 Pseudomonas putida MPE, a manganese-dependent endonuclease of the binuclear metallophosphoesterase superfamily, incises single-strand DNA in two orientations to yield a mixture of 3′-PO4 and 3′-OH termini
Pseudomonas putida MPE, a manganese-dependent endonuclease of the binuclear metallophosphoesterase superfamily, incises single-strand DNA in two orientations to yield a mixture of 3′-PO4 and 3′-OH termini
Nucleic Acids Research



- Altmetric
Pseudomonas putida MPE exemplifies a novel clade of manganese-dependent single-strand DNA endonuclease within the binuclear metallophosphoesterase superfamily. MPE is encoded within a widely conserved DNA repair operon. Via structure-guided mutagenesis, we identify His113 and His81 as essential for DNA nuclease activity, albeit inessential for hydrolysis of bis-p-nitrophenylphosphate. We propose that His113 contacts the scissile phosphodiester and serves as a general acid catalyst to expel the OH leaving group of the product strand. We find that MPE cleaves the 3′ and 5′ single-strands of tailed duplex DNAs and that MPE can sense and incise duplexes at sites of short mismatch bulges and opposite a nick. We show that MPE is an ambidextrous phosphodiesterase capable of hydrolyzing the ssDNA backbone in either orientation to generate a mixture of 3′-OH and 3′-PO4 cleavage products. The directionality of phosphodiester hydrolysis is dictated by the orientation of the water nucleophile vis-à-vis the OH leaving group, which must be near apical for the reaction to proceed. We propose that the MPE active site and metal-bound water nucleophile are invariant and the enzyme can bind the ssDNA productively in opposite orientations.
INTRODUCTION
Nucleases, helicases, and polynucleotide ligases are agents of DNA repair found in all organisms. Multiple nucleases, helicases, and ligases may coexist in the same organism, whereby enzyme paralogs or homologs are dedicated to the repair of particular types of DNA lesions. In bacteria, the grouping of genes encoding nucleases, helicases, or ligases in operons can suggest the existence of a coherent repair pathway. For example, we identified a cluster of four genes encoding predicted nuclease, helicase, and ligase enzymes that is found in many bacteria from diverse phyla (1). The four enzymes are: (i) Lhr-Core, a 3′-5′ DNA helicase; (ii) MPE, a DNA endonuclease; (iii) Exo, a putative exonuclease and (iv) an ATP-dependent DNA ligase. Many other bacteria and archaea have a two-gene cluster encoding Lhr-Core and MPE (1).
To understand how MPE might contribute to nucleic acid repair, we characterized the MPE protein specified by the Exo•Lig•Lhr-Core•MPE gene cluster of Pseudomonas putida (1,2). The 216-amino acid P. putida MPE protein is a member of the metallophosphoesterase superfamily of enzymes that utilize a binuclear transition metal ion center to catalyze phosphomonoester or phosphodiester hydrolysis (2,3). We found that P. putida MPE is a manganese-dependent phosphodiesterase that releases p-nitrophenol from bis-p-nitrophenylphosphate (kcat 212 s−1) and p-nitrophenyl-5′-thymidylate (kcat 34 s−1) but has no detectable phosphomonoesterase activity against p-nitrophenylphosphate (1). Anent its potential nucleic acid repair function, MPE is a manganese-dependent single-strand DNA endonuclease that incises linear single strands and the single-stranded loops of DNA stem–loop structures. MPE has feeble activity on duplex DNA (2).
A crystal structure of P. putida MPE at 2.2 Å resolution showed that the active site includes two octahedrally coordinated manganese ions (2). Seven signature amino acids of the binuclear metallophosphoesterase superfamily serve as the enzymic metal ligands in MPE: Asp33, His35, Asp78, Asn112, His124, His146, and His158 (Figure 1A). A swath of positive surface potential on either side of the active site pocket suggested a binding site for the negatively charged backbone of the single-strand DNA substrate of the MPE endonuclease reaction. MPE amino acids that contribute to the positive surface include Arg83, Arg86, His81, Lys38, Arg172 and His113 (Figure 1A).

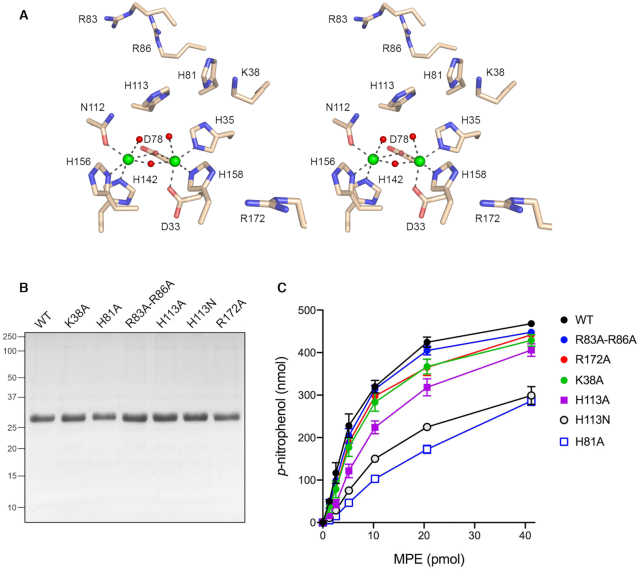
Structure-guided mutational analysis of P. putida MPE. (A) Stereo view of the MPE active site and metal-coordination complexes (from PBD 6NVO). MPE amino acids are depicted as stick models with beige carbons. Mn1 and Mn2 are rendered as green spheres. Waters are depicted as red spheres. Atomic contacts of the manganese ions are denoted by black dashed lines. Six amino acids near the metal complex (Lys38, His81, Arg83, Arg86, His113 and Arg172) that were targeted for mutagenesis in the present study are shown. (B) Aliquots (5 μg) of recombinant wild-type (WT) MPE and the indicated MPE mutants were analyzed by SDS-PAGE. The Coomassie blue-strained gel is shown. The positions and sizes (kDa) of marker polypeptides analyzed in parallel are indicated on the left. (C) Reaction mixtures (50 μl) containing 100 mM Tris–HCl, pH 8.0, 30 mM NaCl, 2 mM MnCl2, 10 mM bis-p-nitrophenylphosphate, and wild-type or mutant MPE as specified were incubated at 37°C for 30 min. The extent of p-nitrophenol formation is plotted as a function of input enzyme. Each datum represents the average of three independent enzyme titration experiments ± SEM.
The structure of MPE highlighted significant differences between MPE and the DNA nucleases Mre11 and SbcD, which are also binuclear metallophosphoesterases (4–12). Mre11 is the nuclease subunit of a eukaryal DNA end-processing complex (which includes an SMC-like ATPase subunit Rad50) that functions in the homologous recombination (HR) and non-homologous end-joining (NHEJ) pathways of DNA double-strand break repair (reviewed in 13). Bacterial SbcD and its SMC-like ATPase partner SbcC form a SbcCD complex (14), a homolog of Mre11•Rad50, that triggers replication-dependent double-strand breaks at palindromic sequences in the bacterial chromosome (15). The Mre11•Rad50 and SbcCD complexes have ATP-dependent double-strand DNA exonuclease activity and ATP-independent single-strand endonuclease activity. Like MPE, SbcCD is adept at cleaving the single-strand loops of DNA hairpin structures (16). The distinctive features of MPE are as follows: (i) MPE is a monomer, whereas Mre11 and SbcD are homodimers; (ii) MPE lacks the so-called capping domain present in Mre11 and SbcD; and (iii) the topology of the β sandwich that comprises the core of the metallophosphoesterase fold differs in MPE vis-à-vis Mre11 and SbcD. Thus, MPE exemplifies a novel clade of DNA endonuclease within the binuclear metallophosphoesterase superfamily.
In the present study, we examine in greater detail the nuclease activity of P. putida MPE, focusing on its ability to incise various DNA structures—e.g., tailed duplexes, nicks and mismatches—that are potentially relevant to DNA repair. An important outstanding issue for MPE is how the scissile DNA strand is engaged in the nuclease active site to promote phosphodiester hydrolysis via proper orientation of a water nucleophile and a DNA leaving group. We take two approaches to this problem. First, we conducted a structure-guided mutational analysis of the basic residues that flank the active site binuclear metal complex, which we view as candidate ligands for DNA phosphates. Second, we interrogated the termini generated by MPE cleavage. Our results reveal unexpected complexity in the MPE mechanism, whereby single-strand DNA can bind productively to the nuclease in either of two opposite orientations to generate either 3′-OH or 3′-PO4 products of phosphodiester hydrolysis.
MATERIALS AND METHODS
P. putida MPE
Recombinant MPE and mutants thereof were produced in E. coli as His10Smt3 fusions and purified from soluble extracts by serial nickel-affinity, tag cleavage, and tag removal steps as described previously (1). Protein concentrations were determined with the BioRad dye reagent using BSA as the standard. For experiments employing wild-type MPE only, the enzyme was subjected to Superose-200 gel filtration (1).
Nuclease substrates
The 5′ 32P-labeled single-strand DNA substrates were prepared by reaction of synthetic oligonucleotides with T4 polynucleotide kinase and [γ32P]ATP. The labeled DNA was separated from free ATP by electrophoresis through a nondenaturing 18% polyacrylamide gel and then eluted from an excised gel slice by overnight incubation at 4°C in 200 μl of 10 mM Tris–HCl, pH 7.5, 1 mM EDTA. To form the 3′ and 5′ tailed DNA duplexes and DNA duplexes with central base mispairs, the radiolabeled pDNA strand and cold complementary DNA oligonucleotides were annealed at a 1:1.5 molar ratio in 10 mM Tris–HCl, pH 7.4, 1 mM EDTA by incubating for 10 min at 65°C, 15 min at 37°C, and then 30 min at 22°C.
MPE hydrolysis of bis-p-nitrophenylphosphate
Reaction mixtures (25 μl) containing 100 mM Tris–HCl, pH 7.5, 10 mM bis-p-nitrophenylphosphate (Sigma), 2 mM MnCl2, and MPE as specified were incubated for 30 min at 37°C. The reactions were quenched by adding 50 μl of 50 mM EDTA and then 0.9 ml of 1 M Na2CO3. Release of p-nitrophenol was determined by measuring A410 and interpolating the value to a p-nitrophenol standard curve.
MPE DNA nuclease assay
Reaction mixtures (10 μl) containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM DTT, 1 mM MnCl2 and 1 pmol (0.1 μM) 5′ 32P-labeled DNA and MPE as specified in the figure legends were incubated at 37°C. The reactions were quenched at the times specified by adding 10 μl of 90% formamide, 50 mM EDTA. The samples were heated at 95°C for 5 min and then analyzed by electrophoresis through a 40-cm 18% polyacrylamide gel containing 7.5 M urea in 44.5 mM Tris–borate, pH 8.3, 1 mM EDTA. The products were visualized by autoradiography.
RESULTS
Structure-guided mutational analysis
A stereo view of the MPE active site highlighting the binuclear metal complex is shown in Figure 1A. The metal-binding mode is characteristic of the metallophosphoesterase superfamily. The Mn1 atom is octahedrally coordinated to Asp33-Oδ, His35-Nϵ, Asp78-Oδ, His158-Nϵ and two waters. The octahedral Mn2 coordination complex includes Asp78-Oδ, Asn112-Oδ, His142-Nϵ, His156-Nδ and two waters. The Mn1 and Mn2 atoms are 3.6 Å apart and are bridged by Asp78 and one of the waters. We reported previously that mutation of Asp78 to alanine squelches MPE phosphodiesterase activity with bis-p-nitrophenylphosphate and its DNA endonuclease activity (1). We proposed that the metal-bridging water is the likely nucleophile in the phosphodiesterase reaction, insofar as its proximity to the two metals will significantly lower the pKa of the water and activate it for attack on the DNA backbone. We suggested that the other two waters of the M1 and M2 coordination spheres occupy the positions of two of the phosphate oxygens of the scissile phosphodiester. The His113 side chain located above the metal complex is conserved in many phosphodiesterase/nuclease enzymes of the metallophosphoesterase superfamily; this histidine may serve as a general acid catalyst, by donating a proton to the R'-OH leaving group (4,17). Because the amino acids comprising the conserved metal-binding site have been mutated extensively in other binuclear metallophosphoesterases and shown to be critical for activity (e.g. see (17–22)), we focused the present structure-function analysis of MPE on His113 and five other basic amino acids near the active site, none of which are involved in metal-binding, that we viewed as potential ligands for either the scissile phosphodiester or the DNA phosphates flanking the scissile phosphodiester. Recombinant MPE mutants K38A, H81A, R83A-R86A, H113A, H113N, and R172A were produced in E. coli and purified from soluble bacterial lysates in parallel with wild-type MPE, as described (1). SDS-PAGE affirmed comparable extents of purification of the MPE polypeptide (Figure 1B).
The MPE preparations were assayed initially by titration for hydrolysis of bis-p-nitrophenylphosphate (Figure 1C). Our reasoning was that an alanine substitution for an amino acid functional group essential for phosphodiesterase chemistry would exert a significant (>10-fold) negative effect on bis-p-nitrophenylphosphatase activity (i.e. akin to the D78A mutation). Because bis-p-nitrophenylphosphate is a generic substrate, with relatively low affinity for MPE (Km of 4.85 mM), and one that lacks phosphates other than the scissile phosphodiester, we expected that alanine mutation of an amino acid that interacts with DNA flanking phosphates during the endonuclease reaction might have little effect on bis-p-nitrophenylphosphatase activity. With respect to a putative enzymic general acid, the low pKa value of the p-nitrophenol leaving group (pKa of 7.15) versus that of a deoxyribonucleotide 3′-OH (pKa of ∼14) (23) engenders the prediction that a mutation of the general acid to alanine or a nonionizable congener might be relatively benign with respect to bis-p-nitrophenylphosphatase activity but disastrous for MPE DNA endonuclease activity. From the results of the enzyme titrations, the bis-p-nitrophenylphosphatase specific activities of the mutants were as follows, normalized to a value of 100% for wild-type: R83A-R86A (89%); R172A (82%); K38A (77%); H113A (51%); H113N (34%) and H81A (23%). We conclude that none of these amino acids is essential for phosphodiesterase chemistry during hydrolysis of bis-p-nitrophenylphosphate.
To gauge mutational effects on DNA endonuclease activity, we reacted 10 pmol MPE with 1 pmol of 5′ 32P-labeled 36-mer single-strand DNA oligonucleotide of mixed nucleobase composition (Figure 2A). Wild-type MPE incised the 36-mer to generate a ladder of shorter 5′ 32P-labeled cleavage products. Note that the complexity of the cleavage ladder (i.e. the number of resolved labeled oligonucleotides) was greater than the number of phosphodiester bonds in the the 36-mer ssDNA substrate. The K38A, R83A-R86A, and R172A mutants generated cleavage ladders of similar complexity to wild-type MPE, though the efficiency of cleavage was reduced versus wild-type MPE, as reflected in the levels of residual uncleaved 36-mer. The salient findings were that the H81A, H113A, and H113N mutants were inert in DNA cleavage (Figure 2A). That neither H113A nor H113N was capable of DNA phosphodiester hydrolysis is consistent with the hypothesis that His113 is the general acid that protonates the terminal oxygen of the leaving DNA strand. We speculate that the essential His81 side chain might engage a DNA phosphate flanking the scissile phosphodiester. Alternatively, His81, which is within hydrogen-bonding distance of His113, could ensure a proper rotamer/tautomer configuration of the His113 general acid and, perhaps, comprise part of a proton relay to the leaving group in the nuclease reaction. His81 is also within hydrogen-bonding distance of the metal ligand His35 and the loss of this contact could contribute to the 4-fold decrement in bis-p-nitrophenylphosphatase activity seen for the H81A mutant.
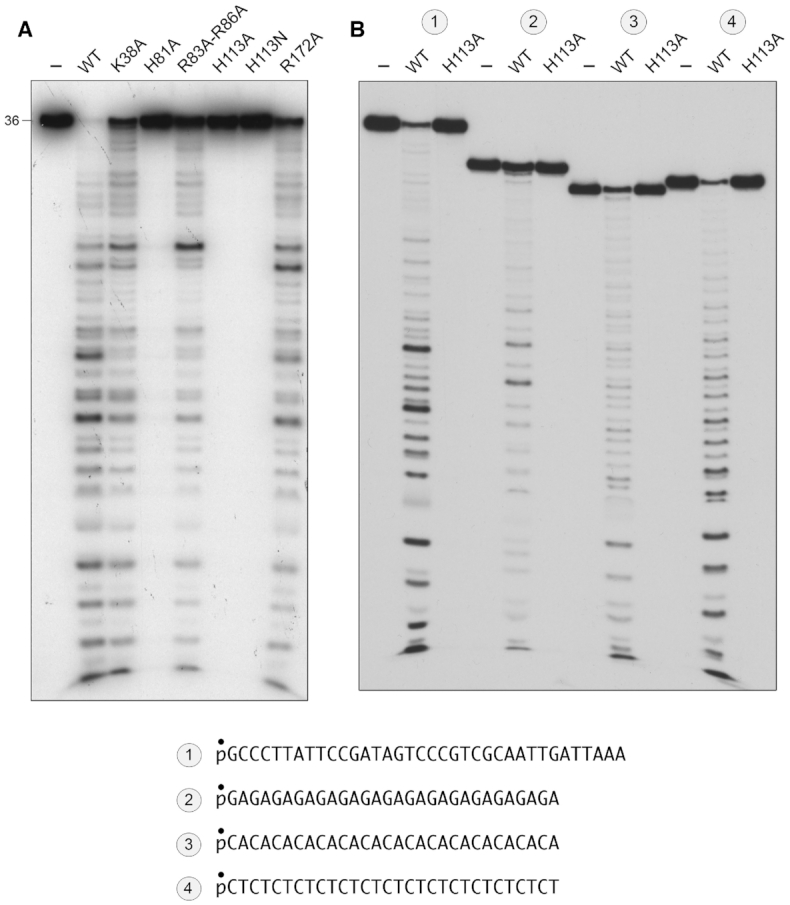
Mutational effects on DNA endonuclease activity. (A) Reaction mixtures (10 μl) containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM MnCl2, 1 mM DTT, 1 pmol (0.1 μM) 5′ 32P-labeled 36-mer ssDNA substrates (shown at bottom), and 10 pmol wild-type or mutant MPE as specified were incubated for 30 min at 37°C. (B) Reaction mixtures (10 μl) containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM MnCl2, 1 mM DTT, 1 pmol (0.1 μM) 5′ 32P-labeled 36-mer ssDNA substrate or 30-mer ssDNA substrates comprising tandem GA, CA or CT dinucleotide repeats (shown at bottom), and 10 pmol wild-type MPE or H113A mutant as specified were incubated for 30 min at 37°C. The products of the reactions in A and B were analyzed by urea-PAGE and visualized by autoradiography.
MPE cleavage of alternating DNA copolymers
In the experiment in Figure 2B, wild-type MPE and the H113A mutant (10 pmol) were reacted with 1 pmol of a series of 5′ 32P-labeled 30-mer DNA substrates consisting of 15 tandem dinucleotide repeats: either GA, CA or CT, which are, in principle, unable to form secondary structures. Wild-type MPE incised the 30-mers to generate ladders of shorter 5′ 32P-labeled cleavage products. Here again, it was noteworthy that the number of resolved labeled oligonucleotides exceeded the number of phosphodiester bonds in the 30-mer DNA copolymer substrates. This issue will be addressed further below. The H113A mutant was unreactive with the three alternating copolymer ssDNA substrates (Figure 2B).
MPE cleavage of tailed duplex and nick substrates
Single-stranded regions of genomic DNA are exposed or generated during DNA replication and DNA repair, an event abetted in many cases by a helicase and/or an exonuclease, which are two of the enzymes encoded in the same bacterial operon as MPE (1). Single-stranded regions can take the form of 3′ or 5′ single stranded tails emanating from duplex DNA or gaps between two segments of duplex DNA. Here we reacted MPE in parallel with a 5′ 32P-labeled 36-mer ssDNA and the same strand annealed to either of two unlabeled 18-mer strands complementary to the 5′ or 3′ halves of the 36-mer, thereby forming the 3′ and 5′ tailed structures shown in Figure 3. Whereas MPE incised the 36-mer ssDNA at sites throughout the strand, the creation of a duplex suppressed cleavages within the duplex segments so that incision was limited to the respective 3′ and 5′ ssDNA tail segments (Figure 3).
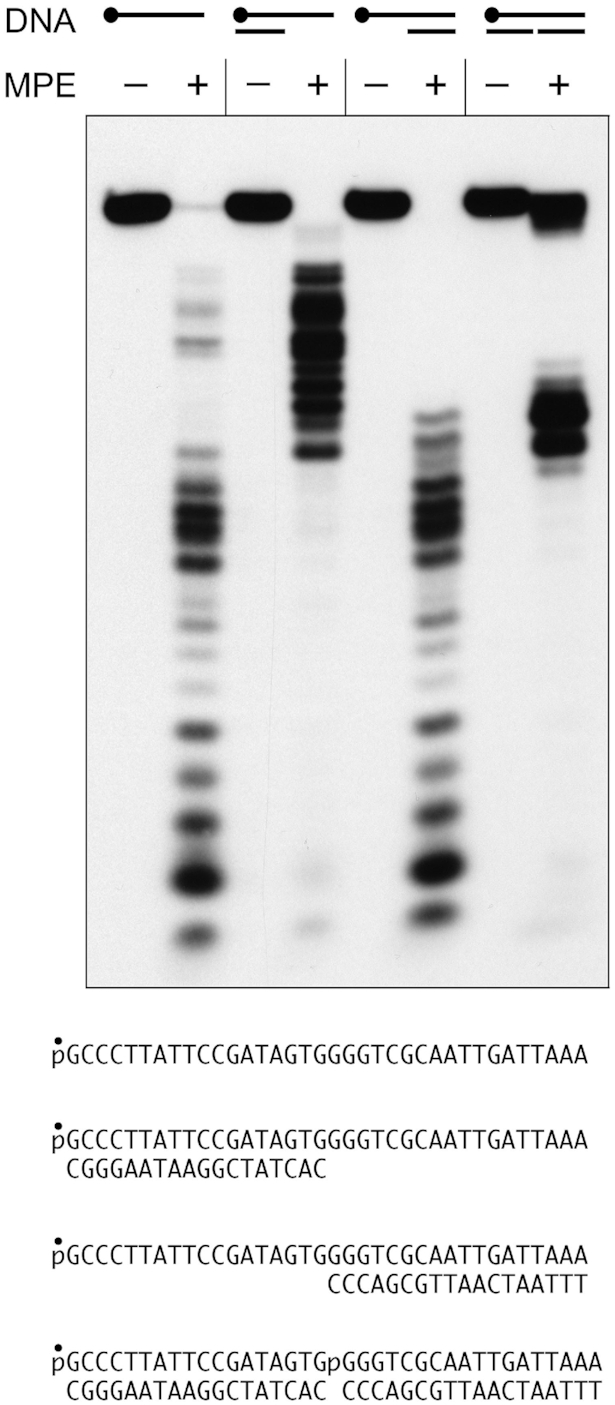
MPE cleavage of tailed duplex and nick substrates. Reaction mixtures (10 μl) containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM MnCl2, 1 mM DTT, 1 pmol (0.1 μM) 5′ 32P-labeled 36-mer ssDNA, 3′ or 5′ tailed duplex DNA, or 36-mer nicked duplex DNA substrates (shown at bottom), and 10 pmol (1 μM) MPE (where indicated by +) were incubated for 30 min at 37°C. The reaction mixtures were analyzed by urea-PAGE and the labeled DNAs were visualized by autoradiography.
Simultaneous annealing 32P-labeled 36-mer to the two unlabeled complementary 18-mer strands generated a nicked duplex DNA substrate depicted in Figure 3. Nicked DNA awaiting sealing by a DNA ligase is the common endpoint of lagging strand DNA replication and gap-filling during DNA repair. Nicks are also generated directly by endonucleases. It was of interest to test if MPE can sense a nick and cleave the intact strand of the nicked duplex. As shown in Figure 3, MPE did incise the intact 36-mer strand at site(s) opposite the nick, generating a tight cluster of 32P-labeled cleavage products of a size that corresponds to the junction of the cleavage patterns seen on the 3′ and 5′ ssDNA flaps. Thus, MPE is capable of converting a single-strand nick into a double-strand break.
MPE cleavage at sites of base mispairing
We tested MPE for its ability to sense and incise a run of three mispaired nucleobases within an intact 36-mer DNA duplex. Two such duplexes were formed by annealing a 5′ 32P-labeled 36-mer ssDNA strand to an unlabeled complementary strand to form a duplex with either a central AAA/GGG mismatch or a TTT/CCC mismatch (as shown in Figure 4). MPE incised the labeled AAA and TTT single strands of the mismatched duplexes at sites within or immediately adjacent to the mismatched segment of the duplex to generate a tight cluster of cleavage products (Figure 4; substrates 2 and 4) that contrasted with the promiscuous cleavages seen with the isolated ssDNA strands (Figure 4; substrates 1 and 3). Duplexes with a central AA/GG mismatch or a single A/G mismatch were not effective substrates for MPE incision (not shown).
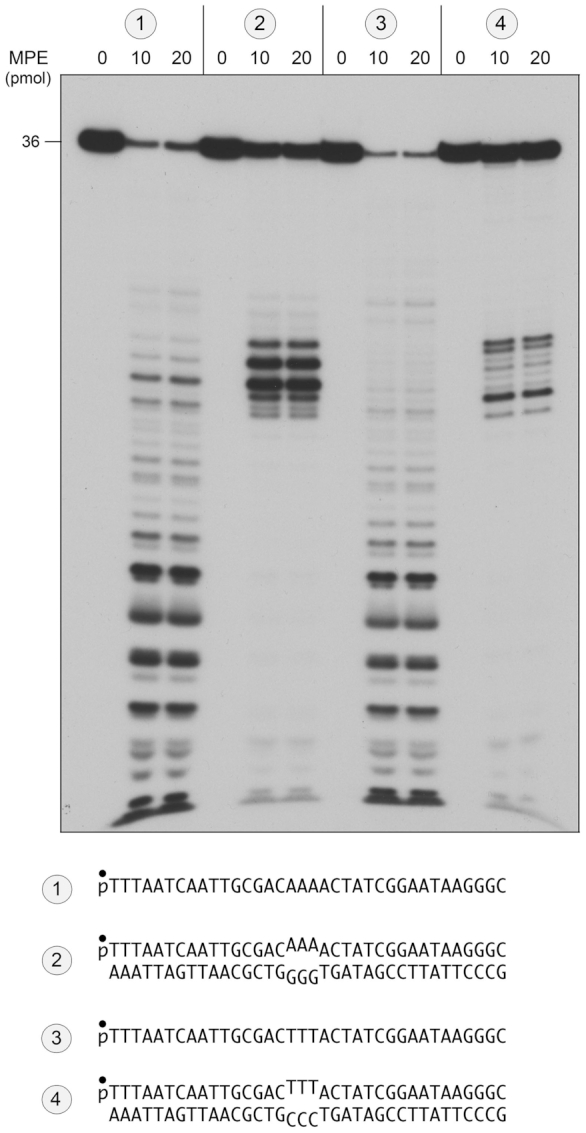
MPE cleavage of DNA mismatch substrates. Reaction mixtures (10 μl) containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM MnCl2, 1 mM DTT, 1 pmol (0.1 μM) 5′ 32P-labeled DNA substrates ( ,
,  ,
,  , or
, or  ; shown at bottom), and 0, 10 or 20 pmol MPE were incubated for 30 min at 37°C. The reaction mixtures were analyzed by urea–PAGE and the labeled DNAs were visualized by autoradiography.
; shown at bottom), and 0, 10 or 20 pmol MPE were incubated for 30 min at 37°C. The reaction mixtures were analyzed by urea–PAGE and the labeled DNAs were visualized by autoradiography.
MPE cleaves ssDNA to generate a mixture of 3′-PO4 and 3′-OH product termini
As noted above, and in previous product analyses of the ssDNA endonuclease activity by P. putida MPE (2), the number of 5′-labeled cleavage products resolvable by urea–PAGE exceeded the number of phosphodiesters in the input DNA substrate. This raised the prospect that the termini of the MPE cleavage products at some or many of the DNA phosphodiesters might be heterogeneous, comprising a mixture of 5′ 32P-labeled oligonucleotides with 3′-OH and 3′-PO4 ends, in which the 3′-PO4 species migrates faster during PAGE than the same DNA with a with 3′-OH end. As an initial test of this idea, we treated the products of MPE cleavage of the 30-mer p(CT)15 substrate with purified bacteriophage T4 polynucleotide kinase-phosphatase (Pnkp), which will convert 3′-PO4 ends to 3′-OH end products. We found that the Pnkp 3′-phosphatase eliminated a subset of the MPE cleavage products (denoted by • in Figure 5A) by converting them to more slowly migrating species. We could assign the cleavage products by electrophoresis in parallel with 5′ 32P-labeled oligonucleotides pCTCTCTCTCTCTCTCOH (15-mer) and pCTCTCTCTCTCTOH (12-mer) that demarcated specific internal incisions generating 3′-COH and 3′-TOH ends. These are labeled C(15) and T(12) in Figure 5A. The complexity of the T4 Pnkp-treated MPE products corresponded to a ladder of incisions at every phosphodiester of the input substrate (excluding the shortest 5′-labeled oligonucleotides that were compressed and therefore unresolved at the electrophoretic front). Figure 5A shows an alternating pattern of stronger and weaker cleavages within the phosphatase-treated product ladder. In a separate experiment in which a mixture of the purified 5′ 32P-labeled 15-mer and 12-mer standards was analyzed in parallel with the MPE products of p(CT)15 cleavage, we scanned the T4 Pnkp-treated MPE product ladder with ImageQuant software and plotted the intensity of 5′ 32P radiolabel signal versus electrophoretic mobility (i.e., product size). The product profile (Figure 6A), wherein all MPE-generated 3′ ends have been converted to 3′-OH, indicated that MPE displayed a preference for incision of p(CT)15 at 5′-TpC phosphodiesters versus 5′-CpT phosphodiesters.
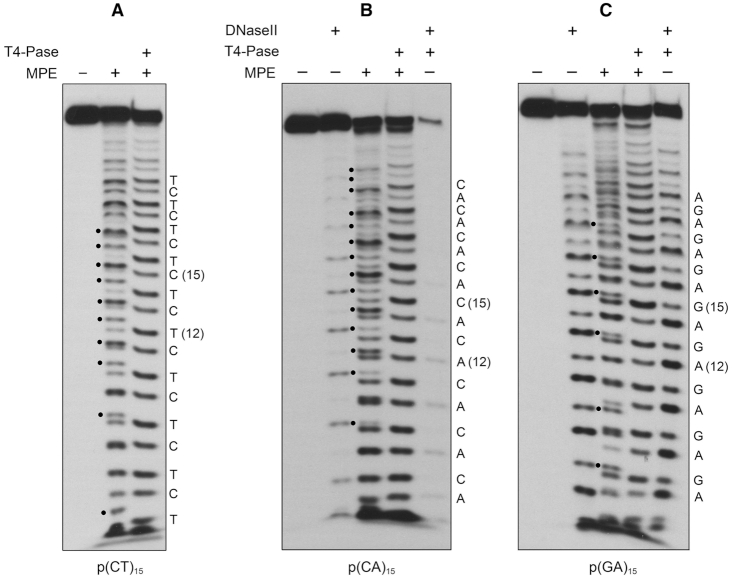
MPE cleaves ssDNA to generate a mixture of 3′-PO4 and 3′-OH product termini. MPE reaction mixtures containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM MnCl2, 1 mM DTT, 1 pmol (0.1 μM) 5′ 32P-labeled 30-mer ssDNA substrates p(CT)15 (panel A), p(CA)15 (panel B), or p(GA)15 (panel C), and 2.5 pmol MPE for p(CT)15 or 5 pmol MPE for p(CA)15 and p(GA)15 (where indicated by +) were incubated for 30 min at 37°C. DNAse II reaction mixtures (10 μl) containing 100 mM sodium acetate, pH 4.6, 1 mM MgCl2, 1 pmol 5′ 32P-labeled 30-mer ssDNA substrate, and 0.02 U porcine spleen DNAse II (where indicated by +) were incubated for 30 min at 37°C. For subsequent T4 Pnkp 3′-phosphatase (3′-Pase) treatment (where indicated by +), the MPE and DNase II reaction mixtures were heated at 95°C for 5 min, adjusted to 100 mM Tris-acetate, pH 6.0, 10 mM MgCl2, 2 mM DTT buffer, supplemented with 5 pmol T4 Pnkp, and incubated for 30 min at 37°C. Reactions were quenched with EDTA/formamide and the products were analyzed by urea-PAGE and visualized by autoradiography. MPE cleavage products with 3′-PO4 termini susceptible to hydrolysis by T4 polynucleotide 3′-phosphatase are indicated by • to the left of the MPE cleavage ladders. The identities of the terminal 3′-OH deoxynucleotides (assigned by reference to 5′ 32P-labeled 12-mer and 15-mer oligonucleotide alternating copolymer markers) are indicated on the right in each panel.
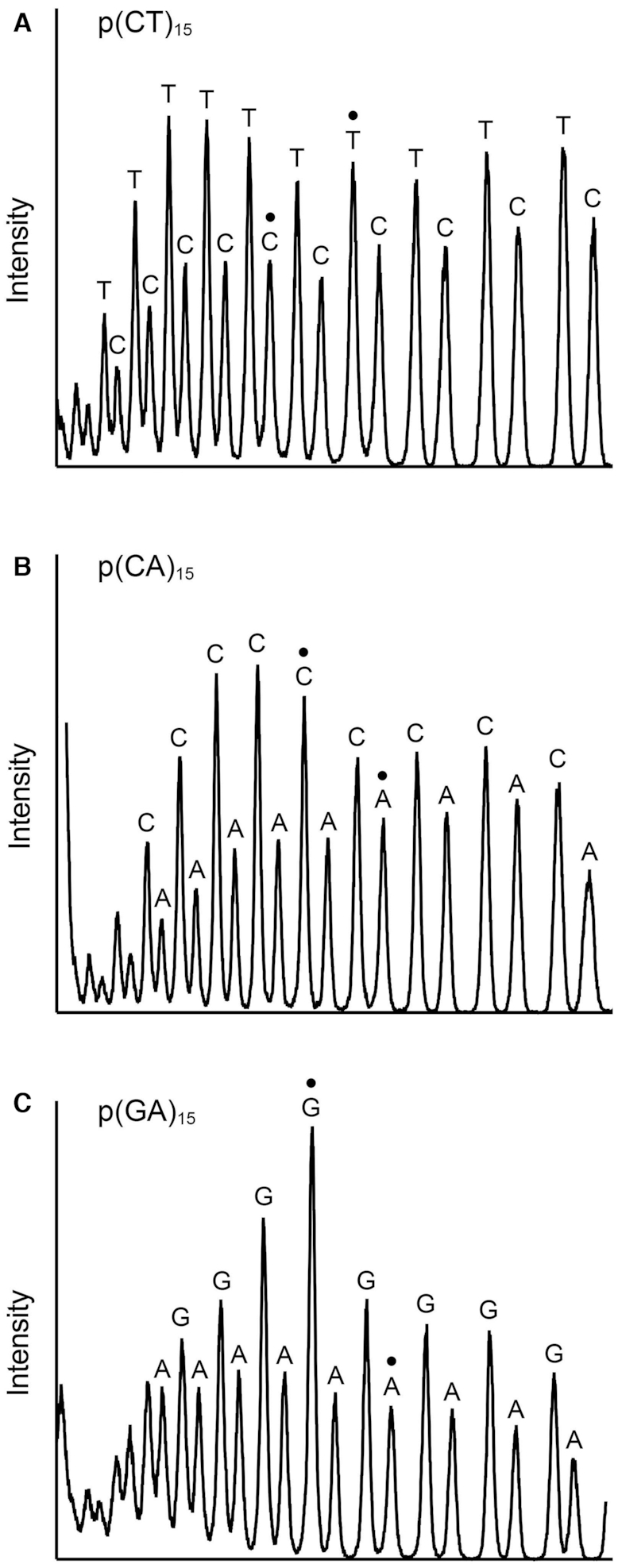
MPE cleavage site preferences. Replicate MPE reaction mixtures (10 μl) containing 20 mM Tris–HCl, pH 8.0, 30 mM NaCl, 1 mM MnCl2, 1 mM DTT, 1 pmol (0.1 μM) 5′ 32P-labeled 30-mer ssDNA substrates p(CT)15 (panel A), p(CA)15 (panel B) or p(GA)15 (panel C), and 2.5 pmol MPE for p(CT)15 or 5 pmol MPE for p(CA)15 and p(GA)15 were incubated for 30 min at 37°C. One set of reaction mixtures was quenched immediately. The other was treated with T4 Pnkp as described in Figure 5. The p(CT)15, p(CA)15 and p(GA)15 cleavage ladders were analyzed by urea-PAGE in parallel with a mixture of two 5′ 32P-labeled 12-mer and 15-mer alternating copolymer markers (that had been individually 5′ 32P-labeled a gel-purified). The markers were: pCTCTCTCTCTCTOH and pCTCTCTCTCTCTCTCOH; pCACACACACACAOH and pCACACACACACACACOH; pGAGAGAGAGAGAOH and pGAGAGAGAGAGAGAGOH. The gel was scanned with a Typhoon FLA 7000 (Version 1.2) Imager and the products in the phosphatase-treated MPE cleavage ladders were quantified using ImageQuant software. The radiolabel signal intensities are plotted in panels A-C, wherein the direction of electrophoresis is from left to right. The peaks corresponding to the 15-mer and 12-mer alternating copolymer markers are indicated by • in each panel. The identities of the terminal 3′-OH deoxynucleotides are indicated above the peaks.
We performed similar analyses of the products of MPE cleavage of the 30-mer p(CA)15 substrate (Figure 5B), as such or after treatment with T4 Pnkp. We included two additional samples in which: (i) the 30-mer p(CA)15 strand was treated with DNase II, a phosphodiesterase that exclusively yields 3′-PO4 and 5′-OH termini at its sites of cleavage and (ii) the DNase II cleavage products were treated with T4 Pnkp to convert the 5′ 32P-labeled 3′-PO4-terminated species to their 3′-OH terminated counterparts. As seen in Figure 5B, DNase II incised every other phosphodiester of the 30-mer p(CA)15 substrate, consistent with its preference for cleavage at purines (24), and, as expected, the DNase II cleavage ladder was shifted upward after treatment with T4 Pnkp. The salient findings were that Pnkp treatment eliminated many of the MPE cleavage products derived from its reaction with p(CA)15 (indicated by • in Figure 5B), converting them to more slowly migrating species. We assigned the MPE p(CA)15 cleavage products by electrophoresis in parallel with 5′ 32P-labeled oligonucleotides pCACACACACACACACOH (15-mer) and pCACACACACACAOH (12-mer) that demarcated specific incisions generating 3′-COH and 3′-AOH ends, labeled C(15) and A(12) in Figure 5B. An alternating pattern of stronger and weaker cleavages within the Pnkp-treated MPE p(CA)15 product ladder was evident in Figure 5B. Quantification of the product distribution was performed in separate experiment, which showed that MPE preferred to cleave p(CA)15 at 5′-CpA phosphodiesters versus 5′-ApC phosphodiesters (Figure 6B). By contrast, DNase II cleaved p(CA)15 preferentially at 5′-ApC phosphodiesters (Figure 5B).
We extended this analysis to MPE cleavage of the 30-mer p(GA)15 substrate (Figure 5C). Again, many of the MPE cleavage products were sensitive to T4 Pnkp 3′-phosphatase treatment (examples indicated by • in Figure 5C), and their conversion to more slowly migrating species was apparent in several cases. We observed alternating stronger and weaker cleavages within the Pnkp-treated MPE p(GA)15 product ladder (Figure 5C). Quantification of the product distribution in a separate experiment showed that MPE preferred to cleave p(GA)15 at 5′-GpA phosphodiesters versus 5′-ApG phosphodiesters (Figure 6C). By contrast, DNase II preferred to incise p(GA)15 at 5′-ApG phosphodiesters (Figure 5C).
Collectively, the results of the experiments in Figure 5 show that MPE hydrolyzed individual ssDNA phosphodiesters in either of two orientations to yield a mixture of 3′-PO4 and 3′-OH product termini. This is in contrast to DNase II and most other DNA phosphodiesterases that have strict specificity with respect to the product termini they generate. From the ladders in Figure 5, we can judge that the relative distribution of MPE-generated 3′-PO4 and 3′-OH ends at any particular scissile phosphodiester can vary within the 30-mer ssDNA strands.
DISCUSSION
P. putida MPE exemplifies a new clade of bacterial DNA endonucleases within the binuclear metallophosphoesterase enzyme superfamily. The present study advances our understanding of MPE in three respects. First, by structure-guided mutagenesis, we identify His113 and His81 as essential for ssDNA nuclease activity, albeit inessential for bis-p-nitrophenylphosphatase activity. We suggest that: His113 contacts the scissile phosphodiester and serves as a general acid catalyst to expel the OH leaving group of the product strand (a function less critical for hydrolysis of bis-p-nitrophenylphosphate); and His81 either contacts a backbone phosphate adjacent to the scissile phosphodiester or promotes a proper rotamer/tautomer of the His113 general acid. Second, by surveying a variety of DNA structures, we affirm that MPE cleavage is suppressed by correctly paired duplex secondary structure. We find that MPE cleaves the 3′ and 5′ single-strands of tailed duplex DNAs and that MPE can sense and incise duplexes at sites of short mismatch bulges and opposite a nick. Such structures are prone to form via DNA damage, replication errors, and/or DNA repair. Third, we show that MPE is an ambidextrous phosphodiesterase capable of hydrolyzing the ssDNA backbone in either orientation to generate a mixture of 3′-OH and 3′-PO4 cleavage products. This ambidexterity is unusual for nucleases. For example, DNase II and DNase I exclusively generate 3′-PO4 and 3′-OH ends, respectively. DNA restriction endonucleases typically form 3′-OH ends exclusively. Transesterifying ribonucleases, e.g. RNase A and RNase T1, exclusively yield cleavage products with 3′-PO4 termini.
The directionality of nucleic acid phosphodiester hydrolysis is dictated by the orientation of the water nucleophile with respect to the R'–O leaving group of the product strand, which must be near apical in the Michaelis complex for the reaction to proceed. Nucleases can ensure a single polarity of cleavage by constraining how the nucleic acid binds to the enzyme so that only one productive orientation of the scissile phosphodiester in the active site is possible. The ambidexterity observed for MPE could, in principle, arise from either of two scenarios: (i) MPE permits a single orientation of ssDNA binding, but the active site is plastic and more than one of the metal-bound waters can serve as the nucleophile, with different leaving group outcomes for each water or (ii) the active site and chemical mechanism are inflexible, but the MPE enzyme can bind the ssDNA in either of two opposite productive orientations. Parsimony leads us to favor the latter model, as illustrated in Figure 7 showing stereo views of the active site metal and metal-bridged water from the MPE crystal structure, with a DNA dinucleotide modeled on the enzyme surface above the active site such that the nonbridging oxygens of the scissile phosphodiester are superimposed on the other two metal-bound waters (see Figure 1A). This places the phosphorus atom adjacent to the metal-bridged water (red sphere). The DNA dinucleotide could be modeled in either of two directions. As depicted in Figure 7A, when the deoxyribose O5′ is oriented nearly apical to the metal-bridged water nucleophile, the cleavage reaction will yield 5′-OH and 3′-PO4 DNA ends. As shown in Fig, 7B, when the deoxyribose O3′ is nearly apical to the water nucleophile, the reaction generates 3′-OH and 5′-PO4 DNA ends. The His113 side chain is in position to donate a hydrogen to the leaving deoxyribose oxygen atom in either orientation.
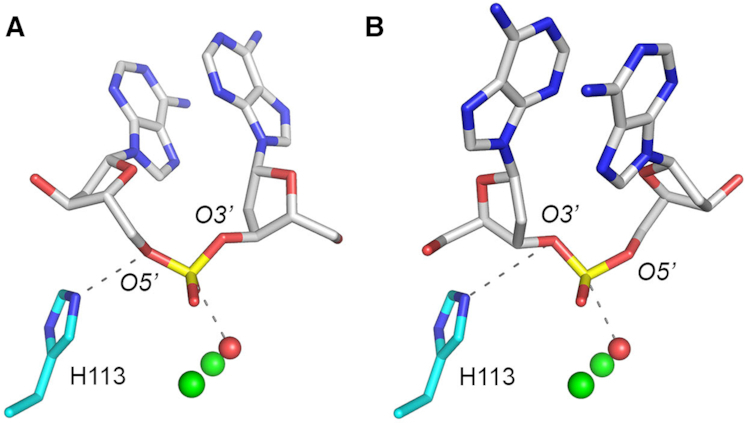
Model for MPE phosphodiesterase activity in alternative DNA-binding orientations. The figure shows views of the MPE active site (from PBD 6NVO) highlighting the two manganese ions (green spheres), the proposed metal-bridged water nucleophile (red sphere), the putative His113 general acid, and a DNA dinucleotide modeled in either of two potential orientations on the exposed surface of the MPE tertiary structure. The nonbridging oxygens of the scissile phosphodiester were superimposed on the two other metal-bound waters (refer to Figure 1A), which were omitted from the models shown here. The phosphorus atom was thereby positioned adjacent to the metal-bridged water. Panel A. When the deoxyribose O5′ is oriented nearly apical to the metal-bridged water nucleophile, the cleavage reaction yields 5′-OH and 3′-PO4 DNA ends. Panel B. When the deoxyribose O3′ is nearly apical to the water nucleophile, the reaction generates 3′-OH and 5′-PO4 DNA ends.
There is precedent for a continuum of strict directionality versus ambidexterity in phosphodiester hydrolysis by members of the binuclear metallophosphoesterase family that act on ribonucleotide 2′,3′-cyclic phosphates, whereby the outcomes of the cyclic phosphate hydrolysis step can differ significantly from one enzyme to another. To wit, Clostridium thermocellum CthPnkp-H189D catalyzes hydrolysis of the P–O3′ bond of 2′,3′ cAMP or cGMP to yield exclusively 2′-AMP or 2′-GMP products (22,25). By contrast, E. coli YfcE-C74H and Deinococcus radiodurans DR1281 catalyze hydrolysis of the P–O2′ bond of 2′,3′ cAMP to yield 3′-AMP as the sole product (26,27). Mycobacterium tuberculosis Rv0805, on the other hand, can hydrolyze either P–O2′ or P–O3′ to yield a mixture of 3′-AMP and 2′-AMP products, with a bias toward generation of 3′-AMP (26). Bacteriophage λ phosphatase also hydrolyzes either P–O2′ or P–O3′ to yield both 3′-AMP and 2′-AMP products, albeit with a preference for 2′-AMP formation (22). It was proposed, based on modeling of 2′,3′ cGMP into the Rv0805 active site in either of two directions depending on whether the ribose O2′ or O3′ was apical to the metal-bridged water nucleophile, that the ambidextrous Rv0805 and λ phosphatase enzymes are less constrained than other family members that hydrolyze 2′,3′-cyclic phosphates with respect to the binding orientations of the cyclic nucleotide, thereby allowing formation of either 2′-NMP or 3′-NMP products (26).
While our MPE studies were in progress, the Hopfner lab reported that E. coli SbcCD cleaves opposing strands of duplex DNA with different product outcomes, yielding a 5′-PO4 terminus on one strand and a 3′-PO4 terminus on the other strand (28). As we do, they proposed that the metal-activated water nucleophile in SbcCD is fixed, but the DNA can engage in either 5′ to 3′ or 3′ to 5′ strand polarity (28). This is sensible for SbcCD in light of its homodimeric quaternary structure and its ATP-dependent cleavage of both strands of duplex DNA (28). In the case of MPE, both types of product termini are generated during action on ssDNA, with no involvement of an ATP cofactor, attesting to ambidexterity as an inherent property of MPE that is not dictated by the antiparallel nature of the DNA duplex.
Understanding the basis for MPE ambidexterity will hinge on obtaining an MPE•(Mn2+)2•DNA co-crystal in a state mimetic of a Michaelis complex. This will, in turn, depend on several enabling steps: (i) identifying a minimal perturbation of the enzyme or the DNA substrate that preserves metal binding and DNA binding but prevents DNA cleavage (the His113 mutants are candidates to meet this test); (ii) honing the DNA ligand so that only a single phosphodiester is positioned in the active site and (iii) constraining the binding of DNA ligand to unique orientations.
FUNDING
NIH [R35-GM126945]; MSKCC is supported by NCI [P30-CA008748]. Funding for open access charge: NIH [R35-GM126945].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.