 The fission yeast Pin1 peptidyl-prolyl isomerase promotes dissociation of Sty1 MAPK from RNA polymerase II and recruits Ssu72 phosphatase to facilitate oxidative stress induced transcription
The fission yeast Pin1 peptidyl-prolyl isomerase promotes dissociation of Sty1 MAPK from RNA polymerase II and recruits Ssu72 phosphatase to facilitate oxidative stress induced transcription
Nucleic Acids Research



- Altmetric
Pin1 is a peptidyl-prolyl isomerase that regulates the structure and function of eukaryotic RNA polymerase II (Pol II) through interaction with the C-terminal domain (CTD) of Rpb1, the largest subunit of Pol II. We demonstrated that this function is important for cellular response to oxidative stress in the fission yeast Schizosaccharomyces pombe. In response to oxidative stress, the Atf1 transcription factor targets Sty1, the mitogen-activated protein kinase (MAPK), to specific stress-responsive promoters. Anchored Sty1 recruits Pol II through direct association with Rpb1-CTD and phosphorylates the reiterated heptad sequence at Serine 5. Pin1 binds phosphorylated CTD to promote dissociation of Sty1 from it, and directly recruits Ssu72 phosphatase to facilitate dephosphorylation of CTD for transcription elongation. In the absence of Pin1, the association of Sty1-Atf1 with Rpb1 persists on stress-responsive promoters failed to generate transcripts of the corresponding genes effectively. The identified characteristic features of the fission yeast Pin1 are conserved in humans. We demonstrated that elevated Pin1 level in cancer cells might help to sustain survival under oxidative stress generated from their altered metabolic pathways. Together, these results suggest a conserved function of Pin1 in cellular response to oxidative stress among eukaryotic cells that might have clinical implication.
INTRODUCTION
Eukaryotic cells have developed response mechanisms to combat harmful effects from changes in surrounded environment. One of the major pathways operates radically reprograming gene expression to increase the proteins with stress protection function. Central to these responses are the stress-activated mitogen-activated protein kinases (MAPK) pathways. In mammalian, two such pathways are identified that lead to the activation of the p38 and JNK kinases and, as a result, a number of targets are phosphorylated including the ATF2 transcription factor (1,2). In the fission yeast Schizosaccharomyces pombe, the Sty1 MAPK is closely related to p38. Like its mammalian counterpart, Sty1 stimulates gene expression via the Atf1 transcription factor, which is similar to the human ATF2 (3). The essential role of Sty1 in activating stress-induced gene expression involves direct association with stress-dependent genes and the phosphorylation of chromatin-bound Atf1 (4). Similarly, in the budding yeast Saccharomyces cerevisiae, the Hot1 transcription factor targets Hog1 MAPK to specific osmotic stress-responsive promoters but not in response to other stressors, such as heat or ethanol (5). Anchored Hog1 directly interacts and recruits RNA polymerase II (Pol II) to the promoter for the stress-induced transcription. The mammalian p38 also interacts with Pol II suggesting a conserved mechanism for the regulation of gene expression by stress-activated MAP kinases among eukaryotic cells.
The Pin1 peptidyl-prolyl isomerase is an evolutionarily conserved enzyme that binds and catalyses cis–trans isomerization of the specific motif comprising a phosphorylated serine or threonine residue preceding a proline (pSer/Thr-Pro) in protein (6). Through this enzyme activity, Pin1 fine-tunes the function of key phosphoproteins including Pol II. Pol II is recruited to the promoter in an unphosphorylated form that becomes extensively phosphorylated at the C-terminal domain (CTD) reiterated hepatapeptide sequence (Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7) of Rpb1 during transcription (7). Phosphorylation of Ser5 is enhanced at the 5′ ends of genes and is associated with initiation complex formation. The level of Ser5 phosphorylation diminishes as Pol II moves into elongation, coincident with Ser2 phosphorylation. In budding yeast, the Ssu72 phosphatase is specific for pSer5 dephosphorylation (8). Ess1, the budding yeast homolog of Pin1, has been shown to preferentially bind to the pSer5 over pSer2 form of the CTD and stimulates pSer5 dephosphorylation by Ssu72 in vitro and in vivo (9).
In this study, we demonstrated that the fission yeast Sty1 interacted and phosphorylated the CTD of Rpb1, the largest subunit of Pol II. A function of the fission yeast Pin1 in promoting the dissociation of Sty1 from Rpb1-CTD to facilitate oxidative stress induced transcription was described. In addition to the binding to Rpb1-CTD to promote the dissociation of Sty1 from it, we found that the fission yeast Pin1 directly interacted and recruited Ssu72 for pSer5 dephosphorylation to facilitate the initiation-elongation transition of transcription, which were important for cellular response to oxidative stress. Similar interaction could be demonstrated for their human counterparts suggesting a conserved function of Pin1 in cellular response to oxidative stress among eukaryotic cells. Pin1 expression in several types of human cancer is frequently found to be higher than that in their normal counterparts (10). A potential role of Pin1 in cancer promotion and therapies mediated by oxidative stress generated reactive oxygen species (ROS) was suggested.
MATERIALS AND METHODS
Fission yeast strains and methods
We constructed all strains according to standard procedures (11). Information of oligonucleotides for gene disruption or modification can be obtained upon requested. The original atf1, sty1 and pin1 strains were gift from P. Russell, T. Humphrey and T. Hunter. A complete list of the strains used in this study was given in Table 1. Conditions for growth, maintenance, and genetic manipulation of fission yeast were as described previously (12). Except otherwise stated, strains were grown at 30°C in yeast extract (YE) or Edinburgh Minimal Medium (EMM2) with appropriate supplements. Where necessary, gene expression from the nmt1 promoter was repressed by the addition of 15 μM thiamine to the culture medium.

| Genotype |
|---|
| h − sty1-GFP::kanr |
| h − pin1-GFP::kanr |
| h − pin1-GFP::hyh ssu72–13Myc::kanr ura4 ura4-D18 |
| h − pin1::hyh |
| h − sty1::hyh |
| h − atf1::kanr |
| h − ssu72::ura4 ura4-D18 |
| h − sty1-GFP::kanr sty1::hyh |
| h − sty1-GFP::kanr pin1::hyh |
| h − sty1-GFP::kanr wis1::ura4 ura4-D18 |
| h − atf1–13Myc::ura4 ura4-D18 |
| h − sty1–13Myc::ura4 ura4-D18 |
| h − atf1–13Myc::ura4 sty1::kanr ura4-D18 |
| h − atf1–13Myc::ura4 pin1::hyh ura4-D18 |
| h − sty1–13Myc::ura4 pin1::hyh ura4-D18 |
| h − sty1–13Myc::ura4 atf1::kanr ura4-D18 |
| h − pREP1-sty1–6xHis leu1–32 |
| h − pREP1-sty1(K49R)-6xHis leu1–32 |
| h − pREP1-pin1–6xHis leu1–32 |
| h − pREP1-pin1(W33A)-6xHis leu1–32 |
| h − pREP1-pin1(S79E)-6xHis leu1–32 |
| h − pREP1-pin1-GST leu1–32 |
| h − pREP1-pin1(W33A)-GST leu1–32 |
| h − pREP1-pin1(S79E)-GST leu1–32 |
| h − pREP1-Rpb1 CTD-GFP leu1–32 |
Plasmid construction and recombinant protein
Plasmids for the expression of Pin1, Sty1, Ssu72 and Rpb1-CTD (1549–1751) were constructed by PCR amplification of the corresponding open reading frame of S. pombe cDNA, and subsequently ligated into plasmids derived from pREP1 or pGEX4T-1 with a GFP, GST, or His-tag inserted at the 3′ end of the multiple cloning sites. The Sty1 (K49R) and Pin1 (W33A, S79E) mutants were created using PCR-based site-directed mutagenesis and verified by DNA sequencing.
The C-terminal His tagged Sty1 (WT and K49R) and Pin1 (WT, W33A and S79E) proteins from yeast can be rapidly purified using Ni-NTA affinity (Qiagen, Hilden, Germany). The C-terminal GST tagged Pin1 (WT, W33A, S79E) fusion proteins from yeast and the fusion protein GST-Rpb1-CTD expressed in Escherichia coli BL21 cells were purified by Glutathione Sepharose™ 4B (170756-01, GE Healthcare) according to the manufacturer's recommendations.
Cell culture and small interference RNA experiments
Human cells were cultured in RPMI medium (Invitrogen, Carlsbad, CA) supplemented with 10% FCS and 100 IU/ml penicillin, 0.1 mg/ml streptomycin and maintained at 37°C in a humidified incubator containing 5% CO2 in air. The ON-TARGET plus SMARTpool siRNA targeting human Pin1 (L-003291) as well as the ON-TARGET plus non-targeting siRNA (siCTL) were purchased from Thermo Scientific, Dharmacon (Illkirch, France). 5 × 105 cells plated and cultured in p-60 dish in RPMI medium (10% FCS serum) at 37°C for 24 h were transfected with siRNA (200 nM final concentration) or control siRNA in RPMI using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) at 37°C for 4 h. One-third volume of RPMI (30% FCS) was added and cells were incubated at 37°C for an additional 2 days before use.
Antibodies and immunoprecipitation
The antibodies against GFP (ab290) and Rpb1 CTD (8WG16, ab817) were purchased from Abcam (Cambridge, UK). Anti-Rpb1 CTD pSer5 (H14, MMS-134R) and pSer2 (H5, MMS129R) was purchased from Covance (Princeton, NJ, USA). Anti-His (YH-8003) and anti-GST (YH-8002) were purchased from Yao-Hong Biotechnology Inc. (New Taipei City, Taiwan). The antibodies against p38 (5F11, 9217), p-p38 (28B10, 9216) and Ssu72 (D3I2D, 12816) were from Cell Signaling Technologies (Danvers, MA, USA). Anti-Flag M2 antibody was from Sigma-Aldrich (Saint Louis, MO, USA). Anti-Pin1 antibody was from R&D Systems (MAB2294, MN, USA). Antibody against α-tubulin (Sigma-Aldrich, Saint Louis, MO) was used as a control.
For immunoprecipitation from yeast extracts, 2 × 108 yeast cells were lysed in 200 μl NP-40 lysis buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 1% NP-40, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 0.1 mM Na3VO4, 1 mM PMSF, 1 mM DTT, cOmplete protease inhibitor cocktail) by vortexing with acid-washed glass beads. The lysate was clarified by centrifugation, and GFP fusion proteins were retrieved using GFP-Trap-coupled agarose beads (Chromotek, Martinsried, Germany) following the manufacturer's instructions.
For immunoprecipitation from Hela cells extracts, 1 mg lysates were incubated with 2 μg anti-Rpb1-CTD antibody (8WG16) and protein G–Sepharose beads (GE Healthcare, Piscataway, NJ) for 4 h at 4°C and washed three times in lysis buffer before elution in SDS sample buffer. Equal amounts of cellular proteins or immunoprecipitates were subjected to Western immunoblotting as described. Five percent of the input was loaded onto the gel.
Chromatin immunoprecipitation (ChIP) and real time PCR
ChIP was performed as described previously (4). Yeast cultures grown to early log phase were exposed to 2 mM H2O2 for the time specified in the figure legends. For crosslinking, yeast cells were treated with 1% formaldehyde for 20 min at room temperature. After sonication, soluble chromatin fragments were obtained by spinning for 30 min in a microcentrifuge. The anti-myc, anti-Rpb1 CTD (8WG16) and anti-pS5-Rpb1 CTD (H14) antibodies were used for immunoprecipitation.
For real time PCR quantifications, the relative enrichments of gpd1, hsp9 and ctt1 were measured over a stress-independent promoter (cdc2). Primers used are denoted relative to translation start codons and correspond to –214 to –192 and –177 to –144 for the cdc2 promoter, +471 to +498 and +518 to +541 for cdc2 ORF, –446 to –421 and –388 to –363 for the ctt1 promoter, +1379 to +1399 and +1423 to +1439 for the ctt1 ORF, –402 to –380 and –360 to –340 for gpd1 promoter, +241 to +261 and +305 to +324 for gpd1 ORF, –264 to –245 and –220 to –199 for the hsp9 promoter, +1 to +20 and +49 to +68 for hsp9 ORF. Experiments were performed on three independent chromatin preparations, and quantitative PCR analysis was performed in real time by using an AB7900HT instrument (Applied Biosystems, Darmstadt, Germany). Immunoprecipitation efficiencies were calculated in triplicate by dividing the amount of PCR product in the immunoprecipitated sample by the amount of PCR product in the input.
GST pull down assay
Interaction between Pin1 and Ssu72: The Pin1-GST fusion protein (10 μg) was incubated with Ssu72-His (10 μg) and 50 μl 50% glutathione-Sepharose 4B resin (GE Healthcare, Piscataway, NJ) at 4°C for 4 h in binding buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1% NP-40, 10% glycerol, protease inhibitors and 1 mM PMSF.
Interaction between Sty1 and Rpb1-CTD: The Rpb1-CTD GST-fusion protein (10 μg) was incubated with Sty1-His (WT and K49R) (10 μg) and 50 μl 50% glutathione-Sepharose 4B resin at 4°C for 4 h in binding buffer.
Interaction between p38 and Rpb1-CTD: Total lysate of HeLa cells transfected with pcDNA3-p38-Flag were incubated with Rpb1-CTD GST-fusion protein (10 μg) and 50 μl 50% glutathione-Sepharose 4B resin over-night at 4°C in binding buffer.
After three washes with the binding buffer, the samples were subjected to SDS–PAGE and Western immunoblotting analysis.
In vitro kinase and phosphatase activity assays
Yeast cells expressing Sty1-His and HeLa cells expressing p38-Flag were subjected to treatment with 2 and 0.5 mM H2O2 respectively for 15 min before purifying fusion protein for kinase activity with 10 μg of Rpb1-CTD GST-fusion protein from Escherichia coli in 200 μl of kinase reaction buffer (1 mM ATP, 25mM Tris, PH7.5, 10 mM MgCl2, 0.1mM Na3VO4, 2 mM DTT) at 30°C for 1 h. Reactions were terminated by the addition of Laemmli sample buffer and products were resolved by 10% SDS-PAGE. Phosphorylated proteins were determined by Western immunoblotting using anti-phospho-Rpb1 (H14) antibodies.
For phosphatase activity assays, the immunoprecipitates of Rpb1-CTD GFP-fusion protein from yeast cells were washed with CIP buffer (100 mM NaCl, 50 mM Tris–HCl, 10 mM MgCl2, 1 mM dithiothreitol, Protease inhibitor cocktail, EDTA-free, pH to 7.9) three times before treated with Ssu72 recombinant protein (5–10 μg) or 20 units calf intestinal alkaline phosphatase (CIP) (M0290S, NEW ENGLAND BioLabs, Ipswich, MA, USA) in CIP buffer at 30°C for 1 h.
Measurement of intracellular ROS Levels
Exponential growing yeast (5 × 107) were harvested and washed twice with PBS buffer before incubation with 10 μM fluorescent dye CM-H2DCFDA (C6827, Invitrogen, Carlsbad, CA, USA) in PBS buffer for 1 h in the dark. Yeast cells were collected by centrifugation and re-suspended in PBS buffer before fluorescence determination using Tecan Infinite M200 Pro Microplate Reader (Männedorf, Switzerland). The excitation and emission wavelengths for CM-H2DCF-DA were 492 and 525 nm, respectively.
Human cells (5 × 105 cells/well) seeded in 24-well plates and incubated at 37°C for 24 h were treated with fresh medium containing 10 μM CM-H2DCFDA. After 30 min, the treated cells were washed twice with PBS and harvested for fluorescence determination.
RESULTS
Pin1 mutant is defective in oxidative stress induced transcription response at the initiation to elongation transition
To gain more insight into the biological function of Pin1, we have performed a phenotypical analysis of the pin1 deletion mutant in S. pombe. We found that fission yeast cells lacking Pin1 were hypersensitive to oxidative stress (H2O2) (Figure 1A). Genetic analysis demonstrated that no additional effects were observed for pin1Δ atf1Δ double mutant (Figure 1A and B). These results suggested that Pin1 acted on the same pathway with Atf1 in cellular responses to oxidative stress. In line with this idea, genes whose expression modulated by Sty1 and Atf1 upon oxidative stress, such as hsp9, gpd1 and ctt1, were compromised in the absence of Pin1 (Figure 1C). Experiments were performed to determine the underlying molecular mechanisms.

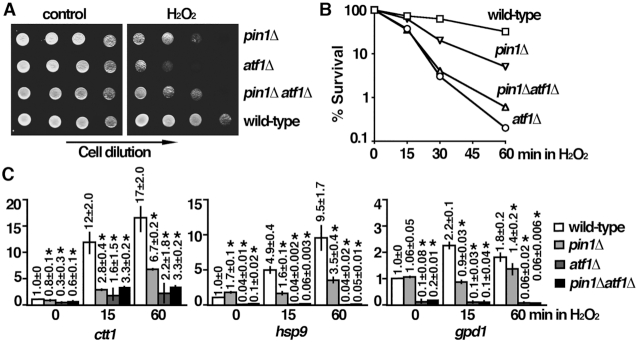
Pin1 mutant is defective in oxidative stress induced transcription. (A) Tenfold serial dilution of strains indicated were spotted onto YES-agar plates without (control) or with 2 mM H2O2 and photographed after 3 days incubation at 30°C. (B) H2O2 (2 mM) was added to an asynchronous culture of strains indicated growing at 30°C in YES medium. Cell viability was measured at the time indicated by plating appropriate dilution of cells onto YES agar plate without drug and scoring colony formation after 3 days incubation at 30°C. Viability (% survival) is expressed as a percentage of the number of colonies obtained before the addition of drug. (C) Expression of stress-responsive genes indicated upon oxidative stress in wild type, pin1Δ, atf1Δ and pin1Δatf1Δ cells. Total RNA from cultures of the indicated strains grown at 30°C was reverse transcribed using an oligo(dT) primer. The cDNA was amplified using quantitative PCR and SYBR green with primers specific for the indicated genes. The amount of RNA normalized to cdc2 mRNA relative to the wild type was expressed as means ± SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type.
The phosphorylation-dependent specificity suggested a potential role of Pin1 in the kinase-mediated signal transduction pathways. Pin1 might play a role in modulating MAPK-mediated signal transduction pathway. However, we found that, similar to wild type cells, pin1 mutant responded to oxidative stress (Figure 2A). Sty1 was phosphorylated and thus activated to phosphorylate Atf1 following H2O2 stress. The increase in level and retardation of mobility due to phosphorylation of Atf1 upon oxidative stress also occurred in the absence of Pin1. In fact, a low level phosphorylated Sty1 and Atf1 were observed in pin1 mutant before H2O2 stress, which could be due to elevated intracellular level of ROS in these cells (see figure below). Together, these results suggested that MAPK-mediated signal transduction pathway was functional in the absence of Pin1.
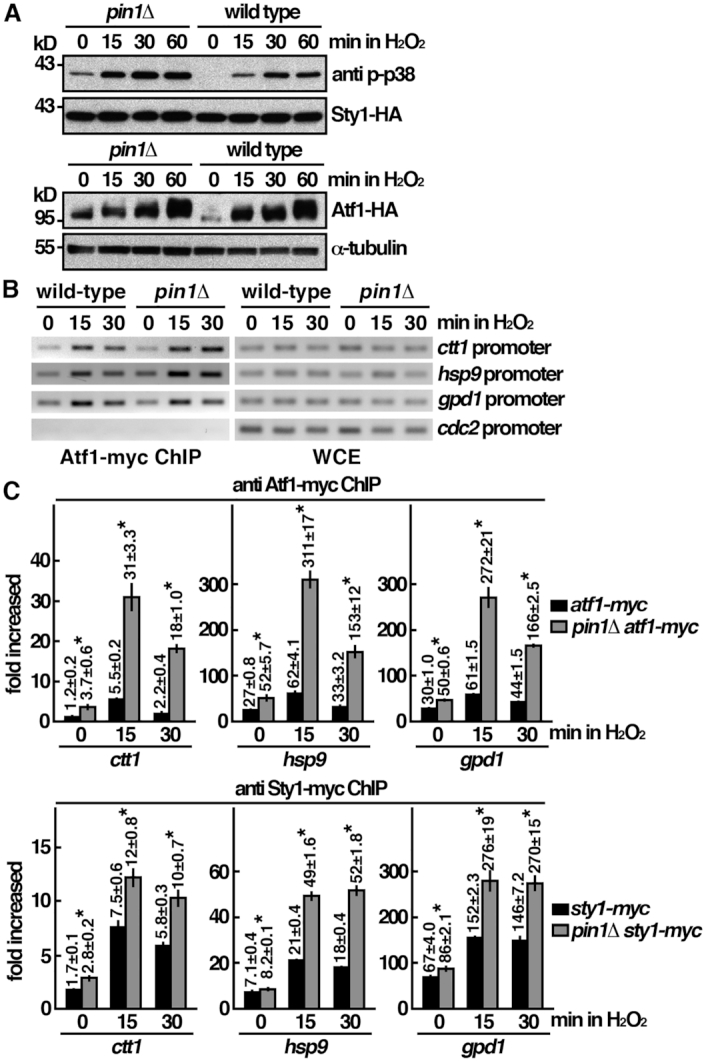
Pin1 mutant is not defective in MAPK-mediated signal transduction pathway. (A) Whole-cell protein extracts of the strains indicated were prepared by alkaline extraction followed by trichloroacetic acid precipitation. The extracts were separated by SDS-PAGE and subjected to Western immunoblotting using anti-HA antibodies to reveal proteins of interest. Anti p-p38 antibody cross-reacted with phosphorylated Sty1. Antibody against α-tubulin was used as the loading control. (B) ChIP assays showing recruitment of Atf1-myc to the promoter of ctt1, gpd1, hsp9 and cdc2 genes upon oxdative stress. The latter was not induced upon stress. Samples were prepared from atf1-myc and atf1-myc pin1Δ cells treated with 2 mM H2O2 for the time points indicated. The DNA recovered from the IP was assayed by PCR using primers specific to the promoter of genes indicated. The control lanes show DNA amplified from whole cell extracts (WCE) prior to performing the IP. (C) ChIP assays showing recruitment of Atf1-myc and Sty1-myc to the promoter of ctt1, gpd1 and hsp9 genes upon oxdative stress in pin1Δ mutant and wild type cells. Samples were prepared from strains indicated treated with 2 mM H2O2 for the time points indicated. The DNA recovered from the IP was amplified using quantitative PCR and SYBR green with primers specific to the promoter of genes indicated normalized to that of the non-stress responsive cdc2 gene. Data were expressed as means ± SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type.
The essential role of Sty1 in transcription activation requires its direct recruitment to stress-induced genes targeted by Atf1 (4). Next we determined the effect of pin1 deletion on these activities using a ChIP assay analyzed by quantitative PCR (4). Association of Atf1 and Sty1 with the promoters of hsp9, gpd1 and ctt1 genes were assessed by an increase in the ratio of the PCR products generated by primers specific for their promoters and the control products that were amplified by primers recognizing the non-stress-specific promoter of cdc2 gene (Figure 2B). As shown in Figure 2C, Sty1 recruitment was elevated 15 min after stress, which matched the kinetics of its activation (Figure 2A) and accumulation of hsp9, gpd1 and ctt1 transcripts (Figure 1C). We found that the associations of Atf1 and Sty1 with the promoter of hsp9, gpd1 and ctt1 genes were accumulated in pin1 mutant (Figure 2C), but failed to generate transcripts of the corresponding genes effectively (Figure 1C). These results suggested that, in the absence of Pin1, Atf1 and Sty1 were recruited to the promoter regions but were unable to proceed to the next step of the transcription cycle.
A function of MAPK for recruitment and activation of Pol II has been suggested (5). Experiments were performed to determine the phosphorylation status of Rpb1-CTD (Figure 3A and B) and its recruitment to targeted promoter (Figure 3C and D). As shown in Figure 3C and D, upon H2O2 stress, Rpb1 was recruited to the promoter and extensively phosphorylated at Ser5. In the absence of Pin1, Ser5 phosphorylated Rpb1 was associated and accumulated at the promoter region following H2O2 stress but was defective in entering elongation to generate transcripts of the corresponding genes (Figure 1C). In line with these results, Ser2 phosphorylation of Rpb1-CTD, which facilitated transcription elongation, was reduced in pin1 mutant as a secondary effect derived from defect in transcription initiation to elongation (Figure 3A and B).
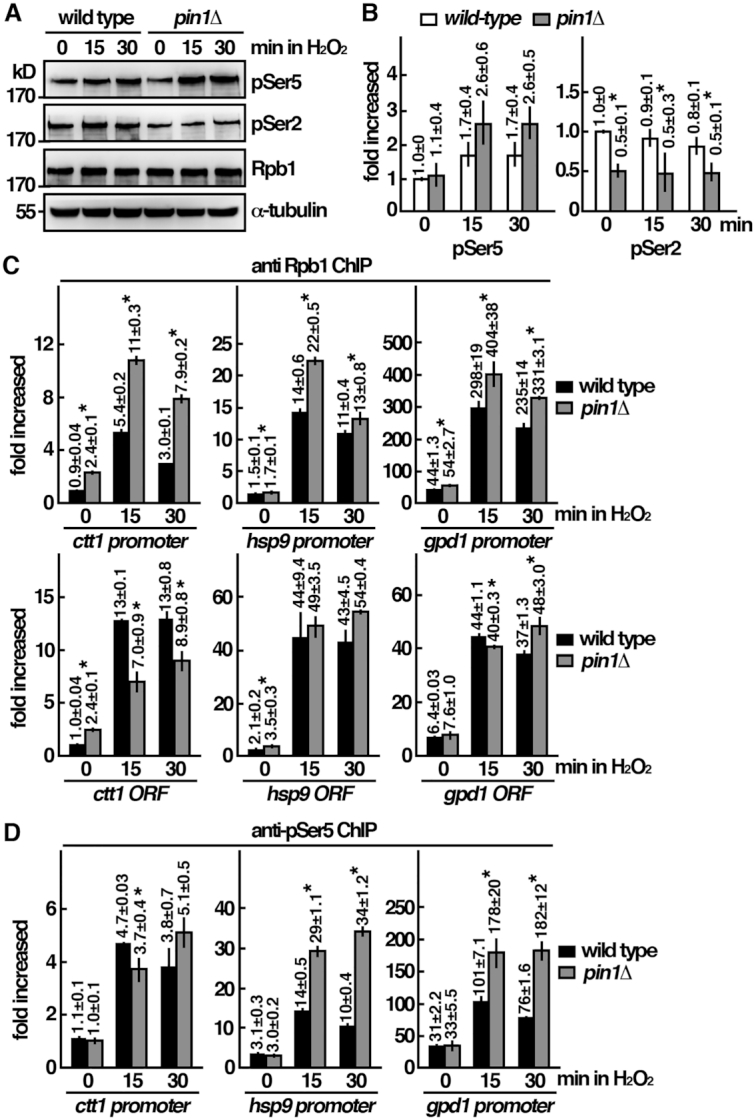
Pin1 mutant is defective in the oxidative stress induced transcription response at the initiation to elongation transition. (A) Whole-cell protein extracts of the strains indicated were prepared by alkaline extraction followed by trichloroacetic acid precipitation. The extracts were separated by SDS-PAGE analyzed phosphorylation by western immunoblotting with CTD phospho-specific antibodies. Antibody against α-tubulin was used as the control. The relative level of phosphorylated protein was shown in (B). Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P<0.05, t-test unpaired) verse wild type. (C) ChIP assays showing the recruitment of Rpb1 to the promoter and the gene coding regions (ORF) of ctt1, gpd1 and hsp9 genes upon oxidative stress in wild type cells and pin1Δ mutant. Samples were prepared from the indicated strains treated with 2 mM H2O2 for the time points indicated. The DNA recovered from the IP was amplified using quantitative PCR and SYBR green with primers specific to the promoter of the indicated genes normalized to that of the non-stress responsive cdc2 gene. Data were expressed as means ± SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type. (D) ChIP assays showing the recruitment of pSer5-Rpb1 to the promoters of ctt1, gpd1 and hsp9 genes upon oxdative stress in wild type cells and pin1Δ mutant as in (C). Data were expressed as means ± SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type.
Furthermore, primer pairs in the gene coding regions were used to determine whether elongation was affected. As shown in Figure 3C, the association of Rpb1 with the gene-coding region was significantly reduced in pin1 mutant for ctt1 gene as compared with the wild type cells. No significant changes were observed for hsp9 gene. Intriguingly, the association of Rpb1 with the gpd1 gene-coding region was significantly reduced at 15 min but was elevated at the later time point of 30 min. Among these three genes, Pin1 deletion affected elongation to a different extends. Together, the results suggest that Pin1 mutant is defective in oxidative stress induced transcription response at the initiation as well as elongation stages.
Sty1 interacted with and phosphorylated Rpb1-CTD at Ser5
The results described above suggested a closed interaction between Sty1 and Rpb1 modulated by Pin1. To gain more insight into the underlying molecular mechanism, experiments were performed to determine the interaction between these molecules following H2O2 stress. As shown in Figure 4A, we found that, similar to human p38 and Hog1 in S. cerevisiae, Sty1 associated with Rpb1 in S. pombe as demonstrated by co-immunoprecipitation before H2O2 stress. This result suggested that the interaction between Sty1 and Rpb1 did not require the activation of Sty1. Intriguingly, upon oxidative stress, the association between Rpb1 and Sty1 was decreased in wild type cells and up regulated in the pin1 mutant with reduced phosphorylation of Ser2 (Figure 4B). Pin1 also interacts with Rpb1. These results suggested a mutual exclusive binding mode of Rpb1 to Pin1 over Sty1. In line with this idea, the interaction between Pin1 and Rpb1 was increased following H2O2 stress (Figure 4C). Pin1 binds phosphorylated CTD of Rpb1 (Figure 4C). To determine whether Sty1 and Pin1 bound the same region of Rpb1, we performed an in vitro binding assay using purified GST-CTD from Escherichia coli with Sty1-His from yeast. As shown in Figure 4D, Sty1 specifically interacted with CTD, but not with the GST control indicating that Sty1 and Pin1 interacted with the same region of Rpb1. Similar result was observed with the kinase-dead (K49R) His-tagged derivative of Sty1, which was in line with the data described above that the interaction between Sty1 and CTD was independent of the activation of Sty1.
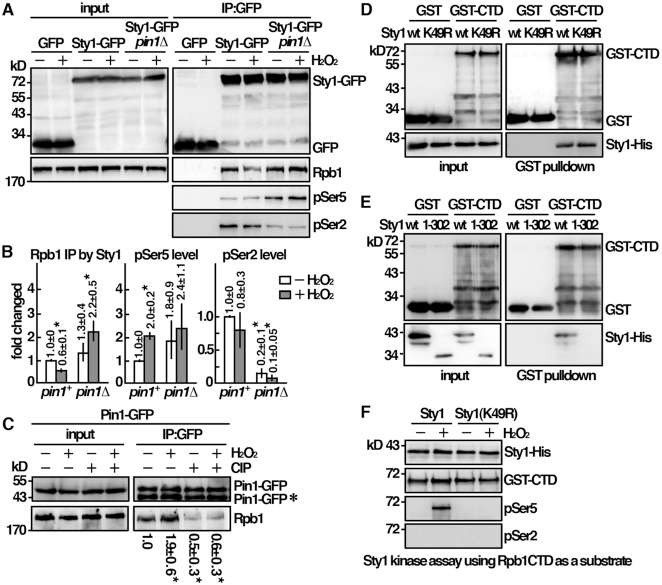
Sty1 interacted and phosphorylated Rpb1-CTD at Ser5. (A) Coimmunoprecipitation was performed with extracts prepared from indicated strains treated with 2 mM H2O2 for 30 min. GFP-Trap affinity resin was used to pull down GFP proteins. Immunoprecipitates were analyzed by Western immunoblotting using antibodies against GFP and Rpb1-CTD. The relative level of Rpb1 pull down by Sty1 and its phosphorylation status was shown in (B). Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type. (C) Coimmunoprecipitation was performed with extracts prepared from Pin1-GFP tagged strains treated with 2 mM H2O2 for 30 min. GFP-Trap affinity resin was used to pull down GFP proteins. Immunoprecipitates were treated with/without CIP and analyzed by Western immunoblotting using antibodies against GFP and Rpb1-CTD. *Degradation product of Pin1-GFP. The relative level of Rpb1 pull down by Pin1 was shown. Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P<0.05, t-test unpaired) verse control of no treatments. (D, E) Purified GST-CTD from Escherichia coli and recombinant protein of Sty1 from yeast cells were subjected to GST pulldown assay followed by Western immunoblotting analysis with antibodies against GST and His. (F) In vitro kinase assay using Sty1-His extracted and immunoprecipitated from activated cells. Following incubation with GST-CTD, proteins were resolved by SDS-PAGE and probed with antibodies indicated.
Furthermore, we found that deletion of the C-terminal region of Sty1 (residues 302–349), which contained a hypothetical docking site, affected the interaction between Sty1 and CTD (Figure 4E). The interaction of MAPKs with substrates is mediated via conserved docking site. This result led us to hypothesize that Sty1 might phosphorylate CTD. An in vitro kinase assay was performed to test this possibility. Recombinant Sty1-His protein purified from yeast cells (after activating the kinase through the exposure to H2O2) was incubated with GST-CTD from Escherichia coli, and analyzed the phosphorylation by western immunoblotting with CTD phospho-specific antibodies (Figure 4F). We found that Sty1 can indeed phosphorylate Ser5 but not Ser2 whereas Hog1 in S. cerevisiae possessed both Ser5 and Ser2 kinase activity (14). Ser5 phosphorylation is associated with initiation complex formation (7). These results suggested a function of Sty1 in initiation complex formation. As controls, neither H2O2-untreated nor kinase-dead (K49R) derivative of Sty1 showed any activity. Together, these results were consistent with a function of Sty1 for recruitment and activation of Pol II through interaction and phosphorylation of Rpb1-CTD at Ser5.
Pin1 affects Sty1 binding of Rpb1-CTD
The results described above provided strong evidence that Sty1 possessed Ser5 kinase activity. Pin1 has been shown to preferentially bind the pSer5 form of CTD and thus might modulate the interaction between Sty1 and Rpb1 following H2O2 stress. A competition assay was performed to test this possibility. Recombinant Sty1-His protein was incubated with phosphorylated CTD-GFP purified from yeast cell in the presence or absence of Pin1. As shown in Figure 5A, we found that CTD preferentially bound Pin1 over Sty1 when phosphorylated. Similar result was observed with the isomerase-dead (S79E) derivative of Pin1 but not with the mutant defective in the substrate-binding activity (W33A). These results suggested that it was the physical binding rather than the isomerase activity of Pin1 affecting the association of Sty1 to CTD. Following H2O2 stress, Pin1 bound phosphorylated CTD to promote Sty1 dissociation from it to facilitate oxidative stress induced transcription.
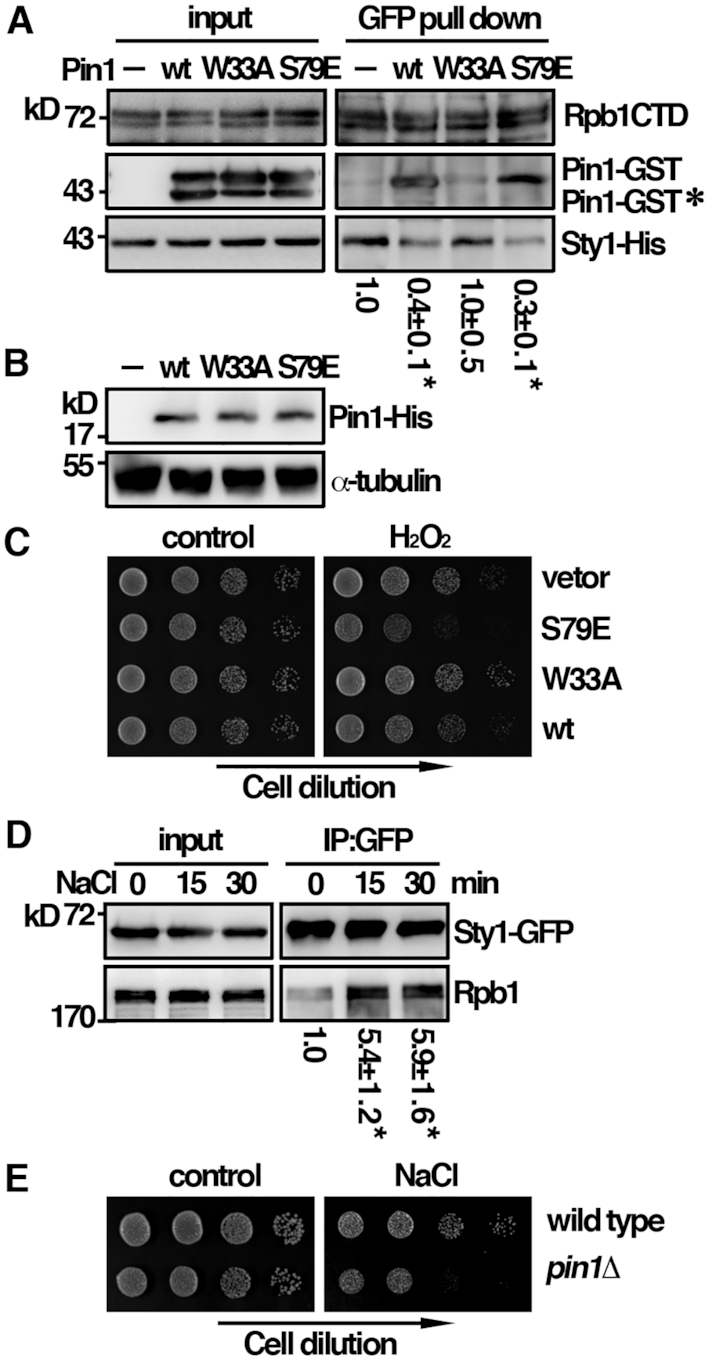
A mutual exclusive binding mode of Rpb1 to Pin1 over Sty1. (A) Recombinant proteins of Sty1 and CTD-GFP from yeast cells were subjected to GFP pulldown assay in the presence or absence of Pin1 followed by Western immunoblotting analysis with antibodies indicated. *Degradation product of Pin1-GST. The relative level of Sty1 pull down by CTD-GFP was shown. Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P<0.05, t-test unpaired) verse control without Pin1. (B) Whole-cell protein extracts of the strains indicated were prepared by alkaline extraction followed by trichloroacetic acid precipitation. The extracts were separated by SDS-PAGE and subjected to immunoblotting using anti-His antibodies to reveal Pin1 proteins. Antibody against α-tubulin was used as the control. (C) Tenfold serial dilutions of strains indicated were spotted onto EMM+thiamine agar plates without (control) or with 2 mM H2O2 and photographed after 3 days incubation at 30°C. (D) Coimmunoprecipitation was performed with extracts prepared from Sty1-GFP tagged strains treated with 0.5 M NaCl for the time point indicated. GFP-Trap affinity resin was used to pull down GFP proteins. Immunoprecipitates were analyzed by Western immunoblotting using antibodies against GFP and Rpb1-CTD. The relative level of Rpb1 pull down by Sty1 was shown. Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse control of no treatment. (E) Tenfold serial dilutions of strains indicated were spotted onto YES-agar plates without (control) or with 200 mM NaCl and photographed after 3 days incubation at 30°C.
To gain more insight into the roles of Pin1 in oxidative stress induced transcription, we further determined the phenotype of Pin1 S79E (isomerase-dead) and W33A (substrate binding-dead) mutants under H2O2 stress. Given that the over-expression of Pin1 in S. pombe caused slow growth and a G1 delay, which obscured the effect of Pin1 on peroxide sensitivity (15). We took advantage of the leaky nmt1 promoter to moderately over-express Pin1 in the presence of low concentration of thiamine (Figure 5B). In line with our model, unlike Sty1, the association of Pin1 with Rpb1-CTD was phosphorylation dependent (Figure 4C) that occurred only after the association and activation of Sty1 upon oxidative stress. Moderate over-expression of the wild type Pin1 or W33A mutant did not affect the peroxide sensitivity (Figure 5C). In contrast, Pin1 S79E mutant had a dominant negative effect to potentiate the cytotoxicity of H2O2. These results suggested that, in addition to the binding to the Rpb1-CTD, the isomerization activity was also required for the function of Pin1 in oxidative stress induced transcription.
Our results were different from the published S. cerevisiae data that the interaction of Hog1 with Pol II was more efficient in yeast extracts of cells treated with osmostress (5). Different from under the oxidative stress, we found that the interaction between Sty1 and Rpb1 was indeed increased upon osmostress (Figure 5D) and fission yeast cells lacking Pin1 were also hypersensitive to osmostress (NaCl) (Figure 5E). These results suggested that, despite present in the same complex, the underlying molecular mechanism could be different dependent on the stress type. Unlike Sty1 (4), the function of Hog1 is rather specific to osmostress but not to other stressors. As suggested for Hog1 (16), under osmostress, Sty1 might interact with some other subunits of the polymerase, such as Rpb2, or with different activator to perform its function. Pin1 might play a role in these processes. This phenomenon would require further investigation and was not persuaded here. Nevertheless, these results highlight the complex natures of the transcription complex formation under different stresses.
Pin1 directly interacts and recruits Ssu72 for pSer5 dephosphorylation
During the phenotypical analysis of the pin1 deletion mutant, we noticed that, following H2O2 stress, the pattern of Rpb1 phosphorylation in ssu72 mutant resembling that in pin1 mutant was defective in Ser2 phosphorylation (Figure 6A and B). These results suggested an overlapping function of Ssu72 in Pin1-medicated cellular response to oxidative stress, which was further explored below. In budding yeast, the Ssu72 phosphatase is specific for pSer5 dephosphorylation (8) and a function of Ess1 in stimulating pSer5 dephosphorylation by Ssu72 has been suggested (9). We found that the fission yeast Ssu72 indeed dephosphorylated pSer5 in an in vitro assay with phosphorylated CTD-GFP isolated from yeast cell (Figure 6C). Although we did not observe the effect of Pin1 stimulating pSer5 dephosphorylation by Ssu72 in the in vitro assay, we found that Ssu72 associated with Pin1 in S. pombe as demonstrated by co-immunoprecipitation (Figure 6D). To assess that the association was direct, an in vitro binding assay was performed. As shown in Figure 6E, Pin1 specifically interacted with Ssu72, but not with the GST control in the pull down experiment. These results suggested that Pin1 directly interacted with and recruited Ssu72 for pSer5 dephosphorylation to facilitate progression of transcription important for cellular response to oxidative stress. Next, we asked whether Ssu72 could form a stable complex with Rpb1-CTD. However, as shown in Figure 6F, we failed to detect any association of Ssu72 with CTD-GFP in the in vitro binding assay even in the presence of Pin1. These results suggested that Ssu72 only transiently associated with Pin1 and might be dissociated after the recruitment to Rpb1-CTD for its function. In support of this idea, unlike Pin1, Ssu72 was not highly enriched in the CHIP assay following the treatment of H2O2 (Figure 6G). The association of Ssu72 with the target genes by ChIP shown in Figure 6G could be mediated by other factors such as Rpb2, which interacted with Ssu72 in S. cerevisiae (17). The results described above suggested a function of Pin1 and Ssu72 in oxidative stress. In the absence of Pin1 or Ssu72, cells could be defective in eliminating the toxic effects raised from oxidative stress. In support of this idea, the intracellular level of ROS was elevated in pin1 and ssu72 mutants (Figure 6H), which could be generated from endogenous metabolic defects. Furthermore, a slow growth phenotype of ssu72 mutant was observed before the treatment of H2O2 (Figure 6I) suggesting that, in addition to response to oxidative stress, Ssu72 might have an additional function in normal transcription program.
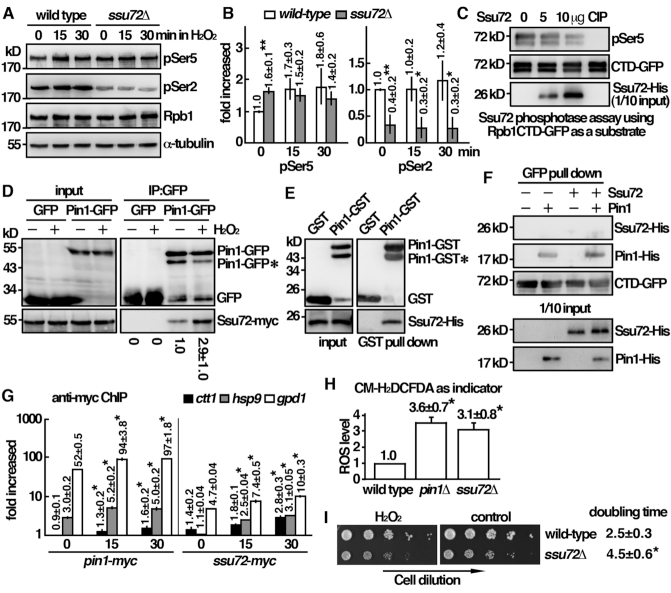
Pin1 directly interacts and recruits Ssu72 for pSer5 dephosphorylation. (A) Whole-cell protein extracts of the strains indicated were prepared by alkaline extraction followed with trichloroacetic acid precipitation. The extracts were separated by SDS-PAGE and analyzed phosphorylation by Western immunoblotting with CTD phospho-specific antibodies. Antibody against α-tubulin was used as the control. The relative level of phosphorylated protein was shown in (B). Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type. (C) In vitro phosphatase activity assay using recombinant protein of CTD-GFP from yeast cells were treated with Ssu72 recombinant protein or 20 units calf intestinal alkaline phosphatase (CIP) at 30°C for 1 h. Reaction mixtures were resolved by SDS-PAGE and probed with antibodies indicated. (D) Coimmunoprecipitation was performed with extracts prepared from Ssu72-myc tagged strains expressing Pin1-GFP and being treated with 2 mM H2O2 for 30 min. GFP-Trap affinity resin was used to pull down GFP proteins. Immunoprecipitates were analyzed by western immunoblotting using antibodies against GFP and myc. *Degradation product of Pin1-GFP. The relative level of Ssu72 pull down by Pin1-GFP was shown. Data were expressed as means±SD in triplicate. (E) Recombinant proteins of Pin1-GST and Ssu72-His were subjected to GST pull down assay followed by Western immunoblotting analysis with antibodies against GST and His. *Degradation product of Pin1-GST. (F) Recombinant proteins of Ssu72-His and Pin1-His were subjected to GFP pulldown assay by CTD-GFP followed by Western immunoblotting analysis with antibodies against GFP and His. (G) ChIP assays showing the recruitment of Pin1-myc and Ssu72-myc to the promoters of ctt1, gpd1 and hsp9 genes upon oxdative stress. Samples were prepared from strains indicated treated with 2 mM H2O2 for the time points indicated. The DNA recovered from the IP was amplified using quantitative PCR and SYBR green with primers specific to the promoters of genes indicated normalized to that of the non-stress responsive cdc2 gene. Data was expressed as means ± SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse control of no treatment. (H) Strains indicated were subjected to measurement of intracellular ROS levels in triplicate using CM-H2DCFDA as indicator. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type. (I) Tenfold serial dilutions of strains indicated were spotted onto YES-agar plates without (control) or with 2 mM H2O2 and photographed after 3 days incubation at 30°C. Growth characteristics of the strains indicated at 30°C (doubling time) were shown on the right. Data were expressed as means ± SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse wild type.
The identified characteristic feature of the fission yeast Pin1 is conserved in humans
Next, we wanted to see whether the identified characteristic features of the fission yeast Pin1 were conserved in humans. HeLa cells expressing flag-tagged p38 were subjected to immunoprecipitation using specific monoclonal antibodies against Rpb1. The presence of p38 in the precipitates was determined by an anti-flag antibody. As shown in Figure 7A, p38 was able to coprecipitate with Rpb1. Similar to that of S. pombe, the interaction between p38 and Rpb1 was decreased following H2O2 stress. To determine whether, similar to Sty1, p38 bound CTD of Rpb1, we performed an in vitro binding assay using p38-flag isolated from HeLa cells with purified GST-CTD from Escherichia coli. As shown in Figure 7B, p38 specifically interacted with CTD irrespective of the treatment of H2O2, but not with the GST control. This result suggested that, similar to Sty1, p38 might phosphorylate CTD. An in vitro kinase assay was performed to test this possibility. Flag-tagged p38 isolated from HeLa cells (after activating the kinase through the exposure to H2O2) was incubated with GST-CTD from Escherichia coli, and analyzed the phosphorylation by Western immunoblotting with CTD phospho-specific antibodies (Figure 7C). Different from Hog1 but similar to Sty1, p38 can indeed phosphorylate Ser5 but not Ser2. The phosphorylation was not detected in the presence of p38 inhibitor (SB203580). These results suggested that the identified characteristic feature between Sty1 and Rpb1 was conserved in human p38, and likely to be modulated by Pin1. Furthermore, we found that Pin1 also interacted with Ssu72 in HeLa cells as demonstrated by co-immunoprecipitation (Figure 7D). The characteristic feature of Pin1 identified in the fission yeast was indeed conserved in humans.
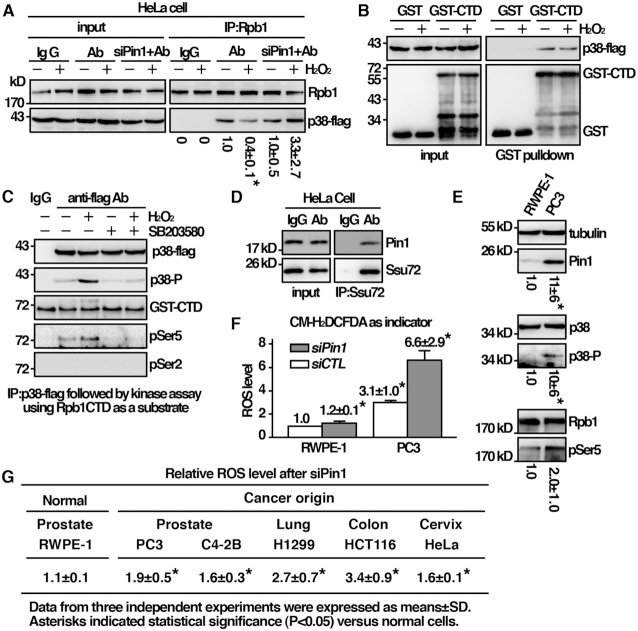
The characteristic feature of the fission yeast Pin1 identified is conserved in humans. (A) Coimmunoprecipitation was performed with extracts prepared from human HeLa cells expressing p38 tagged with flag. Immunoprecipitates were analyzed by Western immunoblotting analysis using antibodies specific for Rpb1 and flag. The relative level of Rpb1 pull down by p38 was shown. Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse control of no treatment. (B) Purified GST-CTD from Escherichia coli and p38-flag precipitated from HeLa cells were subjected to GST pulldown assay followed by Western immunoblotting analysis with antibodies against GST and flag. (C) In vitro kinase assay using p38 extracted and immunoprecipitated from activated cells. Following incubation with GST-CTD, proteins were resolved by SDS-PAGE and probed with antibodies indicated. (D) Coimmunoprecipitation was performed with extracts prepared from human HeLa cells using anti-Ssu72 antibody and IgG as control. Immunoprecipitates were analyzed by Western immunoblotting analysis with antibodies specific for human Pin1 and Ssu72. (E) Total lysates of human cell indicated were subjected to Western immunoblotting analysis with indicated antibodies. The relative level of protein of interest was indicated beneath each lane. Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse RWPE-1 cells. (F) (G) Human cells indicated were subjected to measurement of intracellular ROS Levels in triplicate using CM-H2DCFDA as indicator. Data were expressed as means±SD in triplicate. Asterisks indicated statistical significance (P< 0.05, t-test unpaired) verse normal RWPE-1 cells.
Prostate cancer cells up-regulated Pin1 for adaptation to oxidative stress
The results described above suggest a function of Pin1 in the adaptation to oxidative stress. Cancer cells are continuously exposed to oxidative stress raised from oncogenic stimulation, metabolic defects, hypoxia and treatment of cancer therapies (18). The ability to respond to oxidative stress is thus essential to the survival of cancer cells. The frequent Pin1-overexpression in cancer cells (10) might contribute to improved stress adaptation. Experiments were performed to explore this possibility. As shown in Figure 7E, we found that Pin1 expression in the prostate cancer cell line (PC3) was higher than that in their normal counterparts (RWPE-1). It has been suggested that, in PC3 cells, Pin1 might be negatively regulated at post-transcriptional level by miR-296-5p (19). In line with the alteration and activation of stress response pathway, these results were associated with phosphorylation of p38 and Rpb1-CTD Ser5 in these cells (Figure 7E) and elevated intracellular level of ROS (Figure 7F). Depletion of Pin1 by siRNA led to dramatic increase of ROS in prostate cancer cells (Figure 7F). Similar results could be observed in different types of cancer cells including lung, colon and cervix cancers (Figure 7G). Although Pin1 regulated diverse cellular processes (20), these results suggested a pro-survival function against oxidative stress that could be attributed to the overexpression of Pin1.
DISCUSSION
Pin1 was initially discovered as an important mitotic regulator (6). It targets a number of mitotic proteins with Ser/Thr-Pro dipeptides phosphorylated by cyclin-dependent kinase (CDK). As a consequence, the budding yeast ess1 mutant was not viable exhibiting mitotic arrest and nuclear fragmentation (21). Pin1 knockout mouse also showed cell cycle defects (22). However, unlike ess1, pin1 deletion was not lethal to the fission yeast cells (15). In addition to CDKs, the target of Pin1, pSer/Thr-Pro, matches part of the MAPKs target sites. In this study, we described a function of the fission yeast Pin1 in MAPK-mediated cellular response to oxidative stress. We found that deletion of pin1 affects MAPK-mediated signal transduction pathway. Pin1 played an important role in the process of oxidative stress induced transcription. Ess1 has been reported to affect multiple stages in the transcription cycle including initiation, elongation, 3′-end processing and termination independently (13). The functional role of Pin1 in modulating Pol II transcription via interaction with the Rpb1-CTD has been particularly well studied. We found that this function was important for the initiation to elongation transition of the oxidative stress induced transcription. Our analysis of Sty1-mediated oxidative stress induced transcription suggested a promoter-paused model (Figure 8) resembled that of the first described Drosophila Hsp70 gene (23). As demonstrated by live-cell imaging (24), similar to p38, Sty1 was presented at both cytoplasm and nucleus in unstressed cells. Low level of Atf1 directed Sty1 to specific stress-responsive promoters. Anchored Sty1 directly associated with Rpb1-CTD to recruit Pol II to pause at the promoter. Upon oxidative stress, Sty1 was phosphorylated by upstream kinase Wis1. Phosphorylated Sty1 was activated and accumulated in the nucleus to phosphorylate and stabilize Atf1, which led to further target and recruit Sty1-Pol II to the stress-responsive promoters. Activated Sty1 phosphorylated the CTD reiterated heptad sequence at Serine 5. Pin1 bound phosphorylated CTD to promote Sty1 dissociating from it, and directly recruited Ssu72 to facilitate dephosphorylation of CTD for transcription elongation. In the absence of Pin1, Rpb1 was associated and accumulated at the promoter region following H2O2 stress but failed to enter to elongation to generate transcripts of the corresponding genes. Pin1 might also play a role in elongation. The accumulation of Rpb1 with the gpd1 gene-coding region observed in pin1 mutant (Figure 3C) could be due to elongation defects stalling the translation machinery. In line with this idea, Ess1, the budding yeast homolog of Pin1, has been reported to affect both early and late stages of the transcription cycle (13). Recently, a close relationship among fission yeast Pin1, Ssu72 and Pol II was described (25). In keeping with our results, several Pin1 and Ssu72-dependent genes identified in the study responded to oxidative stress (Table 2) (26). Furthermore, the interaction between Sty1-Rpb1 and Pin1-Ssu72 identified in S. pombe was likely conserved in human p38 and Pin1. Together, these results suggest a conserved function of Pin1 in MAPK-mediated cellular response to oxidative stress, which has significantly expanded our view of the function associated with Pin1. Furthermore, Pin1 overexpression is prevalent in human cancers. We found that elevated Pin1 level might be required to sustain survival under high ROS levels typically seen in cancer cells. Considering that many clinically used cancer therapeutics act by exerting oxidative stress, the level of Pin1 in caner cells may set a threshold determining therapeutic response that appears to be an attractive target for drug development.
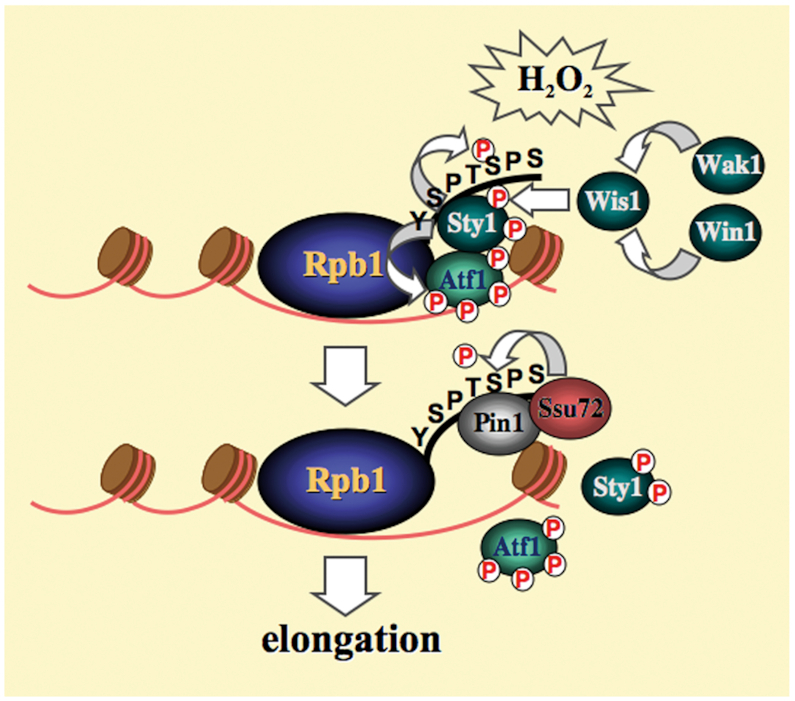
Model of Sty1-mediated oxidative stress induced transcription. Upon oxidative stress, activated Sty1 phosphorylates the CTD reiterated heptad sequence at Serine 5. Pin1 competes the binding of phosphorylated CTD to promote Sty1 dissociating from it, and directly recruits Ssu72 to facilitate dephosphorylation of CTD for transcription elongation.
| Gene | Function |
|---|---|
| SPBC1703.08c | 5-Formytertrahydrofolate cyclo-ligase |
| SPCC794.04c | Amino acid transmembrane transporter |
| dak2 | Dihydroxyacetone kinase |
| SPBPB2B2.01 | Amino acid transmembrane transporter |
| SPAC2H10.01 | Transcription factor |
| SPAC15E1.02c | |
| rds1 | Ferritin related conserved fungal protein |
| SPBC1703.13c | Iorganic phosphate transmembrane transporter |
| qcr8 | Ubiquinol–cytochrome-c reducttase complex subunit |
| SPCC70.08c | Methyltransferase |
| SPACUNK4.17 | NAD binding dehydrogenase |
| gut2 | Glycerol-3-phosphate dehydrogenase |
| anc1 | Mitochondrial ATP:ADP antiporter |
| cdm1 | DNA polymerase delta subunit |
| tpr1 | Paf1 complex subunit |
ACKNOWLEDGEMENTS
We thank Tim Humphrey, Tony Hunter and Paul Russell for yeast strains, Hsiang-Ju Chen and Wei-Ling Wen for technical supports to initiate the project, Yi-Rong Chen for critical review of the manuscript.
FUNDING
National Health Research Institute [MG-108-PP-08]; Ministry of Science and Technology [99-2311-B-400-001-MY3], Taiwan. Funding for open access charge: National Health Research Institutes.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
21.
22.
24.
25.