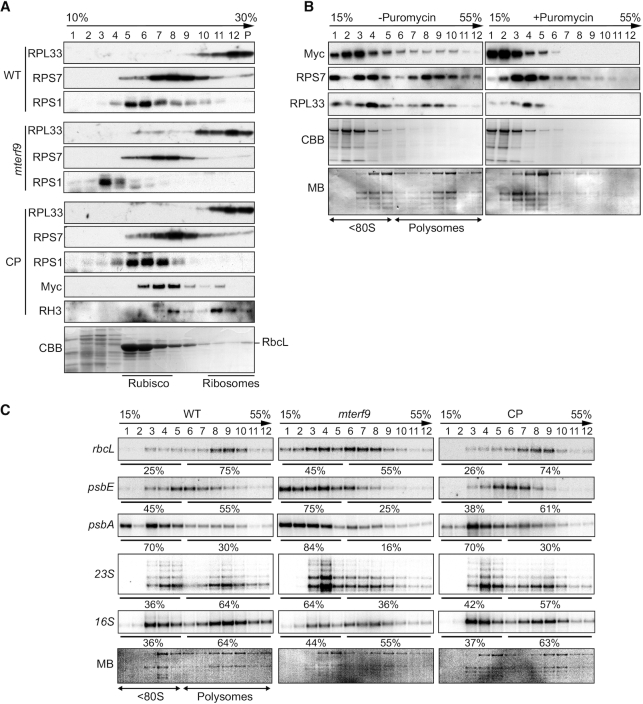
mTERF9 associates with chloroplast ribosomes and promotes mRNA association with ribosomes in vivo. (A) Sucrose gradient fractionation of stroma from the indicated genotypes. An equal volume of each fraction was analyzed by immunoblots with antibodies against ribosomal proteins from the small (RPS1, RPS7) and large subunit (RPL33), mTERF9 (Myc) and the chloroplast rRNA processing factor, DEAD box RNA helicase 3 (RH3) (47). The Coomassie blue-stained membrane (CBB) of the WT fractions is shown below. (B) Polysomal association of mTERF9. Leaf polysomes from complemented mterf9 plants were fractionated on 15–55% sucrose gradients. Fractions were analyzed by immunoblotting with antibodies against mTERF9 (Myc) and ribosomal proteins (RPS7 and RPL33) and RNA electrophoresis on denaturing agarose gel (bottom). The protein and RNA membranes stained with CBB and MB are shown, respectively. The sedimentation of the <80S ribosomes and polysomes on the gradient was confirmed with a puromycin control. (C) Polysome loading of selected chloroplast RNAs in WT, mterf9 and CP plants. Fractions from sucrose density gradients were analyzed by RNA gel blots using gene-specific probes. Signals in the polysomal (fractions 6–12) and monosomes/free RNA fractions (fractions 1–5) were quantified by phosphor-imaging and are displayed as percentage of the total signal over the 12 fractions.
We next determined whether mTERF9 associated with chloroplast ribosomes engaged in translation by polysome analysis from sucrose gradients (Figure 5B). The polysome-containing fractions were identified by immunodetection with the RPS7 and RPL33 antibodies and by visualization of the cytosolic rRNAs by RNA electrophoresis on a denaturing agarose gel. The polysomes were detected in fractions 6–12 and mTERF9 was detected in these fractions as well as in fractions containing monosomes and immature ribosomal particles (fractions 1–5). Treatment of the polysomes with the dissociating agent puromycin prior to their fractionation on sucrose gradient efficiently released mTERF9 from heavy to lighter complexes containing mostly monosomes or immature ribosomal particles, confirming the association of mTERF9 with the polysomes.
Finally, we analyzed the association of chloroplast mRNAs and the 16S and 23S rRNAs with polysomes in the WT, mterf9 and CP plants by sucrose density gradient fractionation and northern blot analyses (Figure 5C). As shown by the levels of mature 16S and 23S in polysomal fractions (fractions 6 to 12), mterf9 contained fewer polysomes than the WT and CP plants. In addition, the RNA gel blot results showed that the rbcL, psbE and psbA transcripts were partially shifted to the top of the gradient in mterf9 compared to WT, indicating that their loading to the polysomes and their translation efficiency were diminished in the mutant. These results correlate well with the lower rate of chloroplast protein synthesis that was observed in mterf9 (Figure 2C).
Altogether, the results show that mTERF9 promotes chloroplast ribosomal assembly and translation. mTERF9 primarily associates with the 30S subunit that assembles with the 50S to form the functional 70S chloroplast ribosome. In addition, mTERF9 association with the polysomes indicates that the protein plays a role during translation.
mTERF9 binds the 16S rRNA in vivo
mTERF proteins are nucleic acid binding proteins that have been predominately involved in DNA-related functions in organelles. However, this paradigm has recently shifted with the reports of two mTERF proteins involved in RNA intron splicing in plant organelles (43,44). The chloroplast rRNA defects in mterf9 and mTERF9 co-sedimentation with the ribosomes suggest that the protein target the rRNAs in vivo. To explore this possibility and identify mTERF9 RNA ligands in vivo, we performed genome-wide RNA co-immunoprecipitation assays (RIP). Stromal extracts from the CP and WT plants were subjected to immunoprecipitation with antibodies raised against the Myc tag and co-immunoprecipitated RNAs were identified by hybridization to tiling microarrays of the Arabidopsis chloroplast genome (RIP-chip) (Figure 6A). The results revealed a prominently enriched peak (>20-fold) in the mTERF9 immunoprecipitate that corresponds to the 16S rRNA and minor peaks (<10-fold) in the 23S, 4.5S and 5S rRNAs as well as atpH, psbC and psbE loci. To quantify mTERF9 binding to these RNA targets, we conducted an independent RIP experiment followed by qRT-PCR analysis of the immunoprecipitated RNAs (Figure 6B). The results confirmed that mTERF9 significantly binds to the 16S rRNAs and to a lesser extent the 23S rRNA. However, atpH, psbC and psbE were not significantly enriched in mTERF9 immunoprecipitate as compared to two negative control genes, rpoB or rbcL indicating that these targets were either false positives or unstable ligands (Figure 6B). Taken together, the results confirmed that mTERF9 primarily binds the 16S rRNA in vivo, which is consistent with its association with the small 30S ribosomal subunit.
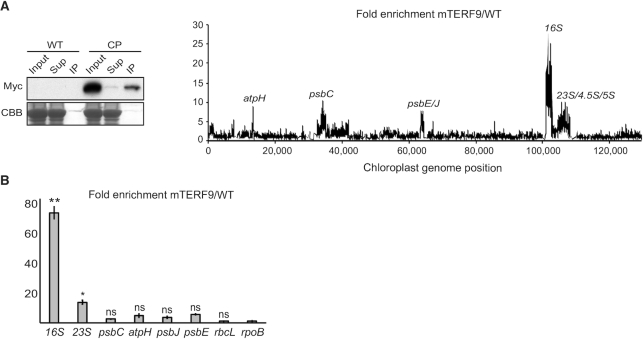
mTERF9 associates with the 16S rRNA in chloroplasts. (A) mTERF9 RNA ligands were identified by co-immunoprecipitation on stromal extract from the complemented mterf9 (CP) or wild-type (WT, negative control) with anti-Myc antibodies, followed by RNA hybridization on a chloroplast genome tiling microarray. The efficiency of mTERF9 immunoprecipitation was confirmed by immunoblot analysis with anti-Myc antibodies. Sup: supernatant, IP: immunoprecipitate. The enrichment ratios (ratio of signal in the immunoprecipitation pellet versus supernatant) are plotted according to position on the chloroplast genome after subtracting values obtained in the negative control immunoprecipitation (WT stroma). The RIP-chip assay revealed the predominant enrichment of the 16S rRNA in mTERF9 immunoprecipitate. (B) Validation of mTERF9 RNA in vivo ligands by qRT-PCR. The levels of immunoprecipitated RNAs were calculated as percent recovery of the total input RNA in control WT IP and mTERF9 IP in triplicate, and the average ratio and standard error are shown. Kruskal–Wallis test; *P < 0.05.
The mTERF9 protein interactome confirms its link to ribosome biogenesis
Our data demonstrated that mTERF9 is involved in ribosomal assembly and chloroplast translation. The recruitment of components of the ribosome biogenesis machinery to the 16S rRNA may be one of the key functions of an rRNA-interacting protein. To understand the protein interactome of mTERF9 in vivo and confirm its in vivo association with the chloroplast ribosome, we performed co-immunoprecipitation of untreated or RNase-treated stromal extracts in biological triplicates and, proteins from the immunoprecipitated fractions were identified by LC–MS/MS. The efficiency of mTERF9-Myc immunoprecipitation between the RNase-treated or untreated samples was similar, allowing a direct comparison of the results (Figure 7A). We identified 158 and 173 proteins significantly enriched by mTERF9-Myc precipitation (log2(FC) > 2 and adj_P < 0.05) in the –RNase and +RNase condition, respectively (Figure 7B and C). The enriched mTERF9-interacting proteins were classified in seven groups according to their functional annotations: ribosomal proteins of the small and large subunits (RPSs and RPLs), CPN60 chaperonins, rRNA processing/ translation factors, RNA binding proteins (RBPs), components of the transcriptional active chromosome (TACs) and finally, the category ‘others’ grouping chloroplast proteins with functions unrelated to gene expression and cytosolic protein contaminants (Figure 7C; Supplementary Data Set 1). Gene ontology term enrichment analyses revealed that the RNA-dependent and -independent protein interactants share over-represented molecular functions in ribosome biogenesis (Figure 7D). As an illustration, the in vivo fishing of mTERF9 in the absence of RNase treatment pulled down 20 out of the 24 proteins that constitute the small ribosome subunit and 27 out of the 33 ribosomal proteins composing the large subunit of chloroplasts (45). Nevertheless, the RNase treatment had differential effects on the accumulation of proteins in mTERF9 co-immunoprecipitates. The treatment reduced the number of chloroplast ribosomal proteins and in particular of the large subunit, rRNA processing/translation factors and TAC components, while it increased the number of RNA binding proteins and proteins from the category ‘others’ (Figure 7C). On contrary, all six subunits (CPN60α1–2, CPN60β1–4) of the chloroplast CPN60 chaperonin complex (46) were constantly retrieved in both conditions as most enriched proteins in mTERF9 co-immunoprecipitates (Figure 7B and E). In total, 92 proteins were commonly found in the untreated and RNase-treated co-immunoprecipitates (Figure 7E) suggesting that the majority of the mTERF9 interactants were not RNA-dependent but rather direct protein interactors. However, this does not exclude the possibility that the remaining co-immunoprecipitated proteins engaged in direct protein-protein interactions with mTERF9. Among the 92 common proteins, none were chloroplast RNA binding proteins, indicating that the RNase treatment efficiently destabilized ribonucleoprotein complexes and that their interaction with mTERF9 was RNA-dependent. Twelve proteins from the small and large ribosomal subunits were respectively enriched under both conditions along with six chloroplast rRNA processing and translation factors (Figure 7E and Supplementary Data Set 1). These include the following rRNA processing factors: RNA helicases, RH3 (47,48) and ISE2 (49), Ribonuclease J RNJ (50), RNA binding protein RHON1 (51) and the translation initiation and elongation factors FUG1 (52) and EF-Tu/SVR11 (53), respectively. Finally, ten TAC components co-immunoprecipitated with mTERF9 under both conditions. The TACs enrichment in mTERF9 co-immunoprecipitates was consistent with their co-localization to the nucleoids, a site known to play a major function in rRNA processing and ribosome assembly in chloroplasts (39,54,55). Interestingly, some ribosomal proteins, rRNA processing factors and RNA binding proteins were exclusively co-immunoprecipitated with mTERF9 by RNase treatment (Figure 7E and Supplementary Data Set 1). The RNase-dependency of these interactors revealed that their interaction with mTERF9 occurred upon mTERF9 dissociation from ribosomal nucleoprotein complexes which suggests that mTERF9 can interact with ribosomal proteins and rRNA processing factors in a spatial and sequential order during the assembly/disassembly of the ribosome subunits in vivo.
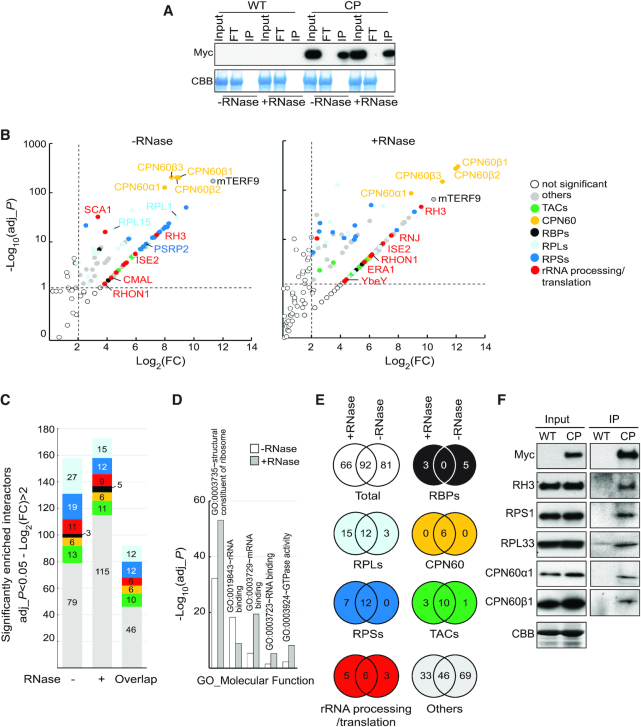
mTERF9 protein interactome is highly enriched with proteins involved in chloroplast ribosome biogenesis. (A) mTERF9 immunoprecipitation. Untreated or RNase-treated stroma extracts from complemented mterf9 (CP) or wild-type (WT) plants were used for immunoprecipitation with anti-Myc antibody. The input, flow-through (FT) and immunoprecipitate (IP) fractions were analyzed by immunoblot with anti-Myc antibody. A portion of the Coomassie blue-stained membrane (CBB) showing the abundance of RbcL serves as loading control. (B) Volcano plots showing the enrichment of proteins co-purified with mTERF9 and identified by mass spectrometry in absence or presence of RNase in comparison with control IPs. IPs were performed on biological triplicate. Y- and X-axis display log10 scale of –log10 adjusted P-values (adj_P) and log2 fold changes (FC) of proteins, respectively. The dashed lines indicate the threshold above which proteins were significantly enriched (P-value < 0.05 and FC > 4). Proteins are color-shaded according to their functional group and the color key is provided to the right. The full lists of mTERF9-associated proteins and their Arabidopsis locus identifiers are available in Supplementary Data Set 1. (C) Bar chart showing the number of significant mTERF9 interacting proteins in the functional groups. The same color code than in (B) is used. The ‘overlap’ bar represents common proteins found in mTERF9 protein interactomes in absence of presence of RNase. (D) Bar chart depicting the functional analysis of the mTERF9 protein interactomes and showing the five terms contained in the top functional annotation cluster identified by DAVID gene analysis online tool using the default parameters (87). GO terms are plotted according to −log10 of their respective adjusted P-values. (E) Venn diagrams showing the significantly enriched proteins in each functional category in mTERF9 immunoprecipitates. (F) Immunoblot validation of mTERF9 interactants identified by co-IP/MS analysis in absence of RNase. Replicate blots were probed with anti-Myc, anti-RH3, anti-CPN60α1/β1, anti-RPS1 and anti-RPL33. A replicate of a CBB-stained membrane is shown as input loading control.
To validate the mTERF9 interactome and its link with ribosome biogenesis, we performed immunoblot analyses of untreated mTERF9 co-immunoprecipitates and confirmed mTERF9 interaction with RH3, a DEAD box RNA helicase involved in rRNA processing (47), RPL33, RPS1 and the two chloroplast chaperonins CPN60α1 and β1 (Figure 7F).
In summary, the mTERF9 protein interactome is in agreement with the function of mTERF9 in chloroplast ribosome assembly. Moreover, the results demonstrate that mTERF9 protein supports protein-protein interaction during ribosome assembly besides its association with the 16S rRNA. Finally, the striking interaction of CPN60 chaperonins with mTERF9 in vivo points towards the potential implication of the CPN60 complex in chloroplast translation.
mTERF9 supports direct protein-protein and protein-RNA interactions
mTERF-repeat proteins are considered to be putative nucleic acid binders and we indeed showed that mTERF9 interacts with the 16S rRNA in vivo. Moreover, our co-immunoprecipitation assays showed that mTERF9 associates in vivo with many proteins that are involved in chloroplast ribosome biogenesis including ribosomal proteins, rRNA processing factors, and unexpected chaperonins from the CPN60 family. Some of these interactions appeared to be RNase insensitive. Together, the protein– and RNA–mTERF9 interactomes indicate that mTERF9 could support both RNA–protein and protein–protein interactions. To test the first possibility, we used mTERF9 as a bait in a modified yeast two-hybrid assay based on split-ubiquitin, called ‘DUAL hunter’ (36). As mTERF9 and many ribosomal proteins partially associate to chloroplast membranes (56,57), this system offered the flexibility to select both membrane and cytosolic protein interactions. We tested the physical interaction of mTERF9 with nine protein candidates that co-immunoprecipitated with mTERF9 in the –RNase or +RNase condition only or in both conditions (Figure 8 and Supplementary Figure S5A). The expression of the mTERF9 bait and the nine prey proteins in the yeast co-transformants were verified by immunoblotting with anti-LexA and anti-HA antibodies, respectively (Supplementary Figure S5B). Out of the nine candidates tested, mTERF9 interacted with five proteins (Figure 8B). These included ERA1, the Arabidopsis ortholog of the bacterial YqeH/ERA assembly factor for the 30S ribosomal subunit (58,59), PSRP2 and RPL1, two proteins of the 30S and 50S ribosomal subunits (60), respectively, and finally, CPN60β1 and β3, two subunits of the CPN60 chaperonin complex (61). These results demonstrated that mTERF9 can directly interact with proteins. The facts that mTERF9 interacts physically with ERA1, a protein that was specifically co-immunoprecipitated by the RNase treatment and with PSRP2 and RPL1 whose in vivo association with mTERF9 was rather sensitive to RNase, reinforced the notion that mTERF9 is likely to sequentially engage in various protein interactions during chloroplast ribosomal assembly and that the rRNAs likely stabilize some of these interactions. The physical interactions between mTERF9 and CPN60 chaperonins revealed that mTERF9 might be a substrate of the CPN60 complex. Alternatively, mTERF9 might recruit the CPN60 complex to ribosomal complexes to assist folding of ribosomal proteins during subunits assembly or neosynthesized proteins during translation. Finally, we demonstrated that mTERF9 had the capacity to self-interact in yeast and that the protein oligomerization likely depends on the mTERF repeats since their truncation abolished the interaction (Figure 8B and Supplementary Figure S5).
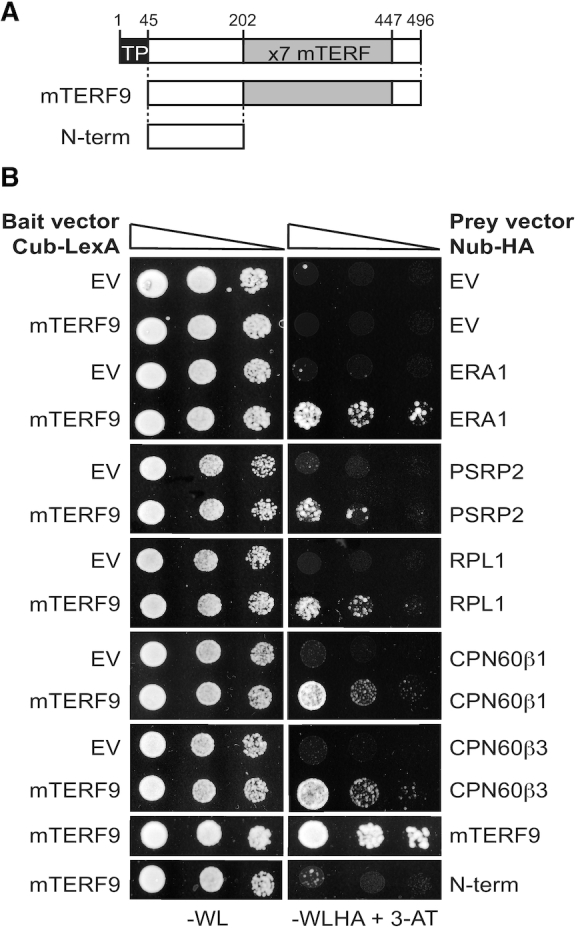
mTERF9 directly interacts with some of its in vivo protein interactants. (A) Schematic representation of mTERF9 used as bait or prey in the yeast two hybrid assay. (B) The yeast two hybrid assay was applied to assess direct interactions of mTERF9 with proteins identified by co-IP/MS analysis and mTERF9 self-association. mTERF9 interacts in yeast with ERA1, a putative 30S ribosomal subunit assembly factor, PSRP2 and RPL1, two plastid ribosomal proteins of the small and large ribosome subunits, CPN60β1 and β3, two subunits of the CPN60 chaperonin complex and finally itself. The bait vector expressing mTERF9 in fusion with the C-terminal half of the ubiquitin and the transcription factor LexA (Cub-LexA) was co-transformed with prey vectors expressing the protein candidates fused to the N-terminal half of the ubiquitin and HA tag (Nub-HA) in a yeast reporter strain. Yeast co-transformants were spotted in 10-fold serial dilutions on plates without Trp, Leu (–WL). Positive interactions allow growth on plates without Trp, Leu, His, Ade in presence of 3-aminotriazol (–WLHA + 3-AT). Negative controls were performed using bait or prey empty vectors (EV).
In a second time, we tested the capacity of mTERF9 to directly bind the 16S rRNA using in vitro protein-RNA interaction assays. To this end, we expressed and purified E. coli recombinant mTERF9 (rmTERF9) fused to a maltose-binding protein (MBP) tag. The purity of the soluble rmTERF9 was visualized on SDS-PAGE and Coomassie Brilliant Blue staining (Figure 9A). A band migrated at the protein expected size (∼96 kDa) but despite several attempts to optimize the purity of rmTERF9, a protein contaminant of ∼60 kDa constantly copurified with rmTERF9. LC-MS/MS analysis confirmed the identity of rmTERF9 in the ∼96 kDa band and identified E. coli GroEL, the ortholog of the Arabidopsis chloroplast CPN60 chaperonins, as the 60 kDa protein contaminant (Figure 9A). This result provides additional evidence for the direct interaction between mTERF9 and the CPN60 chaperonin complex. The specific interaction of rmTERF9 with the 16S rRNA was analyzed by northwestern blotting. This assay detects direct interaction between RNA and proteins that are immobilized on a membrane after their resolution by gel electrophoresis according to their charge and size (62,63), allowing the specific detection of rmTERF9 activity. The binding of mTERF9 to its in vivo target, the 16S rRNA was compared to that for a chloroplast RNA of similar size from the psbC gene (Figure 9B and C). An interaction was detected between rmTERF9 and the 16S rRNA but no binding activity was observed for GroEL nor the purified MBP (Figure 9C), indicating that it is mTERF9 moiety that harbors the RNA binding activity. In addition, no RNA binding activity could be detected for rMDA1, a DNA-binding mTERF protein, that promotes transcription in Arabidopsis (21). In contrast to the 16S rRNA, at similar protein amounts, only residual binding was observed for the psbC RNA confirming that mTERF9 preferentially interacts with the 16S rRNA. The 16S rRNA is predicted to fold into four distinct domains (64,65) (Figure 9D) and we further explored mTERF9 binding specificity for each of the subdomains (Figure 9C). A binding activity was detected for the 16S rRNA domains I, II and III but not for domain IV. At similar protein amounts, the binding of rmTERF9 was higher for domains I and II than for domain III suggesting its binding preference for these two RNA segments. In addition, rmTERF9 showed minimal binding to an unrelated atpH transcript of similar size than domains I-III confirming the protein specific binding towards the 16S rRNA. Altogether, our results demonstrated that rmTERF9 is an RNA binding protein that preferentially binds to the 16S rRNA, which is consistent with the in vivo data and the protein association with the 30S ribosomal subunit.
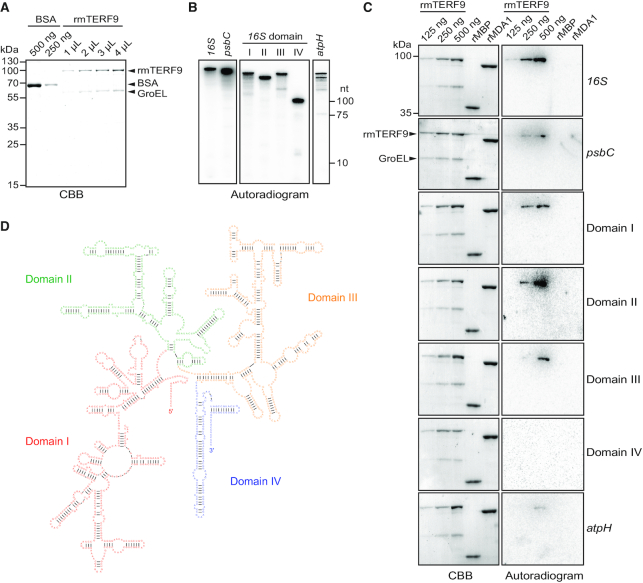
mTERF9 is an RNA binding protein that preferentially interacts with the 16S rRNA in vitro. (A) Purification of rmTERF9. Increasing volumes of the purified rmTERF9 fraction were analyzed along with a BSA standard by SDS-PAGE and staining with CBB. (B) One μl of radiolabeled in vitro transcribed RNAs used in the northwestern assays were electrophoresed on a 7.5% denaturing polyacrylamide gel and autoradiographed. The RNA sizes are: 16S rRNA, 1490 nt; psbC, 1422 nt; domain I, 508 nt; II, 356 nt; III, 478 nt; IV, 148 nt and atpH, 400 nt. (C) RNA binding activity of rmTERF9. The direct interaction between rmTERF9 and RNAs was tested by northwestern blotting. Increasing amount of rmTERF9 were resolved by SDS-PAGE and transferred to a PVDF membrane before hybridization with 0.5 fmole of radiolabeled RNA for 16S and psbC or 1 fmol for domains I–V and atpH. rMBP and rMDA1 (500 ng each) were included as negative controls to show mTERF9 specific RNA binding activity. CBB stained membranes and autoradiograms are shown to the left and right, respectively. (D) Secondary structure of the Arabidopsis 16S rRNA. The domains of the 16S rRNA are labelled and delineated in color. The structure prediction is based on the secondary structure of bacterial 16S rRNA (65) and was obtained from the RNACentral database (88).
DISCUSSION
mTERF9 assists chloroplast ribosome assembly via ribonucleoprotein interactions
We demonstrated in this study that mTERF9/TWIRT1, a member of the mTERF family of transcriptional factors in Arabidopsis has an unexpected function in chloroplast ribosome biogenesis and translation. Our study extends the current functional repertoire of mTERF proteins in plants in a process unrelated to DNA metabolism. We found that the mterf9 knock-out line is defective in chloroplast translation as a result of the reduced accumulation of the 16S and 23S rRNAs, two scaffolding components of the 30S small and 50S large subunits of the chloroplast ribosome, respectively. The decrease of these rRNAs is intricately linked to the reduced assembly of functional chloroplast 70S ribosomes in mterf9. In fact, similar to bacteria, ribosome assembly in chloroplasts is tightly connected to the post-transcriptional maturation of rRNAs (66). For example, the orchestrated assembly of the 50S ribosomal proteins on the 23S rRNA precursor in plants is believed to expose the RNA to endonucleases at particular cleavage sites and to generate the two hidden breaks in the 23S rRNA. Consistent with that, the stability of several isoforms of the 23S rRNA resulting from the hidden breaks processing were impaired in mterf9. mTERF9 associates primarily with the 16S rRNA in vivo and its accumulation is more severely affected than that of the 23S rRNA in the mterf9 mutant. Therefore, the 23S rRNA processing defect in mterf9 might be a secondary effect of the loss of mTERF9 function in the stabilization of the 16S rRNA in chloroplasts. Several auxiliary factors involved in chloroplast ribosomal assembly have been recently characterized and the majority of these are bacterial homologs or harbor RNA binding domains that are conserved in bacteria (47–51,55,67–69). Without any surprise, these protein homologs perform conserved functions in rRNA processing and therefore, ribosome assembly in chloroplasts. On the contrary, mTERF9 belongs to a eukaryote-specific transcription factors family and its function in chloroplast ribosome assembly was unexpected.
To firmly establish the in vivo function of mTERF9, we performed a comprehensive analysis of the in vivo RNA and protein interaction networks of mTERF9. Our co-immunoprecipitation results demonstrated that mTERF9 binds to the 16S rRNA in chloroplasts as well as ribosomal proteins, CPN60 chaperonins and known auxiliary ribosomal factors involved in rRNA processing such as the MraW-like 16S rRNA methyltransferase (CMAL) (68), YbeY endoribonuclease (67), RNase J (50), RNase E-like protein (RHON1) (51) or DEAD/DEAH-box RNA helicases (RH3 and ISE2) (47–49). The fractionation of chloroplast high-molecular-weight protein complexes combined with the comparative mTERF9 protein interactome in presence or absence of RNase together with the 16S rRNA mTERF9 co-immunoprecipitation indicate that mTERF9 preferentially associates with the 30S small ribosome subunit in vivo. These results were highly consistent with the effects caused by the loss of mTERF9 in Arabidopsis, confirming the direct role of mTERF9 in chloroplast ribosomal assembly and translation.
Furthermore, we showed that some, but not all, of the in vivo mTERF9 protein interactions could be reconstituted in a yeast two hybrid assay and we demonstrated mTERF9 capacity to directly interact with ribosomal proteins. These results indicate that both direct and indirect protein interactors constitute mTERF9’s in vivo interactome. Besides supporting direct protein interactions, our RNA-protein interaction in vitro assays showed that mTERF9 is an RNA binding protein that preferentially binds the 16S rRNA. The capacity to interact with proteins and/or nucleic acids are two potent biochemical properties that are intrinsically linked to α-helices structures (reviewed in 70,71), and the ability of mTERF9 to stabilize ribonucleoprotein complexes via physical interactions certainly accounts for its function in ribosomal assembly in chloroplasts. In addition, the capacity of mTERF9 to oligomerize via intermolecular interactions between the mTERF repeats likely confers the protein new opportunities for ligand association by extending the binding surfaces at the dimer (Figure 8B). Finally, mTERF9 association with the polysomes indicates that it plays a function in chloroplast translation after its initiation but how it participates in this process remains elusive at this stage (one possibility is discussed below).
ERA1 and CPN60 chaperonins associate to chloroplast ribosomes in vivo
Our work revealed the presence of proteins in mTERF9 co-immunoprecipitates whose interaction with the chloroplast ribosomes had not been reported so far. The Arabidopsis protein ERA1 has been named after its bacterial homolog, the GTP-binding ERA protein. The protein localizes to the chloroplast nucleoids (72) and was found in stromal megadalton complexes containing ribosomal proteins (73) but its function has never been investigated in plants. In bacteria, the ERA1 homolog associates with the ribosome and binds the 16S rRNA to promote the assembly of the small 30S subunit (58,59,74). Our results confirmed the physical interaction of Arabidopsis ERA1 with mTERF9, a protein involved in the assembly of the 30S ribosomal subunit and therefore, its likely conserved function in ribosome assembly in chloroplasts.
Another surprise in the composition of the mTERF9 protein interactome is the high-enrichment of the six subunits of the CPN60 chaperonin complex, which is related to the bacterial GroEL protein folding machine and has been proposed to share a conserved function in protein quality control in chloroplasts by preventing aberrant protein folding and aggregation during their import into chloroplast or during their synthesis in chloroplasts (61). However, the evidence for such function in plants is scarce and very few ligands of the CPN60 chaperonins have been reported so far. These include the large subunit of Rubisco, RbcL (75), the Ferredoxin NADP+ reductase, FNR (76), the NdhH subunit of the NADH dehydrogenase complex (77), the membrane-bound FTSH11 protease (78) and the Plastidic type I signal peptidase 1, Plsp1 (79). By confirming the in vivo association of the CPN60 complex with the chloroplast ribosomes and the direct interaction of CPN60β1 and β3 subunits with mTERF9, our study provides insights into the molecular function of these chaperonins in chloroplast translation. Based on the mTERF9 protein interactome and mTERF9’s in vivo function, we propose the CPN60 chaperonin complex to be involved in the folding of nascent chloroplast proteins during translation, which would be in agreement with the chaperonin paradigm (61). In this model, mTERF9 would serve as a platform for recruiting the CPN60 chaperonin complex to the chloroplast ribosomes during translation via direct protein–protein interactions. This assumption is tempting as it would explain the functional association of mTERF9 with the chloroplast polysomes besides its role in ribosome assembly. Alternatively, the physical interaction between mTERF9 and the CPN60 complex might reflect the direct involvement of CPN60 chaperonins in the folding of mTERF9 and/or ribosomal proteins during ribosome assembly. In this scenario, the CPN60 complex would play a direct role in chloroplast ribosome biogenesis and translation. This possibility has been foreshadowed in an early study that identified nuclear mutants of maize displaying defects in the assembly of chloroplast polysomes (80). The results showed that the product of a nuclear gene, CPS2 facilitated the translation of various chloroplast mRNAs and the gene was later identified to be the maize orthologous CPN60α1 gene (81).
mTERF proteins as regulators of organellar translation
In metazoans, two out of the four mitochondrial mTERF proteins have been reported to regulate mitochondrial biogenesis and translation. The mTERF3 and mTERF4 proteins both interacts with mitochondrial rRNAs and are required for ribosomal assembly in mitochondria and therefore, translation (82,83). In addition, mTERF4 was shown to directly recruits the 5-methylcytosine methyltransferase, NSUN4 to the large ribosomal subunit to facilitate monosome assembly in mitochondria (83–85). Similar to our observations for mterf9 in Arabidopsis, the loss of organellar translation in mterf3 or mterf4 mutants in mice led to an increase in the steady-state levels of mitochondrial transcripts and de novo transcription which were considered to be a secondary effect of the loss of mitochondrial translation (83,84). On contrary in plants, mTERF9 is so far the only mTERF protein reported to play a direct role in ribosomal assembly and chloroplast translation (86). Two other members, mTERF4 and mTERF6 in maize and Arabidopsis respectively, have been reported to influence chloroplast translation but their effect on translation was rather indirect (22,44). In fact, mTERF4 promotes the splicing of several RNAs encoding ribosome components whereas mTERF6 contributes to the maturation of the trnI.2 in chloroplasts. As a consequence, the loss of function of these proteins caused a reduced accumulation of chloroplast ribosomes and translation in plants. By contrast, our comprehensive analysis of mTERF9 function that combined reverse genetics, molecular and biochemical phenotyping as well as in vitro assays allowed us to make firm conclusion about mTERF9 implication in chloroplast ribosome biogenesis. Our study demonstrated that mTERF9 supports physical in vivo association with RNA and protein components of the ribosome to stimulate ribosomal assembly and chloroplast translation, confirming the conserved function of mTERF-repeat proteins in the regulation of organellar translation in the plant kingdom.
DATA AVAILABILITY
Mass spectrometry proteomic data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018987.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-16-CE20-0007 to K.H.]; IdEx Unistra from the Investments for the future program of the French Government [to K.H.]; Deutsche Forschungsgemeinschaft [ZO 302/5-1 to R.Z., SFB-TRR175 to J.M. and R.Z.]; mass spectrometry instrumentation was funded by the University of Strasbourg, IdEx ‘Equipement mi-lourd’ 2015. Funding for open access charge: Centre National de la Recherche Scientifique.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.