 A fast and robust iterative genome-editing method based on a Rock-Paper-Scissors strategy
A fast and robust iterative genome-editing method based on a Rock-Paper-Scissors strategy
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

- Altmetric
The production of optimized strains of a specific phenotype requires the construction and testing of a large number of genome modifications and combinations thereof. Most bacterial iterative genome-editing methods include essential steps to eliminate selection markers, or to cure plasmids. Additionally, the presence of escapers leads to time-consuming separate single clone picking and subsequent cultivation steps. Herein, we report a genome-editing method based on a Rock-Paper-Scissors (RPS) strategy. Each of three constructed sgRNA plasmids can cure, or be cured by, the other two plasmids in the system; plasmids from a previous round of editing can be cured while the current round of editing takes place. Due to the enhanced curing efficiency and embedded double check mechanism, separate steps for plasmid curing or confirmation are not necessary, and only two times of cultivation are needed per genome-editing round. This method was successfully demonstrated in Escherichia coli and Klebsiella pneumoniae with both gene deletions and replacements. To the best of our knowledge, this is the fastest and most robust iterative genome-editing method, with the least times of cultivation decreasing the possibilities of spontaneous genome mutations.
INTRODUCTION
Escherichia coli is the main workhorse for metabolic engineering, industrial biosynthesis, synthetic biology and many other related fields (1–3). To obtain a strain with the expected phenotype, many strains must be constructed using a suboptimal ‘trial and error’ strategy. This results in a large number of combinations of possible modifications which must be tried and tested. For example, up to 26 genomic modifications were carried out and tested in a project to produce 1,3-propanediol (4), more than 100 strains were required to obtain the best β-carotene producer, which contained 15 genomic modifications (5). And a large number of functional gene modifications were got by various researches including Tn-Seq (6), GREACE (7) and CRISPRi screening (8). Indeed, the outcome after combining these genome modifications may be unpredictable, and can result in both positive and negative epistasis (5). Thus, an efficient, rapid, programmable, combinable and robust genome-editing method would dramatically accelerate related research.
Several methods have been developed to introduce bacterial genome modifications. The main one is recombination, which is completed by either the host enzyme recombinase (9) or by more efficient phage recombinases, such as λ Red recombinase (10) or RecET (11). Almost all these methods contain two recombination steps; in the first step, gene modification is performed, and selected by a selection marker since efficiency is too low for colony PCR screening. This marker is usually an antibiotic resistance cassette introduced together with the recombination. The donor can be linear DNA fragments or suicide plasmids that can only be replicated in specific conditions, e.g. temperature-sensitive plasmids (9), or in specific hosts, like those expressing λ pir (12). In a second step, various strategies are employed to remove the selection marker, either because the number of available markers is limited and must be reused, or because the modification is expected to be scarless. In one of the most widely used methods (10), the selection marker is eliminated by Flp recombinase between the FRT sites introduced in the first recombination. Other site-specific recombinations are also used, like the loxP-mediated recombination by Cre recombinase (13). In any case, these methods require expression of the corresponding recombinase in another helper plasmid that has to be transformed and later cured (10,13,14). The process can be made faster when the two steps are carried out by two different inducers, without plasmid-curing, when the λ Red recombinase and Flp or Cre are expressed by a single plasmid using two different tightly controlled promoters (15,16). Speed is further increased by using two different selection markers that can be eliminated simultaneously, in every two rounds of genome editing (15,16). However, the engineered genome is not scarless. Scarless genome editing is vital when performing iterative genome editing because the scars like the remaining FRT or loxP sites may cause unwanted recombination, and the possibilities increase tremendously with the number of scars. In addition, the control of Flp or Cre expression may be leaky (16).
Although these methods can be further accelerated by integration of all the helper elements into the genome, this has not always been successful (16). Also, elimination of the markers is not 100% efficient, and single clones have to be picked and checked, which is time-consuming.
The removal of selection markers can also be performed by using counter-selectable markers introduced in the first step. One of the most widely used is sacB, which is lethal in presence of sucrose. However, this method may fail if sacB is inactivated (17), therefore single clones must still be picked and checked. Further, sacB-sucrose selection does not work in all bacteria, e.g. O157:H (18) or Pasteurella multocid (19). Another widely used counter-selection marker is double strand break (DSB) induced by homing endonucleases like I-SceI and I-CreI (18,20,21). Similar problems exist for this approach: endonucleases are provided by a helper plasmid that has to be cured later, or which may present leaky expression. Also, counter-selection is not 100% effective and single clones need to be picked and checked.
CRISPR/Cas9 is a recent and powerful genome-editing tool already used in various organisms (22,23). Cas9 can introduce DSB by the guiding of gRNA, and it has been used in E. coli genome editing in combination with λ Red recombinase (5,22–26). Although the method is highly efficient and requires only one step, every gene edited requires its own gRNA. As a result, the gRNA-expressing plasmid has to be cured, and another plasmid must be introduced during iterative genome editing (5,25). The plasmid is typically cured by the CRISPR/Cas9 system itself, using inducible gRNA (5,25). However, this is also time-consuming and not 100% efficient (5,24,27,28), thus a single clone has to be picked and curing has to be confirmed.
One of the most widely used E. coli genome-editing CRISPR/Cas9 systems is a two-plasmid system (25): pCas expresses λ Red recombinase and Cas9 whereas pTarget provides the variational sgRNA with various N20 targeting sequences. In some reports, all the elements (λ Red, cas9 and sgRNA) have been placed in one single plasmid, allowing genome editing in three days (29), although additional plasmid curing was needed during iterative genome editing. Other approaches have completed iterative genome editing using two days per round which included four times of cultivation because plasmid curing and subsequent single clone picking are still needed (5).
In another report, the plasmid was cured by sacB counter-selection (28) during the liquid cultivation of the positive engineered single clone with high efficiency, and prepared the competent cells directly for the next rounds of engineering. The gRNA plasmid curing step is after the single clone picking step for successful genome editing, and the following curing efficiency should be supposed to be 100% if the cells will not be purified by another single clone picking step. In their work, only two rounds of genome editing were performed (28). However, this is subject to the limitations of sacB discussed above. Escape from sacB-sucrose selection increases with more rounds of editing. Escapers carrying the selection marker may be enriched because of growth advantage, whereas only a small portion of cells can be transformed in the subsequent rounds.
Overall, all genome-editing strategies require either separate marker elimination or plasmid curing steps. In addition, to avoid the always likely escapers, single clones have to be picked for subsequent rounds of editing. This is not only time-consuming and laborious, especially for large scale editing rounds, but the probability of unwanted spontaneous genome mutations is increased due to the required lengthy cultivation times. The existence of escapers is pervasive in biology and beneficial for living organisms, as is the basis of adaptation to our environment. However, in biotechnology and artificial biological systems, escapers present an important problem.
To avoid this problem, inspiration for our method comes from a recently reported system that improves robustness and stabilizes functionality in artificial genetic circuits (30). In these sophisticated artificial biological systems (31) runaway mutations may cause loss of function (32). To solve this problem, Liao and colleagues developed a ‘Rock-Paper-Scissors’ (RPS) system consisting of three E. coli strains that can either kill, or be killed by, one of the other strains using three toxin-antitoxin pairs (30). Similarly, we have developed a RPS genome-editing system that consists of three sgRNA plasmids, where each can either cure, or be cured by, one of the other plasmids (Figure 1A). Strains carrying pCas (expressing Cas9 and λ Red recombinase) (25) can be modified by each of these plasmids in ‘RPS order’ iteratively, while curing the previous RPS plasmid simultaneously. In this way, the plasmid used in the previous editing round are expected to be already cured when picking the single successfully edited clones. The curing efficiency by CRISPR/Cas9 is high, but it is further enhanced in our design to reduce escapers, and additionally there is a double-check mechanism (see below). As a result, no separate curing or confirmation step is required. This robust curing-free strategy can dramatically speed up interactive genome editing because only two times of cultivation are needed per round, reducing the probability of spontaneous genome mutation (Figure 1B). To the best of our knowledge, this is the most robust and fast iterative genome-editing strategy reported so far.

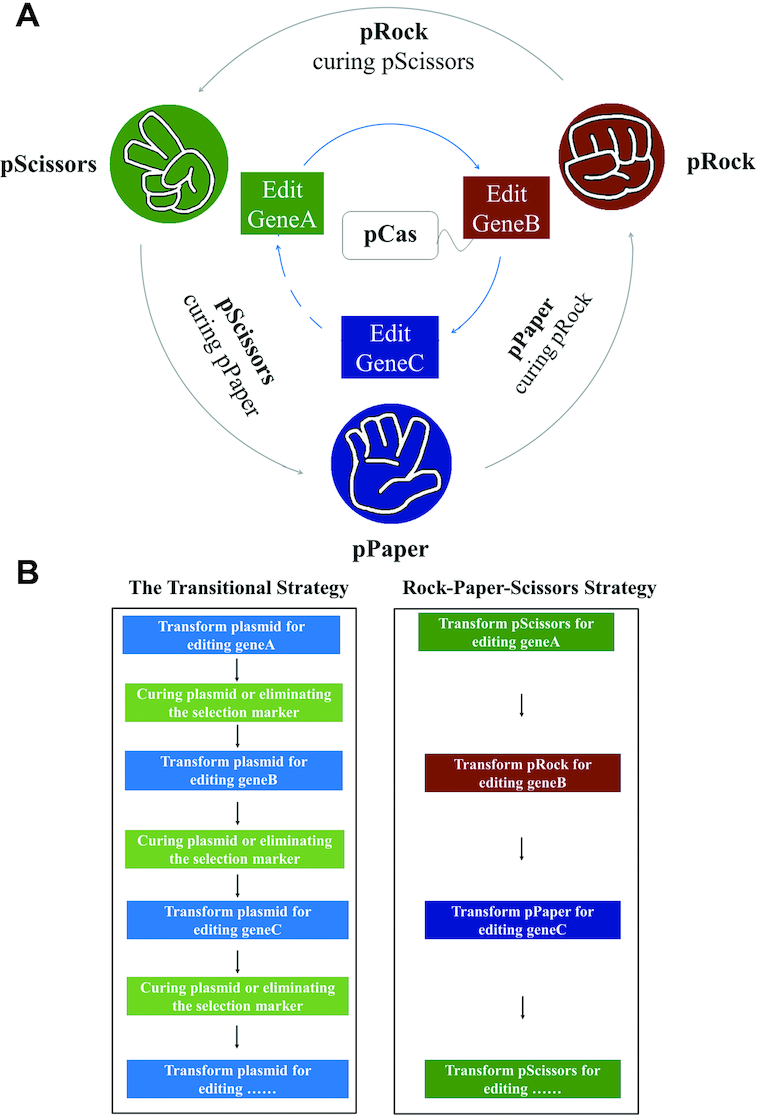
‘Rock-Paper-Scissors’ (RPS) genome-editing strategy. (A) Each of the three plasmids can cure, or be cured by, one of the other two plasmids while editing the genome. (B) No separate steps for curing plasmids or elimination of selection marker are needed.
MATERIALS AND METHODS
Strains, plasmids and cloning methods
All plasmids and strains used in this study are shown in Tables 1 and 2, respectively. Plasmids construction was performed in E. coli TOP10, whereas plasmids curing tests were performed in E. coli W3110. Pyruvate-producing strains were constructed from E. coli strain BW25113. The system was also tested in Klebsiella pneumoniae ATCC25955 and a d-lactate producing strain was constructed.

| Plasmids | Relevant characteristics | Source or reference |
|---|---|---|
| pCas | repA101(Ts) KanR Pcas-cas9 ParaB-Red lacIq Ptrc-sgRNA-pMB1 | MolecularCloud: MC_0000011 (25) |
| pTargetF | pMB1 SpecR J23119-sgRNA | MolecularCloud: MC_0000012 (25) |
| pYTarget | pMB1 SpecR J23119-sgRNA-BsaI | Laboratory stock |
| pSB1A3 | pMB1 AmpR | iGEM Repository |
| pSB1C3 | pMB1 CmR | iGEM Repository |
| pSB1T3 | pMB1 TetR | iGEM Repository |
| pMDIAI | pMB1 ApmR | MolecularCloud: MC_0000009 (21) |
| pBAD30 | p15A AmpR | Laboratory stock |
| pRPS | p15A SpecR PlacIQ-sgRNA-BsaI | This study |
| pRPS-A | p15A AmpR PlacIQ-sgRNA-BsaI | This study |
| pRPS-T | p15A TetR PlacIQ-sgRNA-BsaI | This study |
| pRPS-C | p15A CmR PlacIQ-sgRNA-BsaI | This study |
| pRPS-G | p15A ApmR PlacIQ-sgRNA-BsaI | This study |
| pRPS-kA | p15A SpecR PlacIQ-sgRNA-AmpR | This study |
| pRPS-kT | p15A SpecR PlacIQ-sgRNA-TetR | This study |
| pRPS-kC | p15A SpecR PlacIQ-sgRNA-CmR | This study |
| pRPS-kG | p15A SpecR PlacIQ-sgRNA-ApmR | This study |
| pRPS-CkT | p15A CmR PlacIQ-sgRNA-TetR | This study |
| pRPS-TkG | p15A TetR PlacIQ-sgRNA-ApmR | This study |
| pRPS-GkC | p15A ApmR PlacIQ-sgRNA-CmR | This study |
| pRock | p15A TetR PlacIQ-sgRNA-ApmR J23119-sgRNA-BsaI | This study MolecularCloud: MC_0101139 |
| pPaper | p15A CmR PlacIQ-sgRNA-TetR J23119-sgRNA-BsaI | This study MolecularCloud: MC_0101140 |
| pScissors | p15A ApmR PlacIQ-sgRNA-CmR J23119-sgRNA-BsaI | This study MolecularCloud: MC_0101141 |
| pRock-Δ(focA-pflB) | p15A TetR PlacIQ-sgRNA-ApmR J23119-sgRNA-(focA-pflB) Δ(focA-pflB)(3189 bp):: NotI | This study |
| pPaper-ΔfrdBC | p15A CmR PlacIQ-sgRNA-TetR J23119-sgRNA-frdBC ΔfrdBC(1083 bp):: NotI | This study |
| pScissors-ΔldhA | p15A ApmR PlacIQ-sgRNA-CmR J23119-sgRNA-ldhA ΔldhA(984 bp):: NotI | This study |
| pRock-ΔatpFH | p15A TetR PlacIQ-sgRNA-ApmR J23119-sgRNA-atpFH ΔatpFH(968 bp):: NotI | This study |
| pPaper-ΔadhE | p15A CmR PlacIQ-sgRNA-TetR J23119-sgRNA-adhE ΔadhE(2670 bp):: NotI | This study |
| pScissors-ΔsucA | p15A ApmR PlacIQ-sgRNA-CmR J23119-sgRNA-sucA ΔsucA(2753 bp):: NotI | This study |
| pRock-ΔpoxB | p15A TetR PlacIQ-sgRNA-ApmR J23119-sgRNA-poxB ΔpoxB(1691 bp):: NotI | This study |
| pPaper-ΔackA | p15A CmR PlacIQ-sgRNA-TetR J23119-sgRNA-ackA ΔackA(1197 bp):: NotI | This study |
| pBAD18-ldhA | KnR, pBR322, ParaBAD-ldhA | (49) |
| pPaper-ΔyqhD | p15A CmR PlacIQ-sgRNA-TetR J23119-sgRNA-yqhD ΔyqhD(1143 bp):: NotI | This study |
| pPaper-ΔyqhD::ldhA | p15A CmR PlacIQ-sgRNA-TetR J23119-sgRNA-yqhD ΔyqhD(1143 bp):: ParaBAD-ldhA | This study |
| pScissors-ΔdhaT | p15A ApmR PlacIQ-sgRNA-CmR J23119-sgRNA-dhaT ΔdhaT(1121 bp):: NotI | This study |
| pScissors-ΔdhaT::ldhA | p15A ApmR PlacIQ-sgRNA-CmR J23119-sgRNA-dhaT ΔdhaT(1121 bp):: ParaBAD-ldhA | This study |
KanR: kanamycin resistance; SpecR: spectinomycin resistance; AmpR: ampicillin resistance; CmR: chloramphenicol resistance; TetR: tetracycline resistance; ApmR: apramycin resistance; sgRNA-BsaI: sgRNA with BsaI flanked non-targeting spacer.
| Strains | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli TOP10 | F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL(StrR) endA1 λ- | Laboratory stock |
| E. coli W3110 | F- λ- rph-1 INV(rrnD, rrnE) | Laboratory stock |
| E. coli BW25113 | F- DE(araD-araB)567 lacZ4787(del)::rrnB-3 LAM- rph-1 DE(rhaD-rhaB)568 hsdR514 | Laboratory stock |
| W3Q00 | W3110 /pCas | This study |
| CasA | W3Q00 /pRPS-A | This study |
| CasT | W3Q00 /pRPS-T | This study |
| CasG | W3Q00 /pRPS-G | This study |
| CasC | W3Q00 /pRPS-C | This study |
| BWQ00 | BW25113 /pCas | This study |
| BWQ01 | BWQ00 Δ(focA-pflB)::NotI /pRock-Δ(focA-pflB) | This study |
| BWQ02 | BWQ01 ΔfrdBC::NotI /pPaper-ΔfrdBC (pRock-Δ(focA-pflB) cured) | This study |
| BWQ03 | BWQ02 ΔldhA::NotI /pScissors -ΔldhA (pPaper-ΔfrdBC cured) | This study |
| BWQ04 | BWQ03 ΔatpFH::NotI /pRock-ΔatpFH (pScissors-ΔldhA cured) | This study |
| BWQ05 | BWQ04 ΔadhE::NotI /pPaper-ΔadhE (pRock-ΔatpFH cured) | This study |
| BWS05 | BWQ04 ΔackA::NotI /pPaper-ΔackA (pRock-ΔatpFH cured) | This study |
| BWQ06 | BWQ05 ΔsucA::NotI /pScissors-ΔsucA (pPaper-ΔadhE cured) | This study |
| BWQ07 | BWQ06 ΔpoxB::NotI /pRock-ΔpoxB (pScissors-ΔsucA cured) | This study |
| BWQ08 | BWQ07 ΔackA::NotI /pPaper-ΔackA (pRock-ΔpoxB cured) | This study |
| BWQ08-dN | BWQ08 (pPaper-ΔackA cured) | This study |
| BWQ08-dNK | BWQ08-dN (pCas cured) | This study |
| BWS01 | BWQ00 ΔsucA::NotI /pScissors-ΔsucA | This study |
| KpWT | K. pneumoniae ATCC25955 | ATCC |
| KpQ00 | KpWT /pCas | This study |
| KpT | KpQ00 /pRPS-TkG | This study |
| KpG | KpQ00 /pRPS-GkC | This study |
| KpC | KpQ00 /pRPS-CkT | This study |
| KpQ01 | KpQ00 ΔyqhD::ParaBAD-ldhA /pPaper-ΔyqhD::ldhA | This study |
| KpQ02 | KpQ01 ΔdhaT:: ParaBAD-ldhA /pScissors-ΔdhaT::ldhA (pPaper-ΔyqhD::ldhA cured) | This study |
| KpQ02-dN | KpQ02 (pScissors-ΔdhaT::ldhA cured) | This study |
| KpQ02-dNK | KpQ02-dN (pCas cured) | This study |
All primers (Supplementary Table S1) were synthesized by Sangon Biotech. Colony PCRs were performed by Rapid Taq (Vazyme Biotech, Cat# P222-02), whereas all other PCRs were performed by PrimeSTAR (TaKaRa, Cat# R040A). Ligations were completed by DNA Ligation Kit Ver.2.1 (TaKaRa, Cat# 6022). Competent cells were prepared by Ultra-Competent Cell Preps Kit (Sangon Biotech, Cat# B529303–0200). All plasmid constructions, except the introduction of spacers, were performed by commercial seamless cloning kits ClonExpress Ultra One Step Cloning Kit (Vazyme Biotech, Cat# C115-01) based on in vitro multi fragments recombination. All experiments were performed according to the manufacturer's instructions.
All E. coli strains were cultivated in Luria-Bertani (LB) medium, and 0.4% glycerol was provided after atpFH was knocked out. Sodium succinate (5 mM) was provided when sucA was knocked out, and 10 mM was provided for fermentation. Fermentation was performed in M9CA broth, containing 1 mM MgSO4, 0.1 mM CaCl2 and 20% glucose. To maintain pH, 100 mM MOPS (pH 7.1) was added. Ammonium hydroxide was also used for additional pH control, when required. Appropriate antibiotics were supplied when needed: 25 ng/ml kanamycin, 100 ng/ml spectinomycin, 100 ng/ml ampicillin, 100 ng/ml apramycin, 33 ng/ml chloramphenicol and 17 ng/ml tetracycline.
All K. pneumoniae strains were also cultivated in LB medium. Fermentation was performed in M9CA broth, containing 1 mM MgSO4, 0.1 mM CaCl2 and 20% glycerol. To maintain pH, 100 mM MOPS (pH 7.1) was added, the pH was further adjusted by ammonium hydroxide every eight hours. Appropriate antibiotics were supplied when needed: 50 ng/ml kanamycin, 100 ng/ml spectinomycin, 100 ng/ml apramycin, 50 ng/ml chloramphenicol and 25 ng/ml tetracycline.
Plasmids design and construction
All the spacers were designed by sgRNAcas9 V3.0, as indicated (48), and we selected the best spacers located near the middle of the coding sequences of the targeted genes (Supplementary Table S2).
The representative RPS plasmid design is shown in Figure 2A (see text in Results for details).
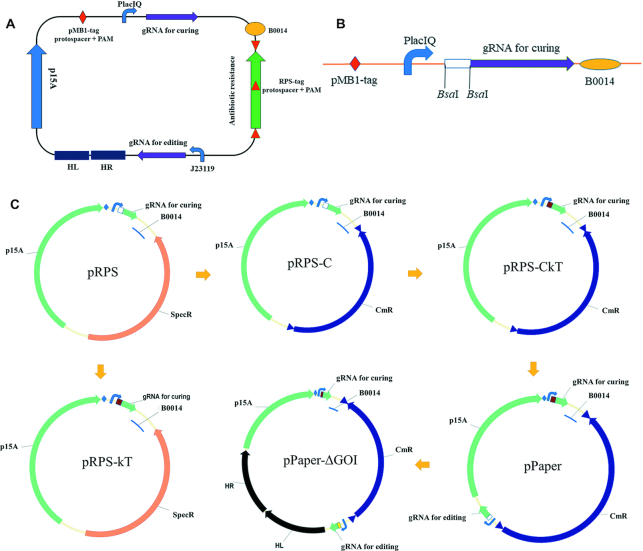
Plasmids design and construction. (A) General design of ‘Rock-Paper-Scissors’ plasmids. The two sgRNAs are under two different constitutive promoters. One targets another RPS plasmid antibiotic resistance gene, for plasmid curing. The other one targets specific genome locus for genome editing. The pMB1-tag contains a protospacer and a PAM sequence, so that all the plasmids can be cured by pCas after IPTG induction (25). The RPS-tag contains a protospacer and a PAM sequence, so that it can be cured by another RPS plasmid. It is duplicated twice by flanking the antibiotic resistance gene on different strands. (B) Synthesized KX part. A non-targeting spacer sequence is flanked by two BsaI sites, so that it can be easily substituted by other designed spacers oligos. C. Constructed plasmid examples. Spec: spectinomycin resistant, Cm: chloramphenicol resistant, GOI: gene of intererest. See the main text for additional details.
In the construction of pRPS, we synthesized the KX part with several genetic parts (Figure 2B) by BGI (Beijing Genomics Institution) (Supplementary Table S3). KX was amplified by H-kX-F and kX-R, p15A was amplified by H-P15AF and P15AR from pBAD30, and spectinomycin resistance cassette was amplified by Spec-R and H-Spec-F from pTargetF. The overlap sequences were added by the primers (Supplementary Table S1) and these three fragments were assembled by ClonExpress Ultra One Step Cloning Kit, according to the manufacturer's instructions.
In the construction of pRPS-C (Figure 2C), the chloramphenicol resistance cassette was amplified by H-V2C-F and H-V2C-R, from pSB1C3. The designed protospacer and PAM sequence was introduced into both ends of the cassette by the primers. The plasmid backbone was amplified by H-P15AF and B0014-R from pRPS. These three fragments were assembled by seamless cloning. Similarly, pRPS-T, pRPS-A and pRPS-G were constructed by amplifying the corresponding antibiotic resistance cassettes from pSB1T3, pSB1C3 and pMDIAI.
For the construction of pRPS-kT and pRPS-CkT (Figure 2C), annealed oligos (BsaI-T.S53F and BsaI-T.S53R) were ligated into pRPS or pRPS-C digested with BsaI, to obtain plasmids pRPS-kT and pRPS-CkT, with curing gRNA targeting the coding sequence of apramycin resistance gene. Similarly, pRPS-kG, pRPS-kA and pRPS-kC were constructed by introducing corresponding spacers targeting different antibiotic resistance genes (Supplementary Table S2). pRPS-GkC and pRPS-TkG were constructed from pRPS-G and pRPS-T, respectively.
For the construction of pPaper (Figure 2C), the ‘for editing’ gRNA cassette was amplified by H-J119F and H-gRNAR4 from pYTarget. The latter contains sgRNA with a non-targeting spacer sequence, flanked by two BsaI sites under J23119. This fragment was assembled with the backbone of pRPS-CkT and amplified by VE-F and VE-R, producing the resulting plasmid designated as pPaper. Similarly, pScissors and pRock were constructed by adding the gRNA cassette to pRPS-GkC and pRPS-TkG, respectively.
Plasmids transformation and curing efficiency test
To obtain the curing efficiency of plasmids by CRISPR/Cas9, CasT was constructed by transforming pCas and pRPS-T into W3110. Plasmids pRPS-kT or pRPS (200 ng) were transformed into CasT, and appropriate portions were spread onto LB plates containing kanamycin and spectinomycin, with or without tetracycline added, to test loss of tetracycline resistance resulting from the curing of pRPS-T by pRPS-kT. Similarly, the curing of pRPS-A, pRPS-C and pRPS-G by pRPS-kA, pRPS-kC and pRPS-kG was tested, with pRPS being used as a control.
To test how efficiently plasmids are cured by other plasmids, 100 ng of pRPS-C or pRPS-CkT were transformed into CasT and appropriate portions were spread onto LB plates, containing kanamycin and chloramphenicol, with or without tetracycline added, to test the loss of tetracycline resistance, i.e. the curing of pRPS-T by pRPS-kT. The number of clones appearing on Kan + Cm and Kan + Cm + Tet plates were counted and taken as a number of successful transformations (Nt) or curing escapers (Ne), respectively. The curing efficiency was calculated as (1-Ne/Nt) × 100%, whereas the transformation efficiency was calculated as Nt/μg plasmid. The transformation and curing efficiencies of the other two groups were determined in the same way.
Similarly, the curing efficiency was also tested in K. pneumoniae. KpT was constructed by transforming pCas and pRPS-TkG into KpWT. pRPS-CkT was transformed into KpT, and appropriate portions were spread onto LB plates containing kanamycin and chloramphenicol, with or without tetracycline added, to test loss of tetracycline resistance resulting from the curing of pRPS-TkG by pRPS-CkT. The curing efficiency was calculated. Similarly, the curing of pRPS-CkT and pRPS-GkC by pRPS-GkC and pRPS-TkG was tested.
Protocol for RPS gene editing plasmids design and construction
The steps are shown in a flow diagram (Figure 3A).
Step 1. Plasmid backbone. Plasmid backbones are amplified by TargetVF2 and TargetVR using pRock, pPaper and pScissors as templates. These three backbones can be reused in different RPS plasmids construction.
Step 2. ‘for genome editing’ sgRNA. Spacers targeting the genes of interest (GOI) are designed by sgRNAcas9 V3.0, as described (48). The spacer is introduced by PCR using primers gRNA5 and corresponding primers named H-GOI-gF, where H stands for overlap sequence of plasmid backbone which is unchanged for different RPS plasmids construction. pTargetF is used as the template and ‘for genome editing’ sgRNA is obtained.
Step 3. Left and right homology arms. Generally, the sequence between initiation and termination codons of GOI should be knocked out, unless the Shine-Dalgarno ribosome binding sequence of the downstream gene is affected. Left and right homology arms are designed of ∼600 bp. This number is variable to produce optimized primers. To improve the PCR specificity and facilitate the following colony PCR, primers upstream the left homology arm and downstream the right homology arm are designed, Pre_GOI and Aft_GOI, respectively. The primers for amplifying left and right arms are also designed. These are named KGOIHLF + KGOIBHLR and KGOIHRF + KGOIHRR, respectively, where K stands for K12 (or Kp for K. pneumoniae), and appropriate overlap sequences for fragment assembly should be included in appropriate primers, then named H-PrimerName. The genome is amplified by Pre_GOI and Aft_GOI and the product is used as template in a second amplification by KGOIHLF + KGOIBHLR and KGOIHRF + KGOIHRR to obtain the left and right arms with appropriate overlap sequences for fragment assembly.
Step 4. Fragment assembly. The four fragments obtained above are assembled by commercial seamless clone kits in one step, and transformed into TOP10. Colony PCR was performed by Y_Target-2F and P15A_endF to obtain positive clones, and clones are confirmed by Sanger sequencing, using primers P15A_endF + P15A_headR + TheadR2/CheadR2/GheadR2, for pRock, pPaper and pScissors, respectively. These steps result in the construction of genome-editing plasmids pRock/pPaper/pScissors-ΔGOI.
Step 5. Fragment insertion. When gene insertion or replacement is needed, the donor fragment is inserted into pRock/pPaper/pScissors-ΔGOI by NotI digestion and following ligation. The resulting plasmids were named pRock/pPaper/pScissors-ΔGOI:: insertion. For instance, the ldhA expressing cassette was amplified from pBAD18-ldhA (49) with primers Pre_BADF and Aft_BADR and then digested by NotI.
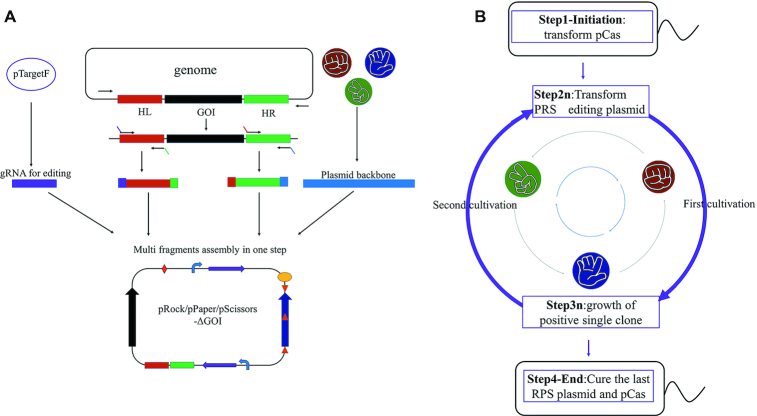
Flow diagram of pRPS editing plasmids construction and iterative genome-editing. (A) Flow diagram of RPS editing plasmids, where four fragments can be obtained by PCR and then assembled in one step (see text in Materials and Methods for details). GOI: gene of interest. (B) Flow diagram of iterative genome editing by RPS editing plasmids, where only 2n times of cultivation were needed for n rounds of iterative genome editing (see text in Methods for details).
Protocol for iterative genome-editing by RPS editing plasmids
The steps followed are shown in a flow diagram (Figure 3B):
Step 1. Initiation. The strain to be engineered is first transformed by pCas.
Step 2. First cultivation. Transform the RPS editing plasmids to obtain a successfully edited single clone. pRock-geneA (depends on the previous round, and the first plasmid can be any of the three types) is then transformed and incubated at 30°C on plates with appropriate antibiotics. Colony PCR is performed with Pre_geneA and Aft_geneA to obtain single clones that are successfully engineered.
Step 3. Second cultivation. Grow the single clone for subsequent steps. A positive single clone is picked and cultivated in liquid LB with Kan and appropriate antibiotic (tetracycline for pRock series). Then the cells are used for preparing competent cells and other purposes.
Step 4. Next round. Go to step 2. pPaper-geneB (depends on the previous round) is then transformed into the competent cells above. The steps are the same as step 2 and 3.
Step 5. End. Cure the last RPS editing plasmids and pCas. After all the genome editing is completed, the last RPS editing can be cured by IPTG induction and pCas can be cured at 42°C, as described (25).
Transformation method
All the transformation of E. coli was by heat shock method using Ultra-Competent Cell Preps Kit (Sangon Biotech, Cat# B529303-0200) according to the manufacturer's instructions.
When constructing BWQ06, electroporation was also tried. All the transformation of K. pneumoniae was by electroporation. When the cell concentration reached OD600 of 0.4–0.9, the culture was washed by twice of ice-cold water and one time of ice-cold 10% glycerol, the electroporation was performed by MicroPulser Electroporator (Bio-Rad, Cat# 1652100) using the Ec1 pre-programmed settings according to the manufacturer's instructions.
When genes need to be edited, 10 mM l-arabinose was added to induce λ Red recombinase before the component cell preparations.
Shake-flask fermentation
Pyruvate production of E. coli BWQ08-dNK was studied using a 500 mL Erlenmeyer flask. The overnight precultured cells were inoculated into 100 mL M9CA, with the additives mentioned above, and cultivated at 37°C under 250 rpm agitation. Every four hours, cell density, glucose, pyruvate and acetate concentrations were measured. Cell density was measured using absorbance at 600 nm. Glucose concentration was measured by a YSI 2900 Series biochemistry analyzer. Pyruvate and acetate concentrations were measured by HPLC monitoring the absorbance at 210 nm, using an Agilent 1200-series HPLC system equipped with an Aminex HPX-87H column and a UV detector. The mobile phase was 5 mM H2SO4. E. coli BW25113 was used as a WT control.
d-lactate production of K. pneumoniae BWQ02-dNK was studied using a 250 mL Erlenmeyer flask. The overnight precultured cells were inoculated into 100 ml M9CA, with the additives mentioned above, and cultivated at 37°C under 100 rpm agitation. 10 mM l-arabinose was added when OD600 reached 0.5–0.8, and was resupplied every 8 h. Lactate, 1,3-PDO and glycerol concentrations were measured by HPLC with a differential refractive index detector. K. pneumoniae ATCC 25955 was used as a WT control.
All experiments were carried in triplicates, and data of mean and standard deviation were determined.
RESULTS
Design and construction of the ‘Rock-Paper-Scissors’ system
The representative RPS plasmid design is shown in Figure 2A. The three RPS plasmids provide different antibiotic resistance and express sgRNA for genome editing compatible with pCas. In order to cure the plasmid from the previous round, an additional ‘for curing’ sgRNA expression cassette was added that targets the previous RPS plasmid. The cassettes for ‘for curing’ sgRNA and ‘for genome editing’ sgRNA were flanked by a p15A replicon and an antibiotic resistance marker ended by a B0014 terminator, so that a recombination between these two homologous cassettes is lethal. The p15A replicon was chosen because of its relatively low copy number and burden to the cell. In order to make the plasmid curable by pCas after induction by IPTG as described (25), the targeted sequence named pMB1-tag (including a protospacer and PAM) was added. In order to reduce the homology between the two gRNA expression cassettes, two different constitutive promoters were used: PlacIQ, for the ‘for curing’ sgRNA cassette and J23119 from pTarget (25) for the ‘genome editing’ sgRNA cassette. The curing sgRNA targets the antibiotic resistance coding sequence of another plasmid.
Due to mutations in either protospacer or PAM, there are always escapers during plasmid curation by CRISPR/Cas9 (24). Therefore, the RPS-tag containing the protospacer to be targeted, and the corresponding PAM sequence, were replicated twice on both strands, flanking the antibiotic resistance cassette. As a result, every plasmid can be targeted by three sites in different strands, which greatly reduces the probability of escape.
Effective curation by CRISPR/Cas9 of plasmids with triple targeted sites
The plasmids construction procedure is shown in Figure 2C. First, a template plasmid pRPS with a non-targeting sgRNA was constructed (Figure 2A). Spacers consisting of 20 nt sequences targeting the coding sequences of ampicillin, chloramphenicol, tetracycline and apramycin resistance genes were designed (Supplementary Table S2) and introduced into pRPS. The resulting plasmids were named pRPS-kA, pRPS-kC, pRPS-kT and pRPS-kG, respectively. The spectinomycin resistance gene of pRPS was replaced by these four antibiotic resistance cassettes, flanked by two corresponding RPS-tags, as described above. The resulting plasmids were named pRPS-A, pRPS-C, pRPS-T, pRPS-G, respectively. These four plasmids were transformed into W3110 carring pCas and the resulting strains were named CasA, CasC, CasT and CasG, respectively.
pRPS-kT was transformed to CasT and spread on an LB plate that contained kanamycin and spectinomycin, with or without tetracycline, to check the curing efficiency of pRPS-kT towards pRPS-T. As a negative control, pRPS was also transformed. (Figure 4) The transformation of pRPS produced less and smaller clones with non-uniform sizes, probably because of the burden caused by the coexistence of two types of pRPS series plasmids. Transformations of pRPS-kT produced more and larger clones, with uniform size, possibly because of the effective curing of pRPS-T. Thus, pRPS-T cannot be cured by pRPS with non-targeting sgRNA, even though the two plasmids are incompatible, which is consistent with previous reports (33).
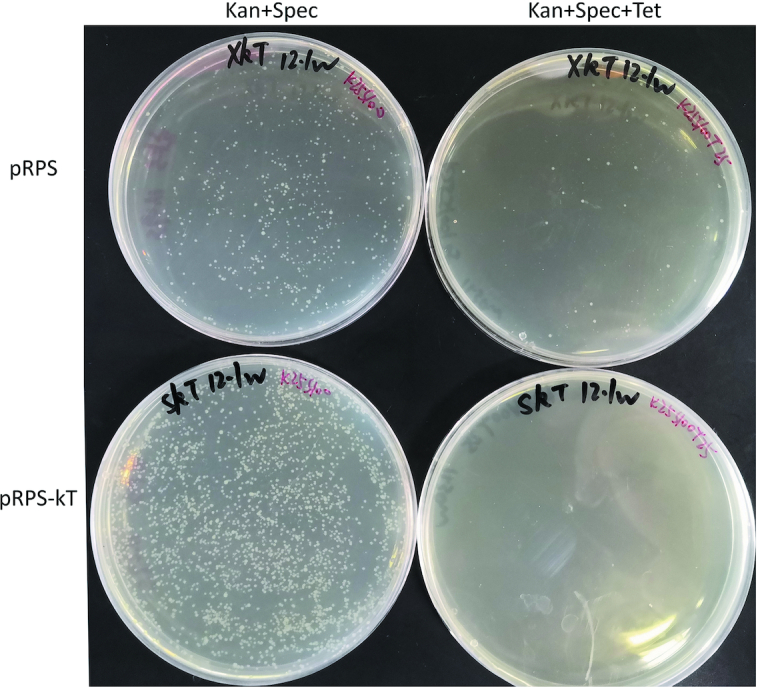
Curing of pRPS-T by pRPS-kT. Curing efficiency was tested by transforming pRPS-kT into CasT, whereas pRPS was transformed as control. An equal volume of transformants was spread onto LB plates containing to either Kan + Spec (successfully transformed clones) or Kan + Spec + Tet (clones that escaped curing). Kan: kanamycin, Spec: spectinomycin, Tet: tetracycline.
Similarly, we tested the ability of pRPS-kA, pRPS-kC and pRPS-kG to cure plasmids pRPS-A, pRPS-C and pRPS-G, respectively (Supplementary Figures S1-S3). Similar to what was found for pRPS-T, pRPS-C and pRPS-G could be cured by pRPS-kC and pRPS-kG, respectively. However, pRPS-A could not be cured for unknown reasons. This problem may be solved by trying different spacers that target the ampicillin resistance gene. Only three different pRPS series plasmids were required to construct our system. Because pRPS-C, pRPS-T and pRPS-G can be cured effectively, they were used in the subsequent steps.
Effective curing of ‘Rock-Paper-Scissors’ plasmids
The N20 sequences present in pRPS-kG, pRPS-kT and pRPS-kC were introduced into pRPS-T, pRPS-C and pRPS-G, respectively. This resulted in plasmids pRPS-TkG, pRPS-CkT and pRPS-GkC (Figure 2C). To test the efficiency of ‘Paper covering Rock’, plasmid pRPS-CkT was transformed into CasT and spread on an LB plate containing kanamycin and chloramphenicol, with or without tetracycline. Plasmid pRPS-C, with non-targeting sgRNA, was also transformed as a negative control. Similar to the curing of pRPS-T by pRPS-kT, the transformation of pRPS-C produced fewer and smaller clones with heterogeneous sizes, whereas the transformation of pRPS-CkT produced many more and larger clones with uniform size (Figure 5A). The transformation efficiency of pRPS-CkT was higher than that of pRPS-C (Figure 5B). These results indicate that successful plasmid curing caused growth advantage possibly because of reduced burden to the cells.
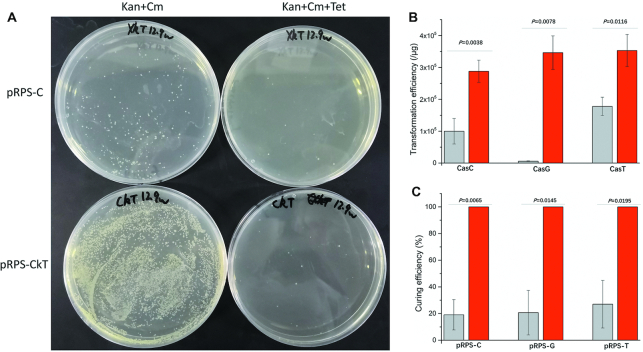
Curing effect of RPS plasmids. (A) Curing of pRPS-T by pRPS-CkT. The curing efficiency of pRPS-T by pRPS-CkT was tested by transforming pRPS-CkT into CasT, whereas pRPS-C was also transformed as control. An equal volume of transformants was spread onto LB plates with Kan + Cm (successfully transformed clones) or Kan + Cm + Tet (clones that escaped the curing). Kan: kanamycin; Cm: chloramphenicol; Tet: tetracycline. (B) Transformation efficiency of plasmids pRPS-GkC, pRPS-TkG and pRPS-CkT (with targeting sgRNA for curing) into CasC, CasG and CasT, respectively (red), and transformation efficiency of control plasmids pRPS-G, pRPS-T and pRPS-C (with non-targeting sgRNA for curing) (gray). (C) Curing efficiency of plasmids pRPS-C, pRPS-G and pRPS-T by RPS-GkC, pRPS-TkG and pRPS-CkT (with targeting sgRNA for curing), respectively (red), and curing efficiencies by control plasmids pRPS-G, pRPS-T and pRPS-C (with non-targeting sgRNA for curing) (gray). Data information: In (B, C), error bars show mean ± standard deviations from three experiments, and p-values are calculated from two tail t-tests.
The curing efficiency by pRPS-C without sgRNA targeting was relatively low, whereas the curing efficiency by pRPS-CkT was 99.99% (1-3/21200) (Figure 5C). After prolonged cultivation, only three clones were found on the three triple antibiotic plates. This high efficiency of plasmid curing may be due to our design of triple RPS-tags in different strands, as this prevents escape by mutations in the protospacers or PAMs. Similar results were obtained when testing the curing effect of pRPS-TkG and pRPS-GkC (Supplementary Figures S4–S5 and Figure 5B and C), with curing efficiencies of 100% in both cases (1-0/20800 and 1-0/17280, respectively) (Figure 5C), i.e. no clones were found on the triple antibiotic plates.
Since all three plasmids can cure each other with an efficiency of about 100%, a ‘for editing’ gRNA cassette, with BsaI flanking a non-targeting spacer sequence, was added to pRPS-TkG, pRPS-CkT and pRPS-GkC. This resulted in plasmids pRock, pPaper and pScissors, respectively. These three plasmids should cure each other while at the same time can edit the genome if specific spacers and homology sequences are introduced (Figure 2C).
When using these three plasmids to edit the genome in the ‘Rock-Paper-Scissors’ order, no separate plasmid-curing step is needed. The curing efficiency was about 100% and the picked successfully engineered single clone was also pure with the plasmid cured.
Rapid construction of a pyruvate-producing strain
TC44 is a strain derived from E. coli W3110 that efficiently converts glucose to pyruvate. TC44 was obtained by eight rounds of gene editing: ΔfocA-pflB::FRT, ΔfrdBC, ΔldhA, Δatp(FH)::FRT, ΔadhE::FRT, ΔsucA::FRT, ΔpoxB::FRT and ΔackA::FRT(34,35).
To demonstrate the efficiency of our RPS gene editing strategy, we attempted to obtain an equivalent strain from E. coli BW25113 by eight successful rounds of genome editing: ΔfocA-pflB::NotI, ΔfrdBC::NotI, ΔldhA::NotI, Δatp(FH)::NotI, ΔadhE::NotI, ΔsucA::NotI, ΔpoxB::NotI and ΔackA::NotI, (Figure 6). NotI was included so that all these editing plasmids could also be reused for insertion. There are quite a large amount of genetic parts that are compatible with BioBrick BBF RFC 10 standards. All these parts are flanked by NotI and can be integrated into the genome by inserting into these RPS editing plasmids. The editing efficiency of every round was high (Table 3) and no plasmid curing or confirmation steps were performed.
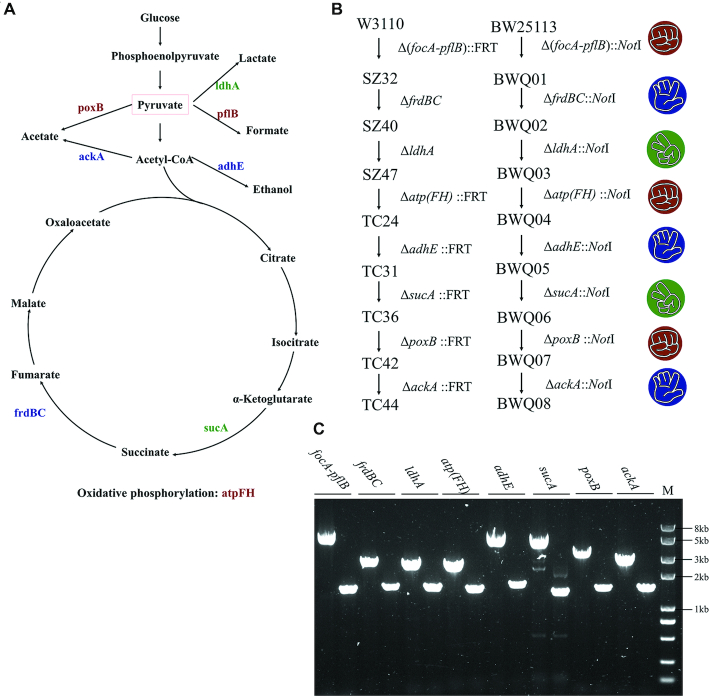
Construction of a pyruvate-producing strain. (A) Genes knocked out to construct the pyruvate-producing strain, edited by pRock (red), pPaper (blue) and pScissors (green). (B) Gene editing order of both TC44(34,35) and the present BWQ08. C. Verification of gene editing of BWQ08-dNK. All genes checked are marked. The left lanes are colony PCR results using WT as templates. The right lanes are results using BWQ08-dNK as templates. M: DNA marker.
| Parent strain | Genes to be edited | Plasmid backbone | Resulting strain | Selected clonea | Positive cloneb | Positive rate |
|---|---|---|---|---|---|---|
| BWQ00 | ΔfocA-pflB | pRock | BWQ01 | 20 | 20 | 100% |
| BWQ01 | ΔfrdBC | pPaper | BWQ02 | 20 | 20 | 100% |
| BWQ02 | ΔldhA | pScissors | BWQ03 | 20 | 19 | 95% |
| BWQ03 | ΔatpFH | pRock | BWQ04 | 20 | 19 | 95% |
| BWQ04 | ΔadhE | pPaper | BWQ05 | 4 | 2 | 50% |
| BWQ04 | ΔackA | pPaper | BWS05 | 20 | 20 | 100% |
| BWQ05 | ΔsucA | pScissors | BWQ06 | 8,20c | 1,19 | 12.5%,95% |
| BWQ00 | ΔsucA | pScissors | BWS01 | 20 | 18 | 90% |
| BWQ06 | ΔpoxB | pRock | BWQ07 | 20 | 14 | 70% |
| BWQ07 | ΔackA | pPaper | BWQ08 | 20 | 16 | 80% |
| KpQ00 | ΔyqhD::ldhA | pPaper | KpQ01 | 46 | 10 | 22% |
| KpQ01 | ΔdhaT::ldhA | pScissors | KpQ02 | 18 | 18 | 100% |
athe number of picked clones was 20 or less: 20 if the total number obtained was more than 20, or all the clones present, otherwise;
bcolony PCR was performed to screen for successful genome editing;
celectroporation was also tried to get more clones.
The successful rates were high, consistent with other CRISPR/Cas9 genome-editing studies performed in E. coli (25). When editing adhE based on BWQ04, less clones and lower positive rates were obtained, whereas editing of ackA on the same strain was as good as expected. This indicates that a lower editing efficiency is due to the genetic background of the strain or to the specific genes to be edited. When knocking out sucA, only eight clones were obtained after trying three times, and only a small clone was found to be positive. This may due to the low transformation efficiency of BWQ05 as transforming the same plasmid into BWQ00 can get plenty positive clones (Table 3). To get higher transformation efficiency, electroporation was tried and get plenty clones with high positive rate (Table 3).
After curing the last RPS editing plasmid and pCas of BWQ08, strain BWQ08-dNK was obtained. We used flask fermentation to compare strain BWQ08-dNK and the wild type (WT) in terms of growth, glucose utilization and pyruvate and acetic acid production (Figure 7). BWQ08-dNK grew slower, but utilized glucose more rapidly and produced more pyruvate (10.66 ± 0.13 g/l versus 0.65 ± 0.25 g/l in WT) whereas acetate production was lower (2.03 ± 0.48 g/l versus 3.72 ± 0.42 g/l in WT). These results show the effectiveness of the strain engineering.
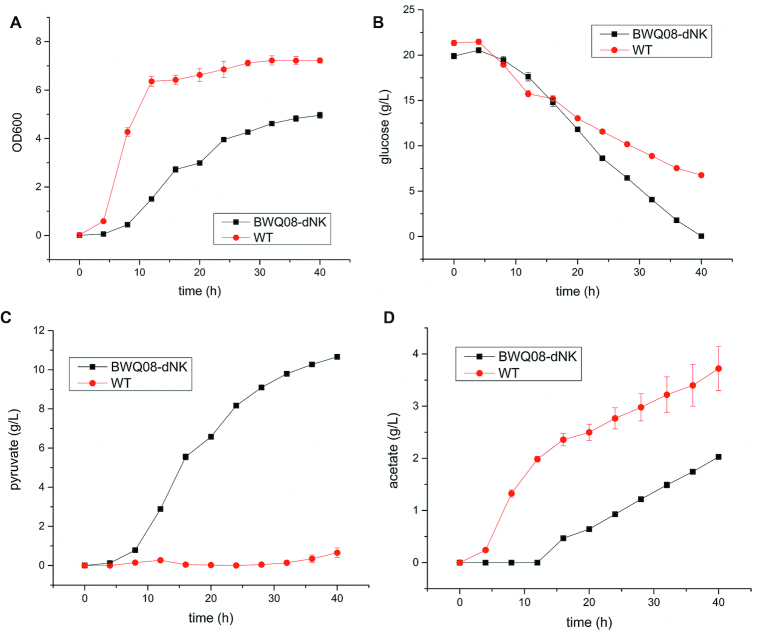
Fermentation results corresponding to BWQ08-dNK and WT strains. (A) Cell growth; (B) glucose utilization; (C) pyruvate production; (D) acetate production. All experiments were repeated three times and the error bars represent mean ± standard deviation.
Production may be further improved by fed batch fermentation. It may also be interesting to compare strains with similar gene editing based on different genetic backgrounds. Different construction orders may also be performed and different intermediate strains can be constructed and tested. As WT BW25113 seemed to produce acetate with high efficiency and TC36 is a high acetate producing strain based on W3310 (34), the equivalent strain BWQ06 based on BW25113 may be a better acetate producing strain.
RPS system also works in K. pneumoniae
To explore the scope of this RPS system, both gene replacement and the performance in another organism were studied. In our previous study, K. pneumoniae was engineered to produce optically pure d-lactate by overexpressing ldhA on a plasmid and knocking out yqhD and dhaT (49).
The curing efficiencies of pRPS-CkT, pRPS-GkC and pRPS-TkG by pRPS-GkC, pRPS-TkG and pRPS-CkT in K. pneumoniae were all 100% (Figure 8A, Supplementary Figure S6). Two copies of ldhA expressing cassette (2895 bp) amplified from pBAD18-ldhA (49) were successfully integrated into yqhD and dhaT locus with high efficiency (Figure 8B, Table 3). Comparing to KpWT, the constructed KpQ02-dNK produced more lactate (14.08 ± 0.52 g/l versus 4.34 ± 0.51 g/l) and less 1,3-PDO (0.17 ± 0.01 g/l versus 0.94 ± 0.12 g/l) (Figure 8C).
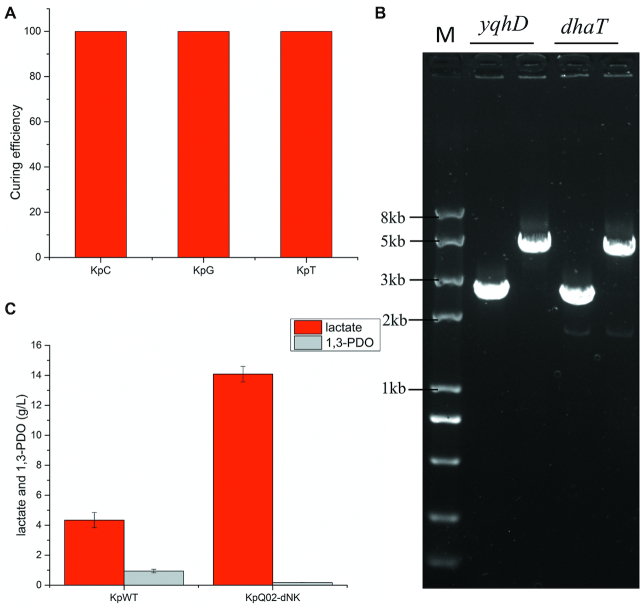
RPS system also works in K. pneumoniae. (A) The curing efficiencies of pRPS-CkT, pRPS-GkC and pRPS-TkG by pRPS-GkC, pRPS-TkG and pRPS-CkT respectively; (B) verification of gene editing of KpQ02-dNK. All genes checked are marked. The left lanes are colony PCR results using KpWT as templates. The right lanes are results using KpQ02-dNK as templates. M: DNA marker; (C) lactate and 1,3-PDO production. All experiments were repeated three times and the error bars represent mean ± standard deviation.
These data implied that RPS strategy worked well in K. pneumoniae. The RPS strategy may be general and can be used to any other organisms where three different selectable markers are available.
DISCUSSION
In this study, we report a fast and robust sequential genome-editing method based on a ‘Rock-Paper-Scissors’ (RPS) strategy. Three sgRNA plasmids were constructed with different antibiotic resistances, where each plasmid can either cure, or be cured by, one of the other plasmids (Figure 1A). When the genome is edited by these three plasmids in an RPS order, no separate plasmid curing or marker elimination steps are required. This is because the plasmids in the previous rounds of genome editing can be cured while current editing is performed. As mentioned above, strains with many edited genes and multiple combinations may be required to obtain a strain with the desired phenotype, and this combination may produce undesirable positive or negative epistatic interactions (5,16), the strains’ designing and engineering may be carried out by rounds of trial and error strategy. As a result, our fast and robust iterative genome editing strategy may contribute dramatically to this kind of researches.
The RPS-CRISPR/Cas9 system is the most robust iterative genome-editing strategy, as it does not require separate selection marker elimination or plasmid curing steps. Since plasmids from the previous round of genome editing are cured simultaneously with the current round of genome editing, single clones picked for successful genome editing are expected to be pure, with the previous plasmid already cured. The possibility of escape from plasmid curing is dramatically reduced by duplicating the targeted sequence on different strands, providing a curing efficiency of 99.99–100%. In addition to their presence being extremely unlikely, escapers cannot be transformed in the next round of genome editing. For example, when editing the genome with pPaper, the pRock plasmid used in the previous round of editing will be cured with high efficiency. Strains carrying escaped pRock cannot be transformed by pScissors in the next round of editing, since pScissors can be cured by the escaped pRock and the transformants cannot grow on the plate for selecting pScissors. In this supposed situation, pRock is of high copy while newly transformed pScissors is of single copy, therefore the second curing efficiency may be even higher. This double check mechanism is independent; the first curing targets the triple RPS-tags on pRock by the ‘for curing’ gRNA on pPaper, whereas the second curing would target the triple RPS-tags on pScissors by the ‘for curing’ gRNA on the - almost impossible - escaped pRock. As a result, escaping from these two highly efficient curing steps requires different mutations. All these mechanisms make this RPS strategy the most robust iterative genome-editing strategy, without separate selection marker elimination or plasmid curing steps.
This is also the fastest iterative genome-editing strategy, since only two times of cultivation are needed for one round of genome editing (Figures 1B and 3B): one cultivation on the plate after transformation to obtain a successfully engineered single clone, and another to grow the single clone for storage and next round of operations. This is probably the lowest possible number of cultivations for feasible iterative genome editing, since picking of single clones and growth are steps always needed in precise genome editing.
Multiplex automated genome engineering (MAGE) is a powerful method for genome editing and works by introducing mutations by short single strand DNA (36). However, a high throughput screening method is needed, or many clones need to be screened, to obtain a successfully engineered clone. MAGE is also inconvenient if intermediate strains need to be tested, and its efficiency is high only for small alterations. Additionally, although efficiency is increased by inactivation of the methyl-directed mismatch repair (MMR) system, this may raise the risk of spontaneous genome mutations.
In our method, since cultivation times are reduced to a minimum, spontaneous genome mutations are reduced. As mentioned above, when many rounds of genome editing are needed, it is crucial to reduce cultivation times to reduce spontaneous mutations which may cause unpredictable results.
Although it is not investigated in our research, this RPS-CRISPS/Cas9 genome-editing system may perform similarly with other CRISPR/Cas9 systems in other aspects, including efficient gene insertion, replacement, or deletion with various lengths (5,24–26). The RPS strategy provides an additional layer of CRISPR/Cas optimization and it can be combined with other Cas proteins like Cpf1 (37). Various strategies have been developed for multi genome editing in one round (5,25) and can also be combined with our strategy. However, multiple rounds of editing are still needed because the number of genes edited in one round is limited, and efficiency decreases dramatically with the number of genes edited (5,25).
The RPS editing plasmids may be combined with powerful automated systems (38,39). The Keio collection (40,41) has greatly facilitated the study of E. coli and single knock-out genes can be transferred or combined by P1 transduction (42). Donath et al. developed an automated platform for P1 phage transduction, and transducted 355 genetic markers from the Keio collection to five different strains (43). The design of our RPS editing plasmids (see text in Materials and Methods) can be easily completed by computational methods, and their construction may also be automated. The collection of all the RPS editing plasmids corresponding to all the genes of E. coli maybe more conveniently used to construct gene editing combinations than the Keio collection. The desired strains may be designed and constructed rapidly, by picking the RPS editing plasmids from the collection and using them in a ‘plug and play’ manner in the future. Such RPS editing plasmids collection would facilitate exploration of genotype space and debugging of our strain design.
In rare instances, some clones still can escape when curing pRock by pPaper. This system may be further improved by trying other antibiotic resistance combinations, and different spacers may be screened to get better performance.
The helper plasmid pCas (λ Red and Cas9 expressing plasmid) may mutate, as it is unchanged during the iterative rounds of genome editing. To reduce the mutation risk, this plasmid can also be integrated to the RPS editing plasmids, and editing can be accelerated by using 37°C, as reported previously (29). The system can be further simplified by using a non-homologous end-joining (NHEJ) mechanism, as introduced into E. coli (44–47), where RPS editing plasmids may be constructed without homologous arms where recombinase is not required. Robustness can also be improved by adding three visible markers to the three RPS plasmids, so that plasmid curing can be confirmed by the naked eye or with a microscope. Despite all the possible improvements, however, we find the RPS-CRISPR/Cas9 system shows good performance for the time being.
DATA AVAILABILITY
pRock, pPaper and pScissors have been deposited in open-source distributor MolecularCloud (www.molecularcloud.org) under the numbers: MC_0101139, MC_0101140 and MC_0101141. Other data supporting the findings of this study are available from the corresponding authors upon request.
ACKNOWLEDGEMENTS
We thank Haiyan Liu for providing plasmids from iGEM distributions, thank Sheng Yang for providing pCas, pTargetF and pMDIAI. We also thank Haiyan Yang for the help in HPLC experiments, thank Tao Cheng, Chao Xu and Xiutao Liu for technical support in fermentation experiments, and thank Ruicun Liu for drawing the symbols of Rock-Paper-Scissors, thank Shengsong Xie for providing the latest version of sgRNAcas9.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31961133014, 31722001 to G.Z., 21807101 to M.L.]; Defence Industrial Technology Development Program [JCKY2018130B005 to G.Z.]; Shandong University [to G.Z.]. Funding for open access charge: National Natural Science Foundation of China [31722001].
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.