 MethHC 2.0: information repository of DNA methylation and gene expression in human cancer
MethHC 2.0: information repository of DNA methylation and gene expression in human cancer
Nucleic Acids Research


The authors wish it to be known that, in their opinion, the first four authors should be regarded as Joint First Authors.

- Altmetric
DNA methylation is an important epigenetic regulator in gene expression and has several roles in cancer and disease progression. MethHC version 2.0 (MethHC 2.0) is an integrated and web-based resource focusing on the aberrant methylomes of human diseases, specifically cancer. This paper presents an updated implementation of MethHC 2.0 by incorporating additional DNA methylomes and transcriptomes from several public repositories, including 33 human cancers, over 50 118 microarray and RNA sequencing data from TCGA and GEO, and accumulating up to 3586 manually curated data from >7000 collected published literature with experimental evidence. MethHC 2.0 has also been equipped with enhanced data annotation functionality and a user-friendly web interface for data presentation, search, and visualization. Provided features include clinical-pathological data, mutation and copy number variation, multiplicity of information (gene regions, enhancer regions, and CGI regions), and circulating tumor DNA methylation profiles, available for research such as biomarker panel design, cancer comparison, diagnosis, prognosis, therapy study and identifying potential epigenetic biomarkers. MethHC 2.0 is now available at http://awi.cuhk.edu.cn/∼MethHC.
INTRODUCTION
DNA methylation is an epigenetic regulator of cell differentiation and development by manipulating gene expression without altering the genomic sequence. This epigenetic change is inheritable and reversible, thus making it a promising therapeutic target (1). Major research advances have furthered the understanding on DNA methylation and its numerous functions, establishment, maintenance and erasure (2). Epigenetics has several roles in fields, such as in viral infections, gene therapy in somatic cells and developmental abnormalities (3). However, the focus has been directed on tumor cells and their comparison with profiles of normal cells. Studies showed that DNA methylation is important in cancer initiation and development. Tumor-specific DNA methylations provide possible biomarkers for cancer diagnostics and monitoring (4).
Research has focused on abnormal DNA hypermethylation and hypomethylation of specific gene sites at promoters, enhancers, and gene bodies that contribute to tumor progression and cancer formation. DNA hypermethylation influences the gene expression at CpG rich promoter regions. These abnormalities can serve as potential biomarkers for various diseases. To date, major clinical programs mainly include diagnostic markers, prognostic markers, tailoring treatment, monitoring treatment efficacy, and epigenetically or genetically targeted therapies (5,6). Epigenetics studies on diseases include TP53 (7) and BRCA1 (8) hypermethylation in breast cancer, WIF-1 hypomethylation in non-small cell lung cancer (9), RGS2 and E-cadherin hypermethylation in prostate and liver cancer, respectively (10,11), and CPNE5 methylation as a biomarker in esophagus cancer (12). Cancer studies over the past 10 years have accumulated a vast amount of DNA methylation results and may contribute to tumor marker or diagnosis and therapy.
Experimental technologies, such as methylation-specific PCR (MSP), quantitative MSP (MethyLight), enzyme digestion-based methods (COBRA, MSRE), methylated DNA immunoprecipitation (MeDIP) and high-throughput microarray and sequencing methods (13) including pyrosequencing (bisulfite-treated DNA), whole-genome bisulfite sequencing, Illumina GoldenGate, and MassARRAY, have been used in the detection and confirmation of DNA methylation. The methods have evolved from gene-specific approaches to genome-wide array and next-generation sequencing (NGS) data to produce methylome data containing comprehensive information on DNA methylation events in human diseases (14).
Large amounts of methylation data and disease information have been collected, integrated, and made available from many sources such as The Cancer Genome Atlas (TCGA) project, Gene Expression Omnibus (GEO), and databases including iMETHYL (15), MethBank (16), DiseaseMeth (17,18), MethyCancer (19), MethDB (20), NGSmethDB (21,22), PubMeth (23) and MENT (24). Most of these sources are constantly updated, including our previously developed MethHC (25). iMETHYL is a multi-omics database that provides DNA methylation, whole genome, and whole transcriptome data for immune cells (15). MethBank 3.0 integrates DNA methylomes across various species with an update of data annotation, detailed methylomes of different developmental stages, and an interactive browser (16). DiseaseMeth has developed a 2.0 version that provides datasets for 88 human diseases in locus-specific and genome-wide form and allows the online automated identification of abnormal DNA methylation in human diseases (17,18). MethyCancer database contains genetic and genomic data in a graphical MethyView of DNA methylation, cancer-related genes and other cancer information specifically from public data sources and experimental sequencing data sets retrieved from the Cancer Epigenome Project in China (19). MethDB is a well-maintained database that unifies experimental data on several 5-methylcytosines (5mC) in DNA to the different methylation status of single nucleotides, especially cell response to modifications in the environment (20). In addition, data including differentially methylated single-cytosines, and genome regions of homogenous methylation (methylation segments), from various animals such as chimpanzees and mice, are integrated into the updated NGSMethDB 2017 (21,22). PubMeth is based on the combined text-mining of published literature and manual reading and expert annotation of preselected abstracts on Medline/PubMed (23). Finally, MENT is one of the initial databases providing data on DNA methylation and gene expression for different tumor tissues (24).
MicroRNAs are 19–24 nucleotide-long small non-coding RNAs that are frequently associated with cancer progression or causation through functions such as RNA silencing and post-transcriptional target gene expression regulator in a sequence-specific behavior. MicroRNA gene expression is important in malignant transformation during oncogenesis. For instance, miR-191, miR-25, miR-34c-5p and miR-34a are useful in determining the histological types of non-small cell lung cancer (NSCLC) (26). Aberrant DNA methylation silences microRNA genes in leukemia (27), liver cancer (28), cervical cancer (29), breast cancer (miR-9 family, miR-335) (30–32) and colorectal cancer (miR-124 family) (33,34). These findings indicate the important role of microRNA deregulation in cancer. DNA methylation and high-throughput approaches have been widely applied for the analysis of genome-wide DNA methylation and are useful in gathering the mRNA/microRNA expression information of normal and tumor tissues. However, no database has combined information on DNA methylation and gene expression including mRNA/microRNA expression. Therefore, MethHC (a DNA methylation and gene expression database for human cancer) was previously developed and is now updated to MethHC 2.0.
Koch et al. mentioned, there's a huge difference between 14 743 articles to 14 DNA methylation-based biomarkers commercially available and reasons are attributed to obstacles such as the complex relationship between DNA methylation and genomic location (35). The MethHC database previously focused on the aberrant methylomes of human cancer, including DNA methylation and gene expression, and consists of information on microRNA methylation, expression, and correlation from TCGA (25). Unlike previously, this paper presents MethHC version 2.0 database, which makes a qualitative leap from the previous version of DNA methylation repository. MethHC 2.0 includes data added from TCGA, GEO and a vast amount of manually curated information including genes/microRNAs, cancer, experimental cell types, experimental techniques, and corresponding methylation expression. MethHC 2.0 also provides clinical-pathological features, mutation and copy number variation, multiplicity of information (gene regions, enhancer regions and CGI regions), and circulating tumor DNA methylation profiles that are helpful in biomarker panel design, cancer comparison, diagnosis, prognosis and therapy study, gene set analysis, primer design, genomic methylation status, identifying novel tumor suppressor genes and potential epigenetic biomarkers. To date, MethHC 2.0 contains methylation data of 28 047 genes, over 1040 microRNAs, 50 118 array and RNA-seq data of 33 cancers, and curated up to 3586 experimental data related to DNA methylation in cancer.
SYSTEM OVERVIEW AND DATABASE CONTENT
On the whole, MethHC 2.0 still integrated two main parts including experimental data source (i.e. TCGA (36) and GEO (37)) and annotated resources (i.e. UCSC Genome Browser (38), and miRStart database (39)). We updated and collected new DNA methylation data from TCGA and GEO to update MethHC. TCGA analyzed the molecular characteristics of >20 000 primary cancers and normal samples from 33 cancer types and was established in 2006 by the joint effort of the National Cancer Institute and National Human Genome Institute. This database provides different genome-wide data including gene expression data, miRNA expression data, methylation data, mutation data, proteomic data and clinical data. GEO is an international database maintained by NCBI and was originally designed to collect and sort out various expression array data. It was later modified to contain various array-based data such as methylation array, lncRNA array, miRNA array, and even high-throughput sequencing data. In addition, circulating tumor DNA methylation profiles are also collected from GEO to enable cancer early diagnosis and prognosis prediction. In summary, MethHC 2.0 integrates 50 118 microarray and RNA sequencing data from TCGA and GEO. For each gene, the relationship between DNA methylation level and gene expression level is explored to investigate the role of DNA methylation in gene expression. Moreover, PubMed was searched, and >7000 articles related to DNA methylation-disease research published since 2010 were downloaded. Our curators continually extracted DNA methylation-cancer information including cancer types, sample types, validation techniques, and methylation sites and regions.
MethHC 2.0 offers the methylation or expression profiles in transcribed genes and microRNAs genes in 33 human cancers. UCSC Genome Browser and the miRStart database are applied to obtain transcription start sites (TSS) information of transcribed genes and microRNA genes. UCSC Genome Browser is a famous web-based viewer presenting all types of information related to the queried region on a genome with alignment annotations in one window (38). miRStart integrates data from cap analysis of gene expression (CAGE), TSS-Seq and H3K4me3 ChIP-Seq data sets to provide direct evidence on miRNA gene TSSs for miRNA-mediated regulatory study (39).
Given that epigenetic dysregulation outside the promoter region is also related to transcriptional changes, MethHC 2.0 investigates the relationship between DNA methylation levels at different regions and CpG islands and gene expression levels (40). Mounting evidence indicates that DNA methylation in the promoter is associated with gene expression decline and thus can be a therapeutic target for some human cancers to reactivate aberrantly silenced genes especially some tumor suppressor genes for example PTEN and Rb (41). However, methylation in the gene body promotes gene expression, but its function remains largely unknown. One theory is that DNA methylation in transcriptional regions can potentially silence functional elements, such as alternative promoters and retrotransposon elements, to maintain transcriptional efficiency (42). MethHC 2.0 offers the methylation level across gene regions (promoter, TSS1500, TSS200, 5′UTR, first exon, gene body, and 3′UTR), CpG islands/CPG island regions, shelves, shores and enhancer region. In addition, single-based DNA methylation site analysis in MethHC 2.0 provides the users with precise methylation site which can help users to further study the target gene.
MethHC 2.0 offers gene information by integrating the UCSC Genome Browser, miRStart, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (43), and Enhancer Atlas2.0. KEGG is important for researchers to integrate and interpret large-scale molecular data generated by genome sequencing and other high-throughput experimental technologies (43). KEGG has a powerful graphic function to introduce many metabolic pathways and their relationship. MethHC 2.0 enables visitors to choose a pathway of interest in KEGG and investigate differentially methylated genes in various cancer types. Enhancer Atlas2.0 archives 13 494 603 enhancers from human, mouse and fly analysed via twelve high-throughput analysis platforms, which enable users to conduct functional analysis of enhancers in different genomes (44). The epigenetic regulation of Super Enhancers (SEs) is a driver of cancer; however, their role in carcinogenesis is still largely unknown (45). Tissue-specific SEs and their target genes can be identified through gene expression and DNA methylation data in MethHC 2.0.
UPDATED DATABASE CONTENT AND STATISTICS
Figure 1 highlights the enhancements of MethHC 2.0. Owing to the importance of DNA methylation to organisms, many web-based DNA methylation data warehouses and functional analysis resources have been developed, including MethDB (20), PubMeth (23), MethyCancer, NGSMethDB (21,22), DiseaseMeth (17,18) and MENT (24). MethHC 2.0 is an online resource that centers on the aberrant methylomes of human cancer by integrating DNA methylation data, gene expression data, and microRNA expression data from TCGA and GEO. The data of MethHC 2.0 include 27 190 Illumina HumanMethylation450 BeadChip DNA methylation data, and 22 928 array or sequencing data for mRNA/microRNA expression in 33 human cancers. Table 1 shows the statistics of sample numbers of each cancer in MethHC 2.0 database. MethHC 2.0 contains 28 047 genes, >1040 miRNAs, 8 gene regions, 5 CGI regions and enhancer regions.

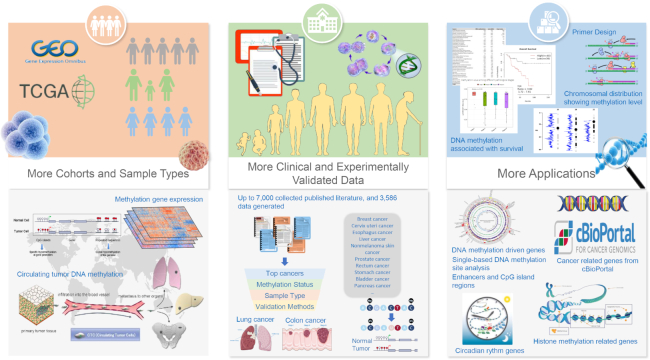
Highlighted enhancements of MethHC 2.0. As a collective and comprehensive expression profile database composed of DNA methylation and mRNA/microRNAs in 33 Homo sapiens tumors and matched normal tissues, this update contains data from GEO, and TCGA and accumulates up to 3586 manually curated data from >7000 collected published literature with experimental evidence.

| Cancer | DNA methylation | Expression | microRNA | CNV | SNV | Circulating |
|---|---|---|---|---|---|---|
| Acute Myeloid Leukemia | 636 | 188 | 238 | 194 | 134 | - |
| Adrenocortical Cancer | 149 | 80 | 79 | 90 | 92 | - |
| Bile Duct Cancer | 272 | 45 | 45 | 36 | 51 | - |
| Bladder Cancer | 700 | 432 | 430 | 413 | 412 | - |
| Breast Cancer | 6571 | 1558 | 1269 | 1104 | 986 | V |
| Cervical Cancer | 362 | 312 | 309 | 297 | 289 | - |
| Colon Cancer | 888 | 461 | 512 | 466 | 399 | - |
| Endometrioid Cancer | 482 | 575 | 583 | 544 | 529 | - |
| Esophageal Cancer | 513 | 198 | 173 | 185 | 184 | - |
| Glioblastoma | 1104 | 5 | 173 | 613 | 390 | - |
| Head and Neck Cancer | 799 | 569 | 546 | 524 | 506 | V |
| Kidney Chromophobe | 66 | 91 | 89 | 66 | 66 | - |
| Kidney Clear Cell Carcinoma | 483 | 592 | 607 | 536 | 336 | - |
| Kidney Papillary Cell Carcinoma | 366 | 326 | 321 | 289 | 281 | - |
| Large B-cell Lymphoma | 145 | 47 | 48 | 48 | 37 | - |
| Liver Cancer | 1646 | 425 | 424 | 378 | 364 | - |
| Lower Grade Glioma | 820 | 530 | 529 | 533 | 506 | - |
| Lung Adenocarcinoma | 968 | 564 | 585 | 531 | 561 | - |
| Lung Squamous Cell Carcinoma | 1020 | 523 | 550 | 503 | 491 | - |
| Melanoma | 475 | 452 | 472 | 472 | 467 | - |
| Mesothelioma | 87 | 87 | 86 | 87 | 80 | - |
| Ocular melanomas | 92 | 80 | 80 | 80 | 80 | - |
| Ovarian Cancer | 1845 | 854 | 379 | 601 | 436 | V |
| Pancreatic Cancer | 195 | 183 | 182 | 185 | 158 | - |
| Pheochromocytoma & Paraganglioma | 211 | 187 | 186 | 169 | 178 | - |
| Prostate Cancer | 867 | 551 | 551 | 502 | 484 | - |
| Rectal Cancer | 584 | 165 | 177 | 166 | 136 | - |
| Sarcoma | 2753 | 263 | 265 | 264 | 237 | - |
| Stomach Cancer | 661 | 477 | 407 | 440 | 433 | V |
| Testicular Cancer | 423 | 156 | 156 | 156 | 145 | - |
| Thymoma | 148 | 126 | 121 | 124 | 122 | - |
| Thyroid Cancer | 802 | 573 | 568 | 512 | 487 | - |
| Uterine Carcinosarcoma | 57 | 57 | 56 | 56 | 57 | - |
| Total | 27 190 | 11 732 | 11 196 | 11 164 | 10 114 |
Table 2 compares MethHC 2.0 with MethHC 1.0. MethHC 2.0 gathers DNA methylation and mRNA/microRNA expression data from 33 human tumor tissues and normal tissues and has been integrated with >50 118 array and RNA sequencing data from TCGA and GEO. In addition, circulating tumor DNA methylation profiles are collected for the early diagnosis, prognosis prediction of cancer. Circulating tumor DNA (ctDNA) is non-invasive, and provides real-time monitoring for cancer in patients and eliminates tumor heterogeneity in solid tumor sampling (46). Integrative analysis of DNA methylation and transcriptional expression has been used in many cancers because it is a cost-effective and reliable method based on multi-omics data to identify and decipher cancer biomarkers (47). Therefore, MethHC 2.0 adds methylation profiles and matches mRNA/miRNA expression profiles from GEO.
| MethHC 1.0 | MethHC 2.0 | |
|---|---|---|
| Publication | NAR Database Issue (2014) | This work for NAR 2021 Database Issue |
| Last update | 2014 | 2020 |
| Support species | Homo sapiens | Homo sapiens |
| Number of samples | 18 cancers | 33 cancers |
| TCGA | TCGA | |
| Methylation: 6548 microarray data, Gene expression: 12 567 RNA sequencing data | Methylation: 9736 microarray data, gene expression: 22 077 RNA sequencing data | |
| GEO | ||
| Methylation: 17 454 microarray, Gene expression: 851 RNA sequencing data | ||
| Number of methylation sites | 482 481 | 486 428 |
| Number of genes | 20 500 genes | 28 047 genes |
| 1040 microRNAs | >1040 microRNA | |
| Data sources | TCGA | TCGA, GEO |
| Experimentally Validated Data | NA | 3586 records |
| Method to build database | Data mining | Data mining |
| Manually collected and up to 3586 curated data | ||
| Correlation analysis | YES | YES |
| microRNA expression | YES | YES |
| Gene regions | 8 Gene regions+ | 8 Gene regions+ |
| 5 CpGIsland regions* | 5 CpG Island regions* | |
| 1 enhancer region | ||
| Other Characteristic | MicroRNA expression, Differential methylation, Correlation analysis | MicroRNA expression, circulating tumor DNA methylation profiles, clinical-pathological indicators from TCGA, gene set analysis, survival analysis, and primer design |
+Including promoter (from −1.5 to 0.5 kb of the transcription start site, TSS), TSS1500, TSS200, 5’UTR, first exon, gene body and 3’UTR gene region.
*Including N shelf, N shore, CpG Island, S shelf and S shore of CpG region.
To help researchers discover novel epigenetic biomarkers for cancer, MethHC 2.0 includes the following rich characteristics. (i) In addition to the newly added single-based DNA methylation site analysis, enhancers and CpG island regions are added for region-based DNA methylation site analysis. (ii) Gene sets analysis is added to our website, including DNA methylation-driven genes, histone methylation related genes, circadian rhythm genes, and cancer-related genes from cBioPortal, which is convenient for users to search for these important genes. (iii) Clinical-pathological features such as the pathological stage from TCGA are also incorporated to facilitate researchers to study the correlation between DNA methylation and tumor stage. We also added the analysis of tumors with or without the presence of mutation and tumors with different copy number variations in MethHC 2.0. Given that not all mutations cause gene dysfunction and lead to cancer, mutation analysis, and copy number variation analysis have great potential to improve the accuracy of cancer detection. MethHC 2.0 also enables users to analyze the survival data to evaluate the diagnosis and guide the therapy of cancer and (iv) MethHC 2.0 adds primer design function. When users identify a single-base DNA methylation site of interest, they can follow the primer design rules for methylation mapping experiments, such as MSP.
For this update, over 7000 research articles related to the methylation in cancer published since 2010 are downloaded from the PubMed database and manually curated to extract DNA methylation-cancer information with experimental evidence. 3586 experimental data related to methylation in cancer have been generated, most of which are related to 10 top cancer with most new cancer cases such as lung, breast, prostate, colon, non-melanoma of skin, stomach, liver, rectum, esophagus, and cervix uteri (48). Presence of primer sequences is also noted from these articles to accelerate cancer methylation research. MethHC 2.0 is greatly enhanced by these data because the methylation level in these articles is validated by experiment method for example MSP, pyrosequencing, bisulfite sequencing, and some enzyme digestion-based methods.
ENHANCED WEB INTERFACE
The web interface has been re-designed to facilitate the analysis of differentially methylated genes and regions among cancers as presented in Figure 2. Users can utilize gene methylation analysis to compare methylation among several cancer types, pathological stages and cancers with or without mutation or with different copy number variations for a given gene. The differentially methylated sites or regions, their chromosomal distribution, and their related genes can be identified in the differential methylation section. Hierarchical clustering is applied to identify cancer-specific co-methylation genes. MethHC 2.0 enables the survival analysis for a CpG or regions located in or around the proximity of a query gene. Curated DNA methylation knowledge base provides information on experimentally validated DNA methylation. These enhancements in web interface can promote MethHC 2.0 as a popular online resource in DNA methylation and cancer research.
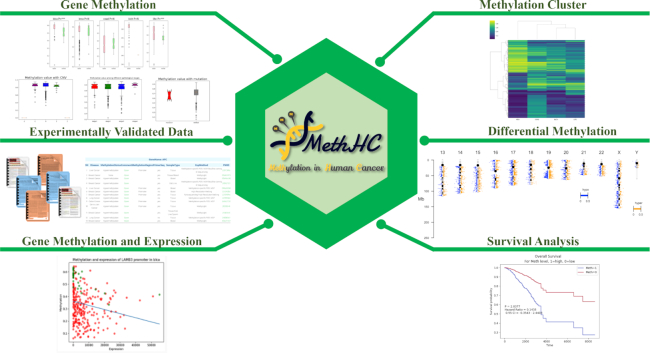
Enhanced web interface of MethHC 2.0. More comprehensive information related to methylation in cancer, such as differentially methylated CpGs/regions and their chromosomal distribution, clinical information, somatic mutation, expression, are provided on the web interface of MethHC 2.0.
SUMMARY AND PERSPECTIVES
More than 10 years ago, biomarkers based on DNA methylation were considered the next ‘big event’ in cancer research. However, the most promising targets in developing powerful biomarkers for diagnosis, prognosis, and disease occurrence have not met expectations. There's a huge difference between 14 743 articles to 14 DNA methylation-based biomarkers commercially available and reasons are attributed to methodological, experimental obstacles, and the complex relationship between DNA methylation and genomic location (35). The new version of the database, MethHC 2.0, can thoroughly evaluate biomarker performance based on DNA methylation and thus support accurate reports on discovery and verification in the future.
MethHC 2.0 is a collective and comprehensive expression profile database composed of DNA methylation and mRNA/microRNAs in 33 Homo sapiens tumors and matched normal tissues. Similar to the previous database, this version uses textual and graphical interfaces when visualizing methylation pattern comparison of normal and tumor tissues. Therefore, users can compare methylation among several cancer types, pathological stage and cancers with or without mutation or with different copy number variations for a given gene.
Previously, MethHC database has been cited and applied in many researches, promoter methylation and determining mechanisms of suppression as well as analysis of DNA methylation of CpG probes, gene expression in large amounts of tumor, and discovery of novel enhancers. Moreover, combined with the functions mentioned above, the prospective applications of the enhanced MethHC 2.0 database include: (i) clinical-pathological features such as tumor stages and survival data that can facilitate study of methylation correlation to stages and evaluation of diagnosis and cancer therapy; (ii) mutation analysis and copy number variation analysis that can potentially improve accuracy of cancer detection; (iii) multiplicity of information (gene regions and CGI regions) facilitating further investigation on genomic methylation status; (iv) identifying novel tumor suppressor genes and potential epigenetic biomarkers based on gene expression profiles; (v) presence of circulating tumor DNA methylation profiles, helping cancer research in diagnosis, prognosis, and therapy, such as non-invasive sample collection compared to surgery; (vi) identifying novel functions of previously known biomarkers in different cancer diagnostic panel through combining biomarker analysis from multiple sources and (vii) presence of gene list allowing visualization of DNA methylation, gene expression and comparison between different cancers. The integration of microRNA expression, circulating tumor DNA methylation profiles, and clinical-pathological indicators from TCGA can contribute to gene set analysis and primer design. These alterations and continuous updates will enhance DNA methylation-based marker performance, experimental reproducibility, clinical settings and reduce current research waste in this field.
DATA AVAILABILITY
The MethHC 2.0 database will be continuously maintained and updated. The database is now publicly accessible at http://awi.cuhk.edu.cn/∼MethHC.
FUNDING
Warshel Institute for Computational Biology funding from Shenzhen City and Longgang District; Ganghong Young Scholar Development Fund of Shenzhen Ganghong Group Co., Ltd.
Conflict of interest statement. None declared.
REFERENCES
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
42.
43.
44.
45.
46.
47.