 A skipping rope translocation mechanism in a widespread family of DNA repair helicases
A skipping rope translocation mechanism in a widespread family of DNA repair helicases
Nucleic Acids Research



- Altmetric
Mitomycin repair factor A represents a family of DNA helicases that harbor a domain of unknown function (DUF1998) and support repair of mitomycin C-induced DNA damage by presently unknown molecular mechanisms. We determined crystal structures of Bacillus subtilis Mitomycin repair factor A alone and in complex with an ATP analog and/or DNA and conducted structure-informed functional analyses. Our results reveal a unique set of auxiliary domains appended to a dual-RecA domain core. Upon DNA binding, a Zn2+-binding domain, encompassing the domain of unknown function, acts like a drum that rolls out a canopy of helicase-associated domains, entrapping the substrate and tautening an inter-domain linker across the loading strand. Quantification of DNA binding, stimulated ATPase and helicase activities in the wild type and mutant enzyme variants in conjunction with the mode of coordination of the ATP analog suggest that Mitomycin repair factor A employs similar ATPase-driven conformational changes to translocate on DNA, with the linker ratcheting through the nucleotides like a ‘skipping rope’. The electrostatic surface topology outlines a likely path for the displaced DNA strand. Our results reveal unique molecular mechanisms in a widespread family of DNA repair helicases linked to bacterial antibiotics resistance.

INTRODUCTION
Organisms in all three domains of life employ nucleic acid-dependent nucleoside triphosphatases (ndNTPases) as motor proteins in DNA replication, recombination and repair as well as in virtually all steps of gene expression and regulation. Many ndNTPases can utilize the energy from nucleoside triphosphate (NTP) hydrolysis to translocate on nucleic acids or to remodel nucleic acids or nucleic acid-protein complexes (1,2). Often, these enzymes can unwind nucleic acid duplexes, i.e. act as nucleic acid helicases (3), with specific ndNTPases exhibiting preferences for DNA:DNA, RNA:RNA or DNA:RNA substrates (4). These activities are supported by a core of RecA-like NTPase domains, and in many cases by additional helicase-associated domains or regions (5). The RecA core domains bind NTPs and the nucleic acid or nucleic acid-protein substrates, and couple NTP binding, hydrolysis and release of the products via conformational changes to nucleic acid/nucleic acid-protein transactions (6). RecA core functions can be modulated by helicase-associated domains in diverse ways (5). Based on their activities, oligomeric state, conserved sequence motifs within the core and assortment of helicase-associated domains/regions, ndNTPases have been classified into six superfamilies (SF1–SF6), most of which comprise several families and subfamilies (7).
DNA damage threatens genome integrity and compromises the fidelity of gene expression and replication. DNA damage repair mechanisms, such as nucleotide excision repair (NER), mismatch repair (MMR) or base excision repair (BER), invariably rely on ndNTPase activities (8). For example during one form of NER in bacteria, the DNA damage is first detected by UvrA and verified by the SF2 helicase UvrB; after UvrA release, the UvrC nuclease is recruited and induces nicks upstream and downstream of the damaged site; subsequently, SF1 helicase UvrD and DNA polymerase I remove the post-incision complex, liberate the damaged DNA section and fill the gap, followed strand ligation by DNA ligase (9). A recent study reported a previously uncharacterized antibiotic-induced DNA excision repair pathway in Bacillus subtilis, that depends on the SF2 ndNTPase, mitomycin repair factor (Mrf) A (formerly YprA), and the metal-dependent exonuclease, MrfB (10). The MrfAB repair factor complex specifically mends DNA damage induced by mitomycin C, a naturally occurring antibiotic that, upon reductive activation, induces guanosine-specific adducts and causes inter-strand and intra-strand DNA crosslinks (11).
MrfA is a representative of a hitherto largely unexplored family of SF2 ndNTPases, characterized by a C-terminal domain of unknown function (DUF) number 1998 (DUF1998, pfam09369). DUF1998 contains four conserved, putatively metal ion-binding cysteine residues and is found with similar spacing to the RecA core in members of the MrfA family (12). One member of the MrfA family, Mycobacterium smegmatis SftH, has been biochemically characterized as a monomeric 3′-to-5′ DNA helicase (12). MrfA homologs are widely distributed in bacteria and are also present in archaea, fungi and plants. The MrfA-homolog in yeast, Hrq1, also reduces mitomycin C sensitivity (13–15). Hrq1 has high similarity to human RecQ4 and was therefore assigned to the RecQ-like helicase family (16,17). While close MrfA homologs appear to be missing in Enterobacteria, certain pathogenic Escherichia coli and Salmonella strains harbor Z5898-like helicases with a DUF1998 (18). Apart from its frequent location in C-terminal extensions of ndNTPases, a DUF1998 was also found encoded in a standalone gene, downstream of putative helicase domain-encoding genes in the context of the bacterial anti-phage defense system DISARM (19).
Presently, the structural basis of DNA-related activities in DUF1998 ndNTPases, and the possible role of the DUF1998 in these activities, are unknown. We therefore determined crystal structures of full-length B. subtilis MrfA alone and in complex with an ATP analog and/or a single-stranded DNA substrate. Our results establish DUF1998 as the major part of a larger Zn2+-binding domain with a novel fold. The Zn2+-binding domain, together with preceding regions, forms a unique array of helicase-associated domains, that sets the MrfA family apart from other ndNTPases. Strikingly, these domains form a canopy across the RecA core, which can undergo large conformational changes upon DNA binding. Apart from entrapping single-stranded DNA, these rearrangements affect an inter-domain linker that is flexible in the apo state but is tautened across the single-stranded DNA upon substrate loading, suggesting that it acts as a ratcheting device during DNA translocation. Our findings further illustrate an astonishing diversity of mechanistic principles, by which nucleic acid remodeling enzymes can operate, and provide insights into a bacterial antibiotics resistance strategy.
MATERIALS AND METHODS
Protein production
The mrfA (formerly yprA) open reading frame (BSU_22220) was PCR-amplified from B. subtilis genomic DNA. Primers employed are listed in Supplementary Table S1. The coding region was inserted into the pETM-11 vector (EMBL Heidelberg) via NcoI and SalI restriction sites to produce plasmid pETM-11-mrfA. Escherichia coli BL21-CodonPlus (DE3)-RIL cells (Agilent) were transformed with pETM-11-mrfA and cultivated in auto-inducing medium (20) at 37°C to an OD600 of ∼0.8, then further incubated at 18°C for 20 h. Cells were harvested at an OD600 of ∼15. Cell pellets were resuspended in 40 mM Tris–HCl, pH 7.5, 500 mM NaCl, 30 mM imidazole, 10% (v/v) glycerol (lysis buffer), supplemented with 20 mM benzamidine and 0.1 mg/ml lysozyme and lysed by sonication using a Sonopuls HD 3100 ultrasonic homogenizer (Bandelin). The lysate was cleared by centrifugation at 75 600 × g and loaded on a 5 ml Ni2+-NTA column (GE Healthcare), washed with 2 M LiCl and eluted with 400 mM imidazole in lysis buffer. The His6-tag was cleaved using recombinant TEV protease during dialysis at 4°C against 20 mM Tris–HCl, pH 7.5, 500 mM NaCl, 15 mM imidazole, 10% (v/v) glycerol, and the sample was again loaded on a 5 ml Ni2+-NTA column. The flow-through was concentrated and subjected to gel filtration chromatography on a Superdex 200 16/600 column (GE Healthcare) in 20 mM HEPES–NaOH, pH 7.5, 200 mM NaCl, 5 mM MgSO4.
For SeMet-labeled protein, transformed E. coli BL21-CodonPlus (DE3)-RIL cells were cultured in minimal auto-inducing medium (20) with methionine replaced by SeMet, and purified as described above, with the addition of 5 mM β-mercaptoethanol during Ni2+-NTA purification and 2 mM DTT during the final gel filtration.
Plasmids encoding MrfA variants were derived from pETM-11-mrfA by the QuikChange II site-directed mutagenesis protocol (Agilent). MrfA variants were produced and purified as the WT protein.
Crystallographic procedures
Crystals were grown using the sitting-drop vapor diffusion technique. Crystals of apo-MrfA were grown at 4°C in drops containing 1.2 μl of protein solution (10 mg/ml) and 0.8 μl 0.1 M Tris–HCl, pH 7.5, 0.2 M MgCl2, 10% (w/v) PEG 4,000. For (SeMet-)MrfA-DNA crystallization, protein was mixed with a 2.1-fold molar excess of a 16-nt ssDNA oligonucleotide (Supplementary Table S1) to a final protein concentration of 10 mg/ml and incubated on ice for 30 min. Crystals of (SeMet-)MrfA-DNA (±AMPPNP) were grown at 18°C for at least two weeks in drops containing 1.2 μl complex and 0.8 μl 0.2 M tri-sodium citrate, 20–24% (w/v) PEG 3350.
Crystals were cryoprotected by transfer into mother liquor containing 20–25% (v/v) glycerol and flash-cooled in liquid nitrogen. Diffraction data for apo-MrfA crystals were collected at 100 K on beamline P11 of the Petra III storage ring (DESY, Hamburg, Germany). Diffraction data for (SeMet-)MrfA-DNA (±AMPPNP) crystals were collected on beamline 14.2 of the BESSY II storage ring (HZB, Berlin, Germany) (21). Diffraction data were collected using the MXCuBE software platform (https://mxcube.github.io/mxcube) and processed with XDS (22) as implemented in XDSAPP (23).
Experimental phases for SeMet-MrfA-DNA data were determined by the single anomalous dispersion strategy using phenix.autosol (24,25). An initial model was generated with phenix.autobuild (26) and iteratively refined by rounds of manual model building with Coot (27) and automated refinement with phenix.refine (28) using the native MrfA-DNA dataset. The crystal structure of MrfA-DNA-AMPPNP was solved by molecular replacement using the MrfA-DNA model. The crystal structure of apo-MrfA was solved in space group P1 by molecular replacement with Phaser (29) using individual domains of DNA-bound MrfA as search models, and converted to space group H3 using Zanuda (30) from the CCP4 software suite (31). MrfA-DNA-AMPPNP and apo-MrfA models were completed manually with Coot and refined as described for MrfA-DNA. All models were validated using MolProbity (32,33). Diffraction data, structure solution and refinement statistics are listed in Table 1.

| SeMet-MrfA-DNA | MrfA-DNA | MrfA-DNA-AMPPNP | Apo-MrfA | |
|---|---|---|---|---|
| Data collection | ||||
| Temperature (K) | 100 | 100 | 100 | 100 |
| Wavelength (Å) | 0.979801 | 0.91841 | 0.91841 | 1.0332 |
| Space group | P41212 | P41212 | P41212 | H3 |
| Unit cell parameters | ||||
| a = b, c (Å) | 140.2, 208.9 | 140.3, 212.0 | 140.6, 210.1 | 191.9, 65.9 |
| Resolution range (Å) | 50.00–3.08 | 50.00–3.16 | 50.00–3.34 | 50.00–3.32 |
| (3.26–3.08) | (3.35–3.16) | (3.54–3.34) | (3.52–3.32) | |
| Unique reflections | 72 416 (11 107) | 37 055 (5841) | 31 095 (4895) | 13 373 (2132) |
| Redundancy | 28.4 (27.8) | 13.5 (14.0) | 14.8 (15.1) | 10.5 (10.1) |
| Completeness (%) | 98.1 (93.0) | 99.8 (99.3) | 99.2 (98.8) | 99.8 (99.0) |
| Intensity [I/σ(I)] | 9.29 (0.79) | 8.90 (1.11) | 8.45 (1.16) | 11.04 (0.98) |
| R meas (%)b | 45.6 (380.9) | 35.9 (242.6) | 37.9 (228.0) | 18.3 (216.8) |
| CC1/2c | 99.5 (35.6) | 99.3 (41.1) | 99.3 (51.0) | 99.8 (36.2) |
| Structure solution | ||||
| Number of selenium atoms | 29 | |||
| FOMd | 0.27 | |||
| BAYES-CCe | 26.1 | |||
| Refinement | ||||
| Resolution range (Å) | 19.32–3.16 | 28.73–3.34 | 47.99–3.32 | |
| (3.24–3.16) | (3.44–3.34) | (3.57–3.32) | ||
| Reflections | 36 845 (2734) | 31 007 (2738) | 13 371 (2628) | |
| R free test set count | 1841 (137) | 1551 (137) | 669 (131) | |
| R work f | 0.219 (0.332) | 0.223 (0.335) | 0.307 (0.512) | |
| R free g | 0.264 (0.353) | 0.282 (0.375) | 0.342 (0.528) | |
| Contents of an A.U.h | ||||
| Residues | 1449 | 1459 | 635 | |
| Non-hydrogen atoms | 12 110 | 12 247 | 5057 | |
| Mean B-factor (Å2) | 78.0 | 80.7 | 128.2 | |
| Rmsd(i) from target geometry | ||||
| Bond lengths (Å) | 0.004 | 0.005 | 0.002 | |
| Bond angles (°) | 0.726 | 0.854 | 0.527 | |
| Model statisticsj | ||||
| Favored regions (%) | 96.01 | 94.46 | 89.29 | |
| Allowed regions (%) | 3.99 | 5.26 | 9.88 | |
| Disallowed regions (%) | 0.00 | 0.28 | 0.82 | |
| MolProbity score | 1.84 | 2.05 | 2.13 | |
| MolProbity clashscorek | 11.26 | 14.75 | 11.09 | |
| PDB ID | 6ZNP | 6ZNQ | 6ZNS |
aValues in parentheses refer to the highest resolution shells.
b R meas(I) = ∑h [N/(N– 1)]1/2 ∑i │Iih – <Ih>│ / ∑h∑iIih, in which <Ih> is the mean intensity of symmetry-equivalent reflections h, Iih is the intensity of a particular observation of h and N is the number of redundant observations of reflection h (54).
cCC1/2 = (<I2> – I2) / (<I2> – I2) + σ2ϵ, in which σ2ϵ is the mean error within a half-dataset (54).
dFOM = mean figure of merit.
eBAYES-CC = overall score.
f R work = ∑h │Fo – Fc│ / ∑ Fo (working set, no σ cut-off applied).
g R free is the same as Rwork, but calculated on the test set of reflections excluded from refinement.
hA.U. – asymmetric unit.
iRmsd – root-mean-square deviation.
jCalculated with MolProbity (33,55).
kClashscore is the number of serious steric overlaps (>0.4) per 1000 atoms.
Structure comparisons
Structures were compared by global superposition or by superposition of selected domains using the Secondary Structure Matching (SSM) algorithm implemented in Coot.
DNA binding assays
DNA binding was monitored by fluorescence anisotropy measurements. 10 nM [5-FAM]-labeled nucleic acid probes (Supplementary Table S1) were titrated with 50–1000 nM protein in 40 μl solutions of 40 mM HEPES–NaOH, pH 8.0, 50 mM NaCl, 5 mM MgCl2, 1.5 mM DTT. Fluorescence polarization was measured in 384-well OptiPlates at room temperature using a VICTOR 3V multilabel plate reader (PerkinElmer; excitation filter 485 nm, emission filter 535 nm). Experiments were performed in technical triplicates. Anisotropy data were plotted as a function of protein concentration (34) and fitted to a single exponential Hill function (fraction bound = A·cproteinn/(cproteinn + Kdn); A, fitted maximum of DNA bound; Kd, dissociation constant; n, Hill coefficient) (35) using Prism 6.0 (GraphPad Software, Inc.). Exemplary data are shown in Figure 1A.
![In vitro activities of MrfA variants. (A) DNA binding monitored by fluorescence anisotropy titrations with a 5-FAM-labeled 16-nt ssDNA. Data were fitted to a single exponential Hill function using GraphPad Prism 6.0. Data represent means ± SD of technical triplicates. Data for the MrfAR332E and MrfAR491A variants could not be fitted; n.d., not determined. (B) Exemplary thin-layer chromatogram monitoring in vitro ATPase activity, tested by incubation of MrfAWT with [α-32P]-labeled ATP for 15 min at room temperature in the absence or presence of a 16-nt ssDNA. (C) Steady-state kinetics for 35-nt ssDNA-stimulated ATPase activity of MrfAWT. KM and kcat were derived by fitting the initial turnover rates at the indicated ATP concentrations to the Michaelis–Menten equation in GraphPad Prism 6.0. Data represent means ± SD of technical duplicates. (D) Fluorescence-based stopped-flow unwinding assays with DNA and RNA duplex substrates, bearing 3′ or 5′-single-stranded overhangs (3′/5′-ovh). Inset, zoom-in on the first 5 s of the trajectories. (E) Comparison of the relative DNA affinities, relative DNA-stimulated ATPase rates (Stim. ATPase) and relative amplitude-weighted unwinding rate constants (kunw) for the indicated MrfA variants. DNA binding data represent means of fold-WT ± SEM for three technical replicates. ATPase data represent means of fold-WT ± SD of two technical replicates. Unwinding data represent means of fold-WT of kunw ± SEM of three technical replicates.](/dataresources/secured/content-1765762253658-0d940087-1a34-4a73-89d8-5876e42f8e25/assets/gkaa1174fig1.jpg)
In vitro activities of MrfA variants. (A) DNA binding monitored by fluorescence anisotropy titrations with a 5-FAM-labeled 16-nt ssDNA. Data were fitted to a single exponential Hill function using GraphPad Prism 6.0. Data represent means ± SD of technical triplicates. Data for the MrfAR332E and MrfAR491A variants could not be fitted; n.d., not determined. (B) Exemplary thin-layer chromatogram monitoring in vitro ATPase activity, tested by incubation of MrfAWT with [α-32P]-labeled ATP for 15 min at room temperature in the absence or presence of a 16-nt ssDNA. (C) Steady-state kinetics for 35-nt ssDNA-stimulated ATPase activity of MrfAWT. KM and kcat were derived by fitting the initial turnover rates at the indicated ATP concentrations to the Michaelis–Menten equation in GraphPad Prism 6.0. Data represent means ± SD of technical duplicates. (D) Fluorescence-based stopped-flow unwinding assays with DNA and RNA duplex substrates, bearing 3′ or 5′-single-stranded overhangs (3′/5′-ovh). Inset, zoom-in on the first 5 s of the trajectories. (E) Comparison of the relative DNA affinities, relative DNA-stimulated ATPase rates (Stim. ATPase) and relative amplitude-weighted unwinding rate constants (kunw) for the indicated MrfA variants. DNA binding data represent means of fold-WT ± SEM for three technical replicates. ATPase data represent means of fold-WT ± SD of two technical replicates. Unwinding data represent means of fold-WT of kunw ± SEM of three technical replicates.
ATPase assays
Reactions were conducted in 10 μl volumes containing 0.5 μM protein in 10 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT with or without 1.0 μM 16-nt ssDNA (Supplementary Table S1), and started by addition of ATP, supplemented with 0.011 mCi/ml [α-32P]-ATP, to a final concentration of 1 mM. After incubation for 15 min at room temperature, reactions were quenched with an equivalent volume of 100 mM EDTA, pH 8.0, separated on PEI-cellulose by thin-layer chromatography plates in 20% (v/v) ethanol, 6% (v/v) acetic acid, 0.5 M LiCl, and visualized by autoradiography with the Storm 860 phosphorimager (GMI, USA). [α-32P]-ADP formation was quantified by densitometry using ImageQuant 5.2 (GE Healthcare). Experiments were performed in technical duplicates. Exemplary data are shown in Figure 1B.
For steady state kinetics, reaction mixtures contained 0.4 μM MrfA and 2.3 μM 35-nt ssDNA (Supplementary Table S1). Reactions were started with 0.25/0.5/1.0/1.5/2.0 mM ATP/[α-32P]-ATP mix. Aliquots of 2 μl were quenched in 6 μl 100 mM EDTA at 10/30/60/90/120/300 seconds. Experiments were performed in technical duplicates. Initial turnover rates were derived by linear regression fitting of the data. To determine KM and kcat, initial rates were plotted against substrate concentration and fitted to the Michaelis-Menten equation using GraphPad Prism 6.0. Quantified data are shown in Figure 1C.
Helicase assays
Nucleic acid unwinding activity was assessed in fluorescence-based stopped-flow experiments on a SX-20MV spectrometer (Applied Photophysics) as described previously (36,37). DNA or RNA substrates contained the same 12-base pair duplex region and either a single-stranded 3′- or 5′-overhang of 31 nts (Supplementary Table S1). For all experiments, 50 nM nucleic acid substrate were mixed with a 20-fold molar excess of protein (1 μM) in unwinding buffer (20 mM HEPES-NaOH, pH 8.0, 73 mM NaCl, 0.5 mM MgCl2), equilibrated at the reaction temperature of 20°C. A 60 μl aliquot was then rapidly mixed with 60 μl 4 mM ATP/MgCl2 in unwinding buffer. The final concentrations in the 120 μl reactions were 0.5 μM protein, 25 nM nucleic acid substrate and 2 mM ATP/MgCl2 in unwinding buffer. The fluorescence change was monitored over the course of 1800 s by acquiring 4000 data points in logarithmic sampling mode. For each experiment, three individual traces were averaged, baseline-corrected by the fluorescence immediately after addition of ATP and normalized to the baseline-corrected maximum fluorescence. Data were plotted using GraphPad Prism 6.0 and fitted to a double exponential equation (fraction unwound = Afast·(1 – exp(–kfastt)) + Aslow·(1 – exp(–kslowt)); A, total unwinding amplitude; k, unwinding rate constants [s−1]; t, time [s]), as described previously (37). The first 340 ms of data acquisition (260 ms for MrfAR322E/E533R) were excluded from curve fitting to account for the initial mixing periods. Amplitude-weighted unwinding rate constants were calculated as kunw = ∑(Aiki2)/∑(kiAi). Exemplary data are shown in Figure 1D, curve fits are shown in Supplementary Figure S1, quantified data are listed in Supplementary Table S2.
RESULTS
B. subtilis MrfA exhibits 3′-to-5′ DNA helicase activity
We recombinantly produced B. subtilis MrfA in E. coli and purified it to near homogeneity by affinity capture, tag cleavage and several column chromatographic steps. Similar to the previously characterized M. smegmatis MrfA homolog, SftH (12) (33% sequence identity), recombinant B. subtilis MrfA (MrfA in the following) exhibited typical properties of a DNA helicase. It bound a 16-nucleotide (nt) single-stranded (ss) DNA (Supplementary Table S1) with an affinity (Kd) of about 0.1 μM, as determined by fluorescence anisotropy titrations with a fluorescein-labeled oligodeoxynucleotide (Figure 1A). MrfA ATPase activity was ssDNA-dependent (Figure 1B). Longer oligodeoxynucleotides stimulated MrfA ATPase more strongly, suggesting a mechanistic coupling between ATPase and translocase activities. Quantification of steady-state, DNA-stimulated ATPase kinetics with non-linear Michaelis-Menten regression curve fitting indicated a KM of 0.32 mM and a kcat of 9.3 s−1 (Figure 1C).
Fluorescence-based stopped-flow unwinding assays revealed that MrfA can efficiently unwind double-stranded DNA duplexes that contain a 3′-single-stranded overhang (Figure 1D). A DNA substrate with a 5′-overhang and a sequence-identical RNA substrate were not unwound, indicating a 3′-to-5′ directionality of unwinding and a specificity for DNA (Figure 1D). Unwinding data followed a two-phase trajectory and were quantified by fitting to a double-exponential equation as suggested previously (37). The curve fits yielded amplitudes, Afast and Aslow, and unwinding rates, kfast and kslow, for the rapid and the slow unwinding phases, respectively (Supplementary Figure S1; Supplementary Table S2). To compare MrfA variants (see below), we calculated amplitude-weighted unwinding rate constants, kunw = ∑(Aiki2) / ∑(Aiki).
Architecture of MrfA
Crystals of full-length MrfA in complex with a 16-nt ssDNA oligomer grew in space group P41212 and diffracted to ∼3.2 Å resolution. The structure was solved by single-wavelength anomalous dispersion using a crystal of selenomethionine (SeMet)-derivatized MrfA in complex with the same oligodeoxynucleotide (Table 1). The crystals contained two highly similar MrfA–DNA complexes per asymmetric unit (root-mean-square deviation [rmsd] of 0.56 Å for 698 pairs of common Cα atoms). The final MrfA-DNA model contains MrfA residues 2–749 without gaps, and 14 nts of ssDNA (two 5′ nts missing). Seven nts at the 3′-end formed six Watson–Crick base pairs and a central non-canonical dC:dC pair with the corresponding DNA region of the second copy of the complex in an asymmetric unit (Supplementary Figure S2A).
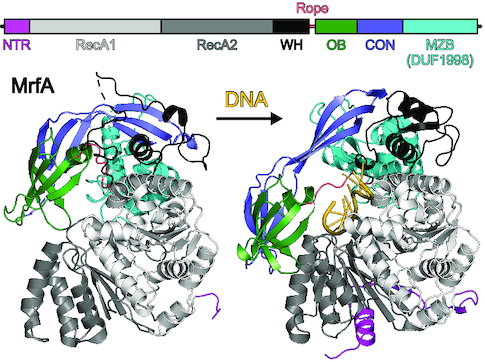
MrfA can be divided into seven domains/regions (Figure 2A), i.e. an N-terminal region (NTR, residues 1–39), two RecA-like domains (RecA1, residues 40–250; RecA2, residues 251–423), a winged-helix (WH) domain (residues 424–490), an oligonucleotide/oligosaccharide-binding (OB) domain (residues 491–556), a connector element (CON; residues 557–630) and a C-terminal MrfA Zn2+-binding domain (MZB, residues 631–749). In the DNA-bound state, the NTR folds across the surface of the RecA2 domain facing away from the DNA substrate (Figures 2B and 3A). It consists of a ten-residue amphipathic α-helix, followed by a seven-residue β-strand that connects laterally to the central β-sheet of the RecA2 domain in an antiparallel fashion, before linking up to the RecA1 domain alongside the connection between RecA1 and RecA2 (Figure 3A). The tandem RecA domains form a groove lined by DNA-binding motifs Ia, Ib, Ic, IV, IVa and V. Nucleotide-binding motifs Q, I, II and VI are positioned at a cleft between the RecA domains. The WH domain exhibits a typical winged-helix fold, flanking the helicase core. Compared to the other domains, the WH sequence is less conserved among MrfA homologs. Most of the few conserved residues, such as N428 and E442, lie on the domain's surface and form polar interactions with MZB and RecA1, respectively, suggesting a conserved domain arrangement in MrfA homologs. The OB domain has a typical β-barrel architecture. In the presence of ssDNA, it folds back onto the helicase core and forms an extended interface with the RecA2 domain, keeping the substrate-bound enzyme in a ring-like conformation (Figure 2B). The CON element forms an elongated, three-stranded, anti-parallel β-sheet, interrupted by proline residues in the centers of two strands. CON connects the OB to the MZB domain and resembles the connector element of the RNA-dependent NTPase, HrpB, from E. coli, which links the HrpB OB domain to a unique C-terminal region (36) (Figure 3B).
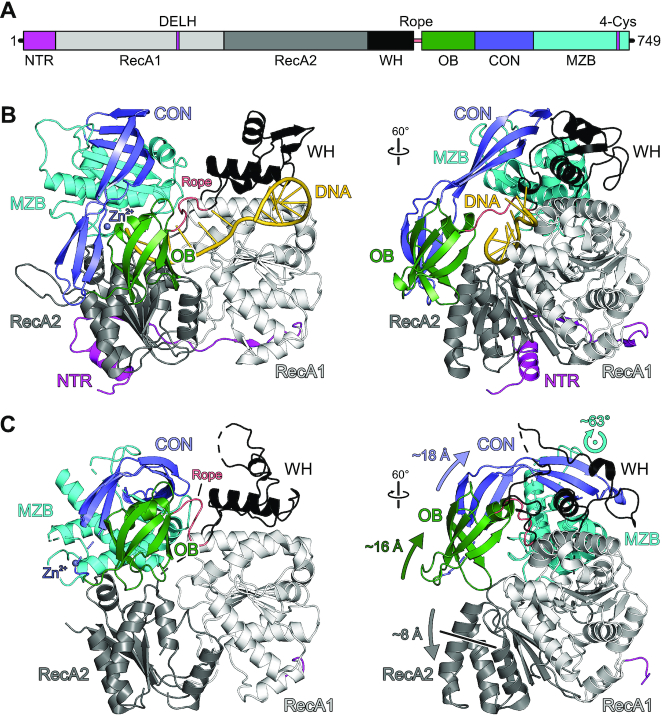
Overall structure of MrfA. (A) Scheme of the MrfA domain organization with indicated signature motifs. DELH, motif II. In this and the following figures: NTR, magenta; RecA1, light gray; RecA2, dark gray; WH domain, black; rope (flexible loop linking WH and OB domains), salmon; OB domain, dark green; CON domain, slate blue; MZB domain, cyan. (B) Cartoon plot of MrfA in complex with a single-stranded DNA substrate. DNA, gold. For an unobstructed view, the five nucleotides at 3′-end are not depicted in the rotated image. Rotation symbols in this and the following figures indicate the view relative to the left panel. (C) Cartoon plot of apo-MrfA in the same orientations as in (B), after superpositioning of the RecA1 domains. Arrows indicate translational and rotational movements of domains/regions relative to the DNA-bound state.
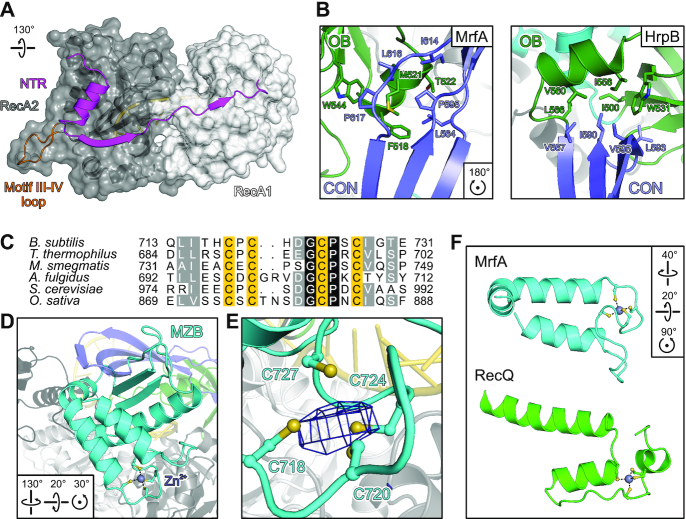
MrfA-specific functional elements. (A) Cartoon representation of the NTR running along RecA1 and RecA2 domains as cartoons and semitransparent surfaces. Pin-like element in RecA2 (motif III-IV loop), orange. (B) The MrfA CON element resembles the CON domain in HrpB (PDB ID 6EUD) and forms a similar hydrophobic interface with the OB domain. Interface residues are shown as sticks and colored by atom type. In this and the following figures: carbon, as the respective domain/region; nitrogen, blue; oxygen, red; sulfur, yellow. (C) Sequence alignment of the four-cysteine motifs of MrfA homologs from the indicated species. B. subtilis, Bacillus subtilis; T. thermophilus, Thermus thermophilus; M. smegmatis, Mycobacterium smegmatis; A. fulgidus, Archaeoglobus fulgidus; S. cerevisiae, Saccharomyces cerevisiae; O. sativa, Oryza sativa. Darker color represents higher conservation. Zn2+-binding cysteine residues, yellow. (D, E) Overall fold of the MrfA Zn2+-binding domain (D) and close-up of the Zn2+-binding site (E). Zn2+-binding cysteines are shown as ball-and-stick models, the Zn2+ ion is shown as a sphere, and colored by atom type. Zn2+, purple. Dashed lines in (D), Zn2+ coordination. Mesh in (E), anomalous Fourier density (Zn peak data; Supplementary Table S3) contoured at the 3 σ level. (F) Comparison of Zn2+-binding regions of MrfA (cyan) and RecQ (green; PDB ID 1OYW). In the MZB domain, Zn2+-coordinating residues are more closely spaced than in the RecQ Zn2+-binding domain. The Zn2+-binding elements exhibit a different topology, and Zn2+-binding cysteines originate exclusively from loops instead of regular secondary structure elements.
The C-terminal MZB domain contains the subfamily-defining DUF1998 with a conserved four-cysteine signature motif (38) (Figure 3C). DUF1998 was previously suggested to constitute a Zn2+-binding motif (39). Indeed, the four closely-spaced cysteines in MrfA reside in a short coil between two α-helices and form a metal ion-binding site (Figure 3D). To elucidate the nature of the bound metal ion, we recorded anomalous diffraction data from an MrfA-DNA crystal using X-rays of 1.27819 Å (Zn peak) and 1.28481 Å wavelengths (Zn low energy; Supplementary Table S3). In contrast to the Zn low energy data, phasing of the Zn peak data using the refined MrfA-DNA model yielded a strong anomalous Fourier density peak (Figure 3E), firmly establishing DUF1998 as a Zn2+-binding fold within the MZB domain.
Zn2+-binding sites comprising four coordinating cysteines are found frequently in ndNTPases, such as bacterial PriA (40), human RIG-I (41) and RecQ-like enzymes (42,43). However, a search using the DALI server (44) retrieved only functionally or structurally unrelated hits with comparatively low Z-scores (Supplementary Table S4). The spacing, arrangement relative to the core and suspension from exclusively irregular structural elements of the Zn2+-coordinating residues are distinct features of the MZB domain (Figure 3F).
DNA substrate binding by MrfA
In the MrfA-DNA crystal structure, the ssDNA ligand is bound exclusively by residues from the RecA1/2, WH and MZB domains (Figure 4A). The DNA enters MrfA through a wide funnel formed by the RecA2, OB, CON and MZB domains, which accommodates six stacked nts at the 5′-end. The funnel leads to a narrow aperture, where WH and OB domains tauten an inter-domain linker across the nucleobase side of the DNA, which we refer to as the ‘rope’. Upon passing this aperture, the DNA undergoes a base-flip and backbone kink, initiating a second, eight-nt stack that further extends along the RecA1 domain. The backbone kink is stabilized by a 310-helix of the WH domain (P484-S486) that diverts the substrate along RecA1 motifs Ib and Ic (residues N157-S163). The DNA exits the protein along a channel formed by RecA1 and WH domains.
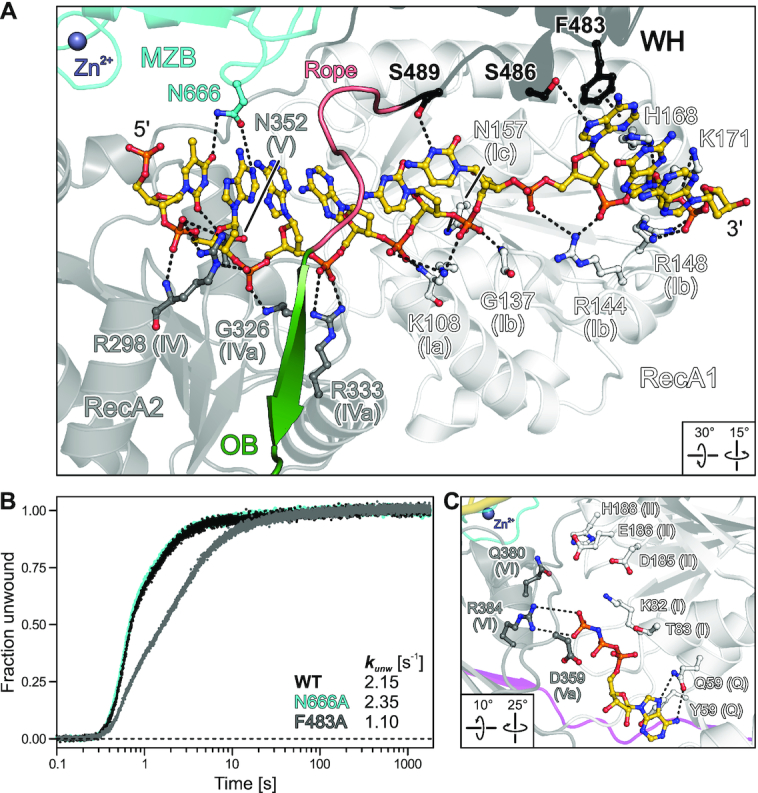
Accommodation of the loading strand and of AMPPNP. (A) Close-up view on the DNA loading strand bound across the RecA domains and underneath the canopy, highlighting DNA-contacting MrfA residues. A flexible loop (rope) links WH and OB domains, running across the bound DNA. DNA and DNA-contacting residues of MrfA are shown as ball-and-stick models and colored by atom type. In this and the following figures: DNA carbon, gold; phosphorus, orange. Purple sphere, Zn2+. For residues pertaining to helicase motifs, motifs are indicated in parenthesis. Dashed lines, hydrogen bonds, salt bridges and aromatic stacking contacts. (B) DNA duplex unwinding by MrfAWT and indicated variants with amplitude-weighted unwinding rate constants, kunw. (C) Close-up view of AMPPNP bound at the nucleotide binding cleft of the MrfA-DNA complex. AMPPNP and selected residues of MrfA are shown as ball-and-stick models and colored by atom type. Helicase motifs are indicated in parentheses. Dashed lines, hydrogen bonds and salt bridges.
The DNA substrate is bound along a basic groove between the RecA domains, predominantly via the sugar-phosphate backbone, by motif Ia/Ib/Ic (RecA1) and motif IV/IVa/V (RecA2) residues (Figure 4A), which are highly conserved among MrfA homologs and across helicase families (17). Two additional RecA1 residues, H168 and K171, coordinate the sugar-phosphate backbone at the 3′-exit site (Figure 4A). There are only spurious contacts to the nucleobases, which are exclusively provided by the auxiliary domains. Residues F483, S486 and S489 of the WH domain bind the two nucleobases bordering the backbone kink by π-π-stacking and hydrogen bonds (Figure 4A). F483, S486 and S489 are not highly conserved among WH domains of MrfA homologs, suggesting that they may support, but most likely are not essential for, helicase activity. Consistent with this notion, an F483A exchange led to a mild reduction in DNA affinity, a fourfold reduction in DNA-stimulated ATPase activity and a twofold reduction in DNA unwinding activity compared to wild-type (WT) MrfA (Figures 1A,E and 4B; Supplementary Figure S1; Supplementary Table S2).
The MZB domain contributes a large surface to the wide DNA entry funnel, but apart from a single asparagine residue, N666, that hydrogen bonds to the nucleobase of the 5′-terminal nt, the MZB exhibits no DNA contacts (Figure 4A). Although the co-crystallized DNA ligand bears two additional nts at the 5′-end that could in principle dock to the MZB, they remain undefined in the electron density. An N666A variant exhibited WT-like DNA duplex unwinding activity (Figure 4B). Hence, MZB/DUF1998 does not seem to be crucial for binding the DNA loading strand.
The DNA-bound conformation represents an ATP-receptive state
To test whether the observed DNA-bound state of MrfA can engage ATP, we co-crystallized the enzyme with DNA in the presence of the non-hydrolysable ATP analog, AMPPNP. Crystals of MrfA-DNA-AMPNP were isomorphous to MrfA-DNA crystals and diffracted to ∼3.3 Å resolution (Table 1). Globally, the MrfA conformation in complex with DNA and AMPPNP closely resembles the nucleotide-free complex (root-mean-square deviation [rmsd] of 0.50/0.81 Å for 746/693 pairs of common Cα atoms). On the local scale, RecA1 residues 76–80, which form a 310-helix in the nucleotide-free MrfA-DNA complex, rearrange to accommodate AMPPNP (Figure 4C). RecA1Y56 and RecA1Q59 (Q motif) bind the adenine base by aromatic stacking and hydrogen bonding, respectively. RecA2R384 (motif VI) contacts the γ-phosphate. However, with the exception of RecA1K82, which also approaches the γ-phosphate, other motif I and II residues (T83, D185, E186 and H188) remain beyond hydrogen bonding distance to the nucleotide phosphates.
The observed AMPPNP binding pose did not involve an Mg2+ ion or catalytic water molecule. Attempts to co-crystallize ADP instead of the ATP-analog failed to produce crystals in our hands, consistent with the γ-phosphate of AMPPNP establishing MrfA contacts. Together, the above findings show that the MrfA-DNA complex can undergo local conformational adjustments to accommodate ATP in a pre-hydrolytic configuration, that ADP is not stably bound after DNA engagement and that additional rearrangements that bring RecA1 motif I/II residues into proximity of ATP are required to initiate ATP hydrolysis-coupled DNA translocation.
The aperture-forming WH-OB linker is essential for helicase activity
A distinctive feature of MrfA is the rope across the loading strand that forms the narrowest aperture through which the DNA is threaded. While the rope has elevated flexibility (Figure 5A) and shows two distinct conformations in the two crystallographically independent complexes, it is stably anchored on both sides. R491 anchors the N-terminal side of the rope through interactions with P107 (motif Ia) and D159 (motif Ic) of RecA1 (Figure 5A,B). Comparison to DNA-bound structures of M. smegmatis Lhr (45), Cronobacter sakazakii RecQ (46) and Archaeoglobus fulgidus Hel308 (47) revealed highly conserved R491-equivalent arginine residues in RecQ or Ski2-like helicases, which are not contributed by a C-terminal accessory domain but reside directly C-terminal of RecA1 motifs II (Figure 5C–E). In the latter enzymes, the R491-equivalent residues position aromatic-rich loops (ARLs) that induce a DNA backbone bend to assist DNA unwinding (46). MrfA homologs lack an ARL and contain a conserved phenylalanine (F194) instead of the conserved arginine C-terminal of motif II in RecQ/Ski2-like enzymes (Figure 5B), reminiscent of DEAD-box helicases (17). Alanine exchange of R491 led to dramatically diminished DNA affinity, ∼20-fold reduced DNA-stimulated ATPase activity and an almost 400-fold reduction in unwinding (Figures 1A, E and 5F; Supplementary Figure S1; Supplementary Table S2).
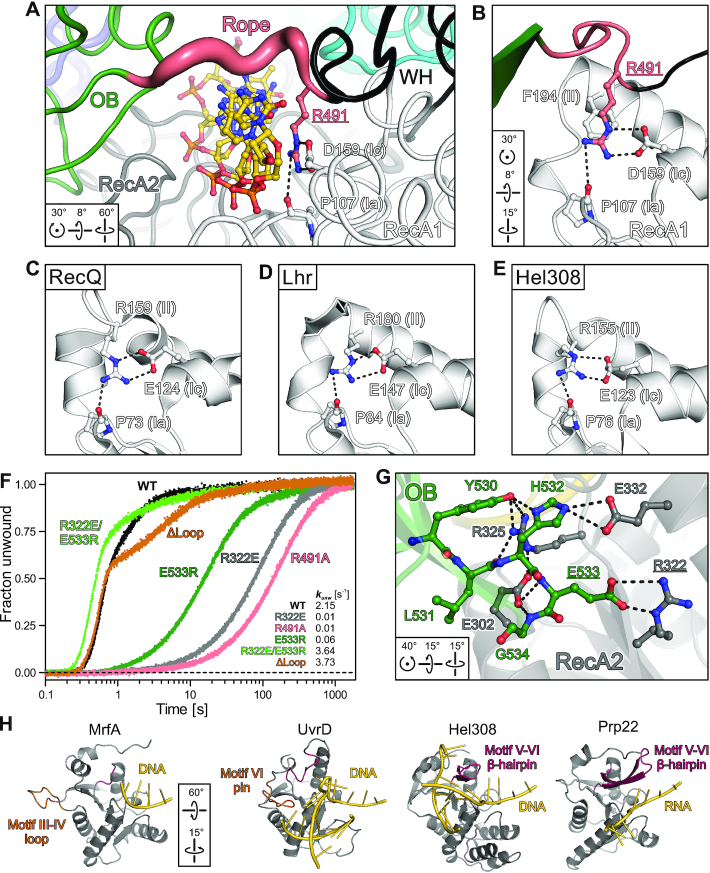
Rope anchoring and DNA separation elements. (A) N-terminal rope anchoring via R491 bridging motifs Ia (P107) and Ic (D159) in RecA1. MrfA is shown in tube representation, with the tube radius scaled to the B-factors. Underlined, subjected to mutational analysis (F). (B–E) Close-up views on the arrangement of conserved helicase motifs Ia, Ic and II (indicated in parentheses) in MrfA (B), Cronobacter sakazakii RecQ (PDB ID 4TMU) (C), M. smegmatis Lhr (PDB ID 5V9X) (D) and A. fulgidus Hel308 (PDB ID 2P6R) (E), after superpositioning of the RecA1 domains. (F) DNA unwinding activity of indicated MrfA variants with amplitude-weighted unwinding rate constants, kunw. (G) C-terminal rope anchoring via a polar interface between OB and RecA2 domains. Selected interface residues are shown as ball-and-stick models. Underlined, subjected to mutational analysis (F). (H) Comparison of the MrfA RecA2 domain to RecA2 domains of nucleic acid-bound structures of the Hel308 (PDB ID 2P6R), UvrD (PDB ID 2IS1) and Prp22 (PDB ID 6I3P) helicases. Prominent loop/pin or β-hairpins, orange or dark red, respectively.
On the C-terminal side, the rope is held in place by OB domain residues 530–534 interacting intimately with RecA2 domain residues E302, R322, R325 and E332 (Figure 5G). MrfA variants containing R322E or E533R exchanges that disrupt a central salt bridge in this interface led to severely diminished DNA affinity, DNA-stimulated ATPase activity and duplex unwinding activity (Figures 1A,E and 5F; Supplementary Figure S1; Supplementary Table S2). To test whether the effects of these residue substitutions are solely due to the disruption of the OB-RecA2 interface, we generated the compensating double variant, R322E/E533R, that is expected to restore the ionic contact. Strikingly, MrfAR322E/E533R unwinding activity not only matched but surpassed MrfAWT helicase activity almost twofold (Figure 5F; Supplementary Figure S1; Supplementary Table S2). Taken together, the above results demonstrate that a stable suspension of the rope across the DNA loading strand is essential for MrfA helicase activity.
Lack of an obvious strand separator in MrfA
The MrfA RecA2 domain lacks a prominent β-hairpin that accompanies helicase motifs Vb in Ski2-like, DEAH/RHA and NS3/NPH-II helicases (17), where it may serve as the wedge for strand separation (47) or provide mechanical support during translocation (48). However, structural comparisons uncovered a morphologically similar loop between motifs III and IV that resembles the separation pin observed in SF1 helicase UvrD (49) (Figure 5H). Replacement of this loop in MrfA (residues 262–270) with an A–G dipeptide (ΔLoop) resulted in 1.7-fold increased ssDNA affinity (Figure 1A, E). MrfAΔLoop still efficiently hydrolyzed ATP and unwound DNA duplexes with slightly elevated kfast and decreased kslow, when compared to MrfAWT (Figures 1E and 5F; Supplementary Figure S1; Supplementary Table S2). The pronounced biphasic reaction trajectory (Figure 5F) may be a consequence of the increased DNA affinity that influences both substrate binding and product release. Irrespectively, efficient duplex separation by MrfAΔLoop suggests that the loop between motifs III and IV is dispensable for unwinding activity.
Detailed inspection of the MrfA surface topology provides insights into a likely trajectory of a displaced strand. With a diameter of at least 24 Å, the entry funnel is wide enough to accommodate double-stranded DNA. Strand separation must occur before the loading strand traverses the aperture formed by the WH-OB rope, as the preceding binding site is also narrow (Figure 6A) and in part electro-negative. An electropositive channel between MZB and RecA2 could provide a suitable exit route for the displaced strand (Figure 6A). In support of this notion, a citrate molecule is bound at this channel in the DNA-bound structure, potentially mimicking displaced DNA (Figure 6A, inset).
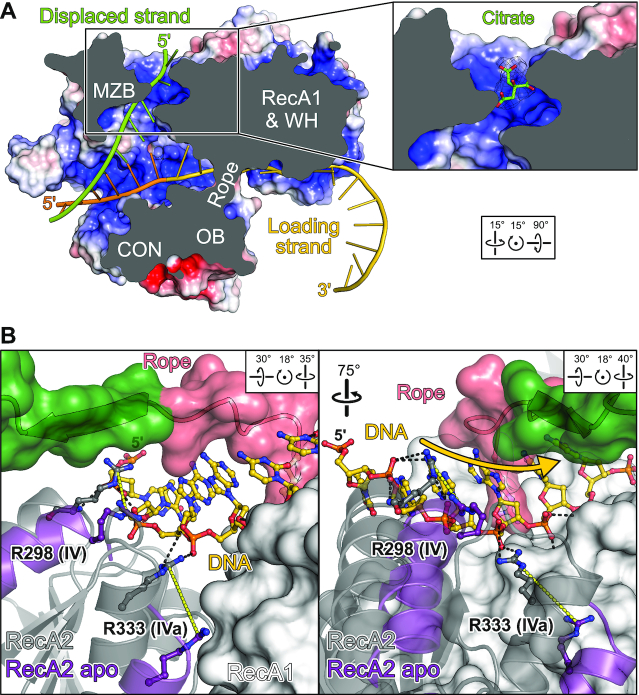
DNA displacement and translocation. (A) Putative exit channel for the displaced strand. MrfA is shown in surface view colored by the electrostatic surface potential (red, negative charge; blue, positive charge). The DNA loading strand (gold; modeled 5′-extension, orange) and a modeled displaced strand (green) are shown as cartoons. Inset, citrate ion bound at the putative displaced strand exit channel, as observed in DNA-bound structures. Mesh, 2mFo – DFc electron density map contoured at the 1 σ level. (B) ‘Shoulder shrugging’ by RecA2. The RecA2 domain in the DNA-bound state (grey) is shown as cartoon, with mobile RecA2 α-helices in the superimposed apo-conformation (purple). The aperture-forming rope and the first β-strand of the OB domain (green) are shown as cartoon with a semitransparent surface, RecA1 as a solid surface. DNA and DNA-contacting arginine residues from helicase motifs IV (R298) and IVa (R333), envisioned to switch between tight and loose DNA contacts, are indicated as ball-and-stick models. Black dashed lines, DNA contacts observed in the DNA-bound structure; yellow dashed lines, repositioning of R298 and R333 in the apo-state. Gold arrow, direction of DNA translocation.
MrfA undergoes major conformational changes upon DNA binding
The observed entrapment of the loading strand in the DNA-bound state poses the question of how MrfA can initially engage a DNA substrate. To address this question, we crystallized MrfA in the absence of DNA. Crystals of full-length apo-MrfA grew in space group H3 and diffracted to ∼3.3 Å resolution (Table 1). The apo-structure could only be solved by stepwise molecular replacement with fragments of DNA-bound MrfA as search models, suggesting major conformational differences to the DNA-bound state. The final model of apo-MrfA encompasses residues 35–745, with residues 246–248, 262–272, 318–324, 346–357, 395, 419–423, 469–471, 558–560, 601–610, 629–638, 672–674, 683–684 and 730–735 lacking well-defined electron density.
In contrast to the closed-ring conformation of the DNA-bound state, apo-MrfA features a more open, lock washer-like conformation, with reduced interactions between OB and RecA2 domains (Figure 2C). Increased B-factors and many regions lacking well-defined electron density (Supplementary Figure S2B, C) suggest that apo-MrfA exhibits elevated flexibility compared to DNA-bound MrfA, which might allow lateral DNA loading between OB and RecA2 domains upon slight additional rearrangements.
While the NTR is undefined in the apo-state, attempts to produce recombinant NTR-deleted MrfA resulted in an unstable protein prone to aggregation. Thus, the NTR appears to remain loosely attached to the RecA domains in the apo-state and reinforces the DNA-binding conformation upon substrate loading. DNA binding also elicits conformational changes in the RecA2 domain: one α-helical edge (the ‘shoulder’; residues 330–342) is lifted by ∼8 Å upon DNA engagement, enabling R298 (motif IV) and R333 (motif IVa) to form direct contacts to the DNA (Figure 6B). RecA2 ‘shoulder shrugging’ supports OB-RecA2 interactions upon DNA binding (Figure 2B), while the OB domain hovers above the DNA binding groove in the apo state (Figure 2C).
Morphing between apo and DNA-bound states revealed that upon DNA binding the MZB domain rotates by ∼63° about a hydrophobic joint formed between MZBY690,I694 and RecA1V193 (motif II); concomitantly, the OB domain slides ∼16 Å across the DNA binding groove towards RecA2 (Supplementary Movie S1). The CON element couples the OB movement to the MZB rotation, with the central proline residues acting as hinges that allow the two halves of the CON β-sheet to remain attached to moving neighboring elements. The overall movement resembles the rolling in and out of a canopy (CON and OB) about a drum (MZB; Supplementary Movie S1). As movement of the canopy interpolates between low and high DNA affinity states, it is likely also involved in DNA translocation and thus DNA unwinding.
DISCUSSION
Our analyses of B. subtilis MrfA provide deep mechanistic insights into the currently largely unexplored but phylogenetically widespread DUF1998-containing family of DNA repair helicases. They unveil a unique assortment of helicase-associated domains, with the family-defining DUF1998 exerting a vital function for helicase activity as part of a larger MZB domain, and suggest unique mechanisms of substrate loading, DNA translocation and strand separation.
Establishment of a MrfA-specific functional architecture
Upon DNA engagement, MrfA OB and CON domains are repositioned by a rotation of the MZB domain to encapsulate the loading strand. Residues involved in inter-domain contacts that stabilize the resulting closed-ring architecture are highly conserved among MrfA homologs. In particular, interaction sites in the WH domain to RecA1 and MZB domains encompass most of the few conserved WH residues. Conversely, DNA-contacting residues in the auxiliary domains are poorly conserved and predominantly engage the nucleobases, in contrast to other helicases, such as Hel308 (47), Lhr (45) or Prp43 (50), which exhibit partially similar auxiliary domains.
In most structures of RecQ-like and Ski2-like helicases, motifs Ia and Ic are bridged by a conserved arginine residue following motif II (Figure 5C-E). Although having been considered a member of the RecQ-like family, MrfA lacks this arginine and the accompanying ARL. Instead, R491 at the N-terminus of the WH-OB rope substitutes as a motif Ia-Ic bridge. This spatial substitution not only preserves the basic function of the conventional motif II arginine, but additionally anchors one end of the aperture-forming rope. The importance of R491 is supported by the dramatically diminished DNA binding, stimulated ATPase and unwinding activities upon its exchange for an alanine. Notably, helicases outside the RecQ-like and Ski2-like families, e.g., DNA helicases RecG (51) and PriA (40) or RNA helicase Prp22 from the DEAH/RHA family (48), do not uphold a bridge between motifs Ia and Ic. We therefore suggest that in the case of MrfA, rope anchoring represents the crucial function of R491. Consistent with this notion, strongly decreased helicase activity also resulted when the C-terminal rope anchor, established by OB-RecA2 interactions, was compromised.
Mechanism of DNA loading
We presently do not know how exactly MrfA initially engages DNA. Although apo-MrfA exhibits a rolled-in canopy with the OB domain above the DNA binding groove, the gap between OB and RecA2 domains remains sealed, albeit via a much reduced interface. To allow lateral passage of a DNA strand into the binding groove, the OB has to detach from the RecA2 domain, e.g. by further rolling in of the canopy and/or by lifting of the OB domain via a lid-opening motion about the proline hinge in the CON β-sheet (Figure 7; state 1). It is conceivable that such rearrangements are induced when a DNA strand initially docks to the surface of MrfA, upon binding of MrfA to the MrfB exonuclease or upon binding of other factors putatively involved in mitomycin-C damage repair (10). After DNA loading, the lid may close (Figure 7; state 2) and rolling out the canopy allows the OB domain to lock into position at the RecA2 domain. Sliding of the OB domain towards RecA2 tautens the WH-OB rope across the DNA loading strand (Figure 7; state 3).
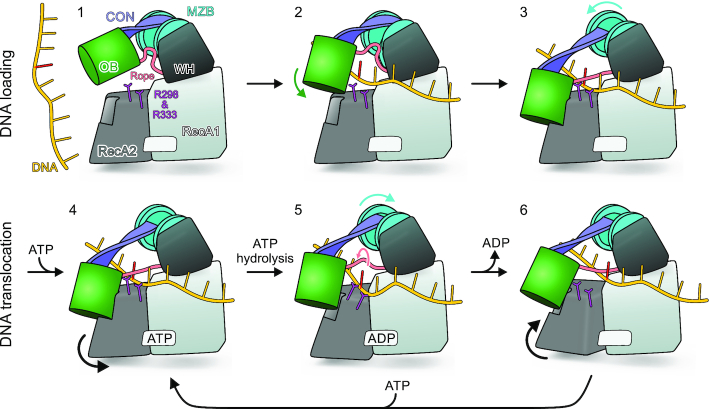
Model for MrfA substrate loading and translocation. State 1 corresponds to the structure of apo-MrfA with an additional tilt about the proline hinge of the CON element, leading to further separation of OB and RecA2 domains. States 2 and 3 correspond to the crystal structures of apo-MrfA and DNA-bound MrfA, respectively, with DNA modeled into the apo conformation. State 4 corresponds to the crystal structure of DNA- and AMPPNP/DNA-bound MrfA. States 5–6 illustrate putative conformational changes during ATP binding, hydrolysis and release of the products that drive DNA translocation. R298 and R333 switching between tight and loose DNA contacts are indicated. Reference nucleotide in the DNA substrate, red.
Mechanism of DNA translocation
Our findings show that MrfA lacks a ratchet helix that is thought to support translocation in the DEAH/RHA and Ski2-like families of helicases (47,48). Moreover, we demonstrated that MrfA does not contain a prominent β-hairpin in the RecA2 domain as a mechanical aid in translocation, as is the case in DEAH/RHA enzymes (48). The conformational changes upon DNA binding and our mutational analyses instead suggest that MrfA relies on a unique structural element, the rope, to support translocation.
Release of the OB-RecA2 interaction would allow the RecA2 domain to transition from the DNA-bound towards the apo conformation, during which its shoulder retracts from the OB domain. This rearrangement would drive DNA-contacting motifs IV (R298) and IVa (R333) in 3′-direction with respect to the bound DNA strand (Figures 6B and 7). Assuming that the bound DNA will tag along, R298/R333 will transport the DNA towards the RecA1 domain and guide the sugar-phosphate backbone underneath the canopy into the funnel (R298) and through the aperture (R333) (Figure 7). R298 and R333 will eventually have to release the DNA and may rebind at equivalent positions further 5′ in the DNA. Thus, R298 and R333 may establish alternating tight-loose contacts that could support an inchworm mode of translocation (52,53).
In support of their relevance for translocation, the observed conformational changes of the MrfA RecA2 domain resemble RecA2 domain movements in DEAH/RHA RNA helicases during the transition from ATP to ADP-bound states, which have been implicated in translocation (48). RecA2 conformational changes may hinge on the apparent elevated flexibility of the domain (Supplementary Figure S2B,C), which was also observed for DEAH/RHA NTPases (48). Our finding that RecA2 in the DNA-bound state can bind AMPPNP, but that conformational changes are required to elicit ATP hydrolysis, suggests that ATP binding followed by accommodation may initiate the translocation cycle.
In the DNA-bound state, the WH-OB rope is tautened across the DNA loading strand, forming an aperture that is too narrow to allow passage of the nucleotides, thus preventing back-slippage of the DNA (Figure 7; state 4). Detachment of the OB domain from RecA2 upon RecA2 conformational changes and rolling in of the canopy, will allow DNA translocation, as the WH-OB rope is loosened (Figure 7; state 5). We envision that for continued translocation, the aperture will periodically dilate and contract due to the rope being recurrently loosened and tautened by rolling-in and rolling-out of the canopy, respectively, correlated with ATP transactions. It thereby ratchets through the nucleobases like a skipping rope that is attached to the WH domain by R491 and controlled by the OB domain repeatedly detaching from and attaching to the RecA2 domain (Figure 7; state 4–6). The above model of translocation is fully in line with our structure-informed functional analyses, including the key importance of WH-OB rope attachment sites for helicase activity.
Mechanism of DNA unwinding
As our structural analyses did not reveal an obvious element involved in DNA strand separation, comparable to the separator loop in Ski2-like helicases (47), and a loop resembling the RecQ-like separation pin in the RecA2 domain (residues 262–270) was dispensable for unwinding activity, the question remains how DNA translocation is coupled to nucleic acid unwinding in MrfA. The electrostatic surface topology of MrfA suggests that the displaced strand is guided between MZB and RecA2 (Figure 6A). With the displaced strand exiting along the MZB surface, rotational movements of the MZB that serves to roll the canopy in and out during translocation could additionally aid in strand separation by also translocating the displaced strand.
Implications for non-canonical DNA damage repair
Our results reconcile findings from previous functional interrogations of the MrfAB excision repair system. While alteration of MrfA motifs I (K82A), II (S222A) and III (DE185–186AA) suggested that MrfA helicase activity is required for DNA damage repair in vivo, a T134V motif Ib variant still complemented mitomycin C sensitivity (10). Consistent with these observations, our structural analyses show that in contrast to motif Ib residues G137 and R144, T134 is not directly involved in DNA substrate binding.
N- and C-terminal MrfA truncations abrogated interaction with the exonuclease MrfB in a bacterial 2-hybrid system (10). Our structures suggest that such MrfA truncation variants are most likely conformationally compromised; consistent with this notion, our attempts to produce and isolate truncated versions of MrfA that lack either the NTR (MrfA39–749) or the C-terminal CON and MZB domains (MrfA1–555) resulted in insoluble protein. Thus, the mode of MrfA interaction with the exonuclease MrfB remains a subject of future studies.
Presently, it is unknown how the MrfAB excision repair machinery is specifically targeted to mitomycin C-induced DNA lesions. Our findings do not suggest any specificity of MrfA for mitomycin C-damaged DNA regions. This observation is consistent with the suggestion that the MfrAB helicase/nuclease machinery may be recruited to mitomycin C-induced DNA lesions via a yet to be identified factor (10).
DATA AVAILABILITY
Structure coordinates and diffraction data have been deposited in the RCSB Protein Data Bank (https://www.rcsb.org/) under accession codes 6ZNS (apo-MrfA; https://doi.org/10.2210/pdb6ZNS/pdb), 6ZNP (MrfA-DNA; https://doi.org/10.2210/pdb6ZNP/pdb) and 6ZNQ (MrfA-DNA-AMPPNP; https://doi.org/10.2210/pdb6ZNQ/pdb). All other data supporting the findings of this study are available from the corresponding author on request.
ACKNOWLEDGEMENTS
We are grateful to Claudia Alings, Laboratory of Structural Biochemistry, Freie Universität Berlin, for help with crystallization and to Christoph Weise, Laboratory of Protein Biochemistry, Freie Universität Berlin, for support in mass spectrometric verification of target proteins. We acknowledge access to beamlines of the BESSY II storage ring (Berlin, Germany) via the Joint Berlin MX-Laboratory sponsored by the Helmholtz Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Universität zu Berlin, the Max-Delbrück-Centrum, the Leibniz-Institut für Molekulare Pharmakologie and Charité – Universitätsmedizin Berlin. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at Petra III and we would like to thank Alke Meents for assistance in using beamline P11.
Author contributions: J.J.R. and S.L. performed experiments. U.N. and B.L. helped with structure solution, model building and refinement. J.J.R. and M.C.W. wrote the manuscript with contributions from the other authors. M.C.W. conceived, coordinated and provided funding for the project.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [RTG 2473-1 to M.C.W.]. Funding for open access charge: Home institution (Freie Universität Berlin).
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
5.
7.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.